

Abstract
Machado-Joseph disease (MJD) is a genetic neurodegenerative disease caused by an expanded polyglutamine tract within the protein ataxin-3 (ATXN3). Despite current efforts, MJD’s mechanism of pathogenesis remains unclear and no disease-modifying treatment is available. Therefore, in this study, we investigated (1) the role of the 3′ UTR of ATXN3, a putative microRNA (miRNA) target, (2) whether miRNA biogenesis and machinery are dysfunctional in MJD, and (3) which specific miRNAs target ATXN3-3′ UTR and whether they can alleviate MJD neuropathology in vivo. Our results demonstrate that endogenous miRNAs, by targeting sequences in the 3′ UTR, robustly reduce ATXN3 expression and aggregation in vitro and neurodegeneration and neuroinflammation in vivo. Importantly, we found an abnormal MJD-associated downregulation of genes involved in miRNA biogenesis and silencing activity. Finally, we identified three miRNAs—mir-9, mir-181a, and mir-494—that interact with the ATXN3-3′ UTR and whose expression is dysregulated in human MJD neurons and in other MJD cell and animal models. Furthermore, overexpression of these miRNAs in mice resulted in reduction of mutATXN3 levels, aggregate counts, and neuronal dysfunction. Altogether, these findings indicate that endogenous miRNAs and the 3′ UTR of ATXN3 play a crucial role in MJD pathogenesis and provide a promising opportunity for MJD treatment.
Keywords: Machado-Joseph disease, SCA3, microRNA, lentivirus, gene therapy, 3′, UTR, mechanism of disease, microRNA machinery dysregulation
Graphical Abstract
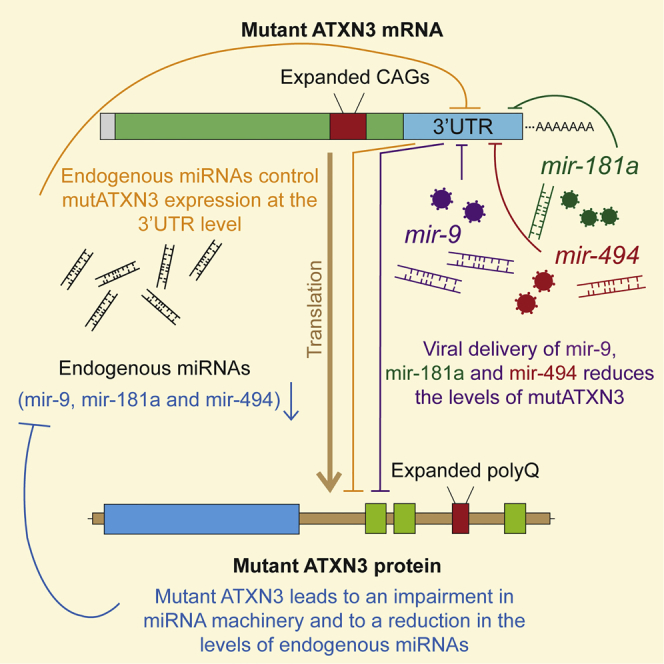
Machado-Joseph disease (MJD) is a fatal neurodegenerative disorder caused by a polyglutamine expansion in the ataxin-3 protein. In this study, Carmona and colleagues identified multiple microRNAs regulating ataxin-3 expression, which are also dysregulated in MJD. Brain delivery of these microRNAs may represent a promising therapeutic approach for MJD.
Introduction
Machado-Joseph disease (MJD), or spinocerebellar ataxia type 3, belongs to the group of polyglutamine (polyQ) diseases, dominantly inherited disorders caused by a similar mutation, an overrepetition of the trinucleotide CAG, which translates into overlong polyQ tracts in different proteins. In the case of MJD, this mutation occurs in the MJD1/ATXN3 gene. MJD is characterized by multiple clinical symptoms such as gait and limb ataxia, peripheral neuropathy, dystonia, and dysarthria, altogether leading to a progressive impairment of motor coordination.1, 2, 3
These symptoms result from severe neuronal dysfunction and neurodegeneration in selective brain regions such as the cerebellum, substantia nigra, and striatum.4 Although the exact pathological mechanisms underlying MJD are still unclear, it is accepted that the polyglutamine expansion in the ataxin-3 (ATXN3) protein results in a toxic gain-of-function, involving protein cleavage, oligomerization and aggregation, dysfunction of cellular quality control mechanisms, and transcriptional and translational dysregulation, among others.5, 6, 7, 8 Currently, there is no available treatment to cure or delay the progression of the disease.
Promising results were observed in animal models of MJD upon in vivo genetic silencing of mutant ataxin-3 (mutATXN3) using viral encoded engineered short-hairpin RNAs (shRNAs), artificial microRNAs (miRNAs), or systemically delivered siRNAs targeting mutATXN3 mRNA.9, 10, 11, 12, 13, 14 These studies took advantage of the siRNA/miRNA machinery and demonstrated that a reduction of mutATXN3 protein levels in MJD animal models reduces disease manifestations. Nevertheless, the contribution of the endogenous miRNA pathway to the regulation of ATXN3 levels and to MJD pathogenesis is still unclear.
Furthermore, despite having been extremely important for the understanding of different pathogenic mechanisms in MJD, most of the animal models that have been developed are based on the expression of only the coding region of human mutATXN3.15, 16, 17, 18, 19, 20, 21, 22 Therefore, further studies in other models are required in order to understand the regulatory role of the human ATXN3 mRNA non-coding regions, particularly the 3′ UTR, a known regulatory hub for miRNAs and RNA binding proteins.23
miRNAs are a class of endogenous small non-coding RNAs (containing about 22 nucleotides in size) that mediate post-transcriptional gene regulation of their targets, mostly through sequence complementarity to the 3′ UTR of mRNAs, negatively controlling their translation or causing mRNA degradation.24 Recent studies have shown that miRNAs are important players in neurodegenerative diseases and in particular in polyQ disorders.25, 26, 27, 28, 29
In the case of MJD, Bilen and colleagues identified a miRNA (bantam) as a modulator of mutATXN3 toxicity in Drosophila.30 Despite the fact that bantam is not conserved between Drosophila and mammals, this study together with more recent studies reporting miRNA dysregulation and ATXN3 targeting in in vitro and in vivo models and in human peripheral serum samples14, 31, 32, 33 are suggestive of an important role of the miRNA pathway in MJD.
Therefore, in this study, we evaluated whether the 3′ UTR of ATXN3 modifies disease progression, in vitro and in vivo, in a modified lentiviral model of MJD that includes the 3′ UTR non-coding region of human ATXN3. Moreover, we investigated whether miRNAs have a relevant physiological role in the regulation of mutATXN3 levels, their implication in the pathogenesis of MJD, and the in vivo therapeutic potential of specific miRNAs as silencing molecules for the reduction of mutATXN3 levels.
Our results suggest that the 3′ UTR of ATXN3 enables specific miRNAs to exert an unsuspectedly strong negative regulation of mutATXN3 levels, with crucial relevance in MJD neuropathology. Furthermore, our results suggest that impairments in the miRNA biosynthesis and machinery contribute to MJD pathogenesis. Importantly, reinstating a specific set of miRNAs targeting the 3′ UTR allows dramatic alleviation of MJD neuropathology.
Results
The 3′ UTR of ATXN3 Reduces Levels and Aggregation of mutATXN3
We previously developed a lentiviral vector system to induce the expression of mutAtxn3 in vitro and in vivo (LTR-SIN-PGK-mutAtxn3-LTR),17 a strategy that is highly effective for disease modeling.11, 17, 34 Therefore, to evaluate the role of the 3′ UTR of the human ATXN3, we inserted this sequence immediately downstream of the mutATXN3 coding sequence (LTR-SIN-PGK-mutATXN3-3′ UTR-LTR; Figures 1A and S1). Furthermore, based on previously published data,5, 35, 36, 37, 38, 39 in order to potentiate mutATXN3 aggregation in HEK293T cells, we generated two lentiviral vector constructs encoding a C-terminal fragment of mutAtxn3 with 72 glutamines, with (LTR-SIN-PGK-TmutATXN3-3′ UTR-LTR) and without the 3′ UTR (LTR-SIN-PGK-TmutATXN3-LTR), respectively (Figure S2). As expected, TmutATXN3 led to extensive aggregate formation in in vitro cells when compared to full-length mutATXN3 (Figure S3). This has been previously associated with increased nuclear translocation of C-terminal mutATXN3 fragments, which will then escape cytoplasmic quality control leading to their further accumulation and aggregation.38, 39 Moreover, direct expression of TmutATXN3 will bypass the requirement of the proteolytic cleavage of full-length mutATXN3, which has been described as essential for aggregate formation.37
Figure 1.

Inclusion of the 3′ UTR of Human Ataxin-3 Reduces Expression and Aggregation of Mutant Ataxin-3 in HEK293T Cells
(A) Schematic representation of the lentiviral constructs used for the evaluation of the role of ataxin-3 3′ UTR in vitro. Human ataxin-3 3′ UTR (1–3,434 bp) was cloned immediately downstream of the mutATXN3 (72Q) coding sequence. The truncated versions of mutATXN3 with and without the 3′ UTR were constructed in order to promote aggregation of mutATXN3 in vitro. (B) Western blot analysis of HEK293T transfected with equimolar amounts of TmutATXN3 and TmutATXN3-3′ UTR constructs showing aggregated (stacking gel) and soluble forms of TmutATXN3 (∼36 kDa). (C and D) Optical densitometry analysis of ATXN3 fractions normalized with actin showing a significant reduction in aggregated and soluble TmutATXN3 protein levels (n = 3). (E and F) Representative images from laser confocal microscopy of HEK293T transfected with equimolar amounts of TmutATXN3 and TmutATXN3-3′ UTR constructs. The analysis was performed by staining mutATXN3 (red), ubiquitin (green), and nuclei (blue). (F) Quantification of mutATXN3 aggregates per cell was normalized by nuclei counts, confirming western blot results (n = 4). (G) Dual-luciferase activity evaluation from HEK293T transfected with a control construct encoding for firefly luciferase and renilla luciferase (FLuc) or a construct encoding for firefly luciferase bound to ATXN3 3′ UTR and renilla luciferase (FLuc-3′ UTR). The quantitative analysis presented as firefly/renilla ratio (FL/RL) relative to control shows a reduction in FLuc-3′ UTR luminescence activity (n = 4). (H) MutATXN3 mRNA stability assay from Neuro2A cells stably expressing mutATXN3 or mutATXN3-3′ UTR after treatment with actinomycin D for 0, 2, and 4 hr. The qPCR analysis for mutATXN3 mRNA levels normalized with endogenous control (18S) and relative to t = 0 showing a faster decay rate for mutATXN3-3′ UTR mRNA (n = 4 for each time point) is shown. The statistical significance was evaluated with paired Student’s t test (C–G) and unpaired Student’s t test (H) (*p < 0.05 and **p < 0.01). The data are expressed as mean ± SEM. The scale bars represent 5 μm.
HEK293T cells were transfected with equimolar amounts of TmutATXN3 or TmutATXN3-3′ UTR plasmid DNA. Immunoblotting of total protein extracts revealed, upon incorporation of the 3′ UTR, a striking reduction not only in aggregate levels (Figures 1B and 1C; TmutATXN3: 2,063 ± 0.1455 versus TmutATXN3-3′ UTR: 0.8594 ± 0.2843), but also in soluble TmutATXN3 (Figures 1B and 1D; TmutATXN3: 1.917 ± 0.1906 versus TmutATXN3-3′ UTR: 1.181 ± 0.3388). Confocal microscopy analysis confirmed this result showing a marked decrease in the number of ubiquitinated TmutATXN3 inclusions numbers upon inclusion of ATXN3 3′ UTR (Figures 1E and 1F; TmutATXN3: 32.10 ± 5.147 aggregates/100 cells versus TmutATXN3-3′ UTR: 6.426 ± 0.3043 aggregates/100 cells).
In order to evaluate whether the effect of the ATXN3 3′ UTR was linked exclusively to the ATXN3 coding sequence (CDS), HEK293T cells were transfected with a construct encoding for firefly luciferase (FLuc-CTRL) or firefly luciferase bound to the ATXN3 3′ UTR (FLuc-3′ UTR). As expected, the 3′ UTR induced a reduction in the expression of a luciferase reporter gene, even in the absence of ATXN3 CDS, suggesting the involvement of independent endogenous regulators. Moreover, internal normalization with Renilla luciferase control gene confirmed that the observed results were not due to differences in transfection efficiencies (Figure 1G; FLuc: 100.0 ± 4.95 versus FLuc-3′ UTR: 77.80 ± 4.70).
Taking into account that both constructions have the same coding sequence, leading to the expression of the same protein, these results led us to hypothesize that the ATXN3 3′ UTR was promoting a reduction in the expression of mutATXN3, either by promoting mRNA decay or through translational inhibition. To investigate whether the inclusion of the ATXN3 3′ UTR changed mutATXN3 mRNA stability, Neuro2A cells stably expressing either mutATXN3 or mutATXN3-3′ UTR were treated with actinomycin D (ActD) to block transcription. As shown in Figure 1H, there was a significantly faster decay of mutATXN3-3′ UTR mRNA compared to mutATXN3, which persisted until 4 hr post-treatment with ActD (Figure 1H; mutATXN3: 0.57 ± 0.018 versus mutATXN3-3′ UTR: 0.46 ± 0.04).
Overall, these data indicate that mutant ataxin-3 mRNA and protein levels are strongly negatively regulated by its 3′ UTR.
Genetic and Pharmacologic Blockage of miRNA Biogenesis Prevents ATXN3 3′ UTR Regulatory Effects
Given that miRNAs are among the most relevant class of molecules capable of inducing mRNA degradation and inhibition of translation, we next assessed whether they could mediate ATXN3 expression regulation through its 3′ UTR. We simultaneously silenced two key enzymes for the biogenesis of miRNAs, Dicer and Drosha, to deplete cells from endogenous miRNAs. At 48 hr after co-transfecting TmutATXN3-3′ UTR and either Dicer and Drosha shRNA-encoding vectors or two control shRNA-encoding vectors (Figures 2A and 2B), we observed, in the knockdown condition, a significant decrease in the levels of both Drosha and Dicer mRNA levels (Figure 2C; hDrosha shCTRL: 1.00 ± 0.05 versus hDrosha shDrosha/Dicer: 0.50 ± 0.01 and hDicer shCTRL: 1.02 ± 0.16 versus hDicer shDrosha/Dicer: 0.38 ± 0.03). Simultaneously, we observed a significant increase in TmutATXN3 mRNA (Figure 2D; shCTRL: 1.00 ± 0.01 versus shDicer/Drosha: 1.15 ± 0.02) and protein levels (Figure 2E; aggregated TmutATXN3 shCTRL: 1.00 ± 0.19 versus aggregated TmutATXN3 shDicer/Drosha: 1.43 ± 0.32 and soluble TmutATXN3 shCTRL: 1.00 ± 0.03 versus soluble shDicer/Drosha: 1.72 ± 0.21). The comparatively small increase of 15% in TmutATXN3 mRNA relative to the robust increase of 72% in soluble protein levels upon Dicer/Drosha inhibition suggest a dominant effect of miRNAs on TmutATXN3 3′ UTR through translational inhibition and to a lesser extent through mRNA degradation, although implicating both mechanisms.
Figure 2.

Genetic and Pharmacological Blockage of miRNA Biogenesis Blocks ATXN3 3′ UTR Regulatory Effects
(A) Schematic representation of the lentiviral constructs used for the modulation of endogenous hDrosha and hDicer through H1 mediated expression of shRNAs targeting hDrosha and hDicer. The GFP or lacZ expression cassettes were inserted in order to follow gene expression in both targeting shRNAs and negative shRNAs (predicted not to target any known human or mouse gene). (B) HEK293T co-transfected with TmutATXN3 and either shRNAs against hDrosha and hDicer or neg shRNAs are efficiently co-transfected as can be observed via reporter gene expression of GFP and lacZ. (C) qPCR analysis of endogenous hDrosha and hDicer mRNA levels 48 hr after co-transfection with shRNAs against hDrosha and hDicer compared to control showing efficient gene knockdown (n = 3). (D) qPCR analysis of TmutATXN3 mRNA levels after hDicer and hDrosha knockdown displays a significant increase in TmutATXN3 mRNA levels (n = 3). (E) Western blot evaluation of TmutATXN3 protein levels (n = 6) in the knockdown conditions when compared to control. The TmutATXN3 aggregated and soluble levels were increased after hDrosha/hDicer knockdown. (F) Western blot analysis of TmutATXN3 protein levels in HEK293T cells stably expressing TmutATXN3-3′ UTR. The cells were maintained during 48 hr in complete DMEM as the control condition or in complete DMEM containing kanamycin at a final concentration of 100 nM (n = 7). The TmutATXN3 protein levels were significantly increased in kanamycin treated cells. The qPCR analysis was normalized with endogenous control (ACTB). The statistical significance was evaluated with paired Student’s t test (*p < 0.05 and **p < 0.01). The data are expressed as mean ± SEM.
We next took advantage of the previously described properties of aminoglycoside antibiotics, reported to inhibit Dicer-mediated pre-miRNA processing to pharmacologically reproduce the previous results.40, 41 In accordance with the genetic silencing results, Kanamycin A increased TmutATXN3 protein levels in cells stably expressing TmutATXN3-3′ UTR, as compared to PBS (vehicle) treated cells. (Figure 2F; vehicle: 1.00 ± 0.07 versus Kanamycin A: 1.26 ± 0.13).
Altogether, these results confirm an important role for endogenous miRNAs in controlling TmutATXN3 expression through interaction with its 3′ UTR.
The 3′ UTR Reduces ATXN3 Aggregation and Neuropathology in a Lentiviral-Based Mouse Model of MJD
Even though our in vitro results suggest an important role for the 3′ UTR of ATXN3 enabled through the action of endogenous miRNAs, they were performed in cell models. As endogenous miRNA profiles can vary between different cell types and tissues,42 and taking into account the neurodegenerative profile of MJD, we then addressed whether these results occurred in an in vivo context. For this, we employed our previously characterized lentiviral-based mouse model of MJD.6, 17 In this experiment, lentiviral vectors encoding for mutATXN3 or mutATXN3-3′ UTR (Figure 3A) were stereotaxically injected in each hemisphere of the striatum of 5-week-old mice (Figure 3A) that were later sacrificed at 5 weeks post-injection. Importantly, no differences were observed between viral productions regarding their transduction efficiencies after infection of HEK293T cells with equal p24 titers (Figure 3B; LV-mutATXN3: 1.001 ± 0.029 versus LV-mutATXN3-3′ UTR: 1.035 ± 0.1295).
Figure 3.

The 3′ UTR of ataxin-3 Reduces mutATXN3 Inclusions and Associated Neuronal Dysfunction and Neuroinflammation in a Lentiviral Mouse Model of Machado-Joseph Disease
(A) Schematic representation of the lentiviral vectors used for the production of lentivirus used in the evaluation of the role of ATXN3 3′ UTR in vivo and schematic representation of mice stereotaxic procedure. Lentiviral particles encoding for mutATXN3 and mutATXN3-3′ UTR were injected bilaterally in the striatum of 5-week-old C57/BL6 mice at the following coordinates (x: +0.6; y: ±1.8; and z: −3.3). (B) qPCR analysis of gDNA transduction rates of both LV-mutATXN3 and LV-mutATXN3-3′ UTR after infection of HEK293T cells (n = 3). (C–E) Immunohistochemical peroxidase staining using anti-Ataxin3 antibody (1H9 ab), 5 weeks post injection. The control mutATXN3 injected animals displayed a large number of mutant ataxin-3 inclusions (C), which were significantly decreased in the mutATXN3-3′ UTR transduced striatum (D), as quantified in (E) (n = 4). (F–H) Immunohistochemical analysis using an anti-DARPP-32 antibody and lesion identification. The mutATXN3-3′ UTR injected hemisphere displayed a marked reduction in DARPP-32 depleted volume as quantified in (H) (n = 4). (I–K) Fluorescent immunohistochemical analysis of IBA-1 immunoreactivity showing a decrease in mutATXN3-3′ UTR injected animals as quantified in (K) (n = 4). qPCR analysis was normalized with endogenous control (Albumin). The statistical significance was evaluated with paired Student’s t test (**p < 0.01 and ***p < 0.001, n = 4). The data are expressed as mean ± SEM. The scale bars represent 40 μm (C and D) and 200 μm (F, G, I, and J).
We observed that the inclusion of the ATXN3 3′ UTR drastically reduced the number of mutATXN3 inclusions when compared to the control hemisphere (Figures 3C–3E; LV-mutATXN3: 45,914 ± 2,084 aggregates versus LV-mutATXN3-3′ UTR: 5,234 ± 306 aggregates).
This model is also characterized by early neuronal dysfunction as shown by depletion of the marker dopamine and cyclic adenosine monophosphate-regulated phosphoprotein (DARPP-32).6, 17, 34 In accordance with the observed reduction of mutATXN3 inclusions, immunostaining for DARPP-32 revealed a proportionally much smaller area of depleted staining (Figures 3F–3H; LV-mutATXN3: 0.382 ± 0.041 mm3 versus LV-mutATXN3-3′ UTR: 0.031 ± 0.007 mm3). Moreover, ionized calcium-binding adaptor molecule 1 (Iba-1) immunoreactivity which, as previously reported,43 was strongly exacerbated in the hemisphere injected with LV-mutATXN3, was reduced in the hemisphere injected with LV-mutATXN3-3′ UTR (Figure 3I–3K; LV-mutATXN3: 1.382 ± 0.013 versus LV-mutATXN3-3′ UTR: 1.160 ± 0.030).
These results indicate that, similar to the previously presented in vitro data, the 3′ UTR of ATXN3 controls the expression of ATXN3 itself in vivo, resulting in a massive decrease of mutATXN3 inclusions, associated neurodegeneration, and neuroinflammation.
Transcriptional Dysregulation of miRNAs Predicted to Target ATXN3 3′ UTR and of RISC Associated Genes
Building on the hypothesis that miRNAs modulate ATXN3 mRNA levels through binding sequences in the 3′ UTR, we screened target prediction databases to identify miRNA candidates.44, 45, 46, 47 After identification of a first set of candidates, this information was crossed with expression data of miRNAs in the brain to pick those with potential relevance for MJD pathogenesis.48
Three distinct miRNAs (hsa-mir-9-5p, MIMAT0000441; hsa-mir-181a-5p, MIMAT0000256; and hsa-mir-494-3p, MIMAT0002816) were identified as conserved miRNAs targeting the ATXN3 3′ UTR, with simultaneously high expression in brain (Figure 4A; Table S1). qPCR analysis of mature miRNA levels in post-mortem samples from the dentate nucleus of a small number of patients with different age and disease progression stages revealed a tendency for decreased miRNA expression in MJD patient samples (Figure 4B; mir-9 CTRL: 1.138 ± 0.306 versus mir-9 MJD: 0.736 ± 0.129; mir-181a CTRL: 1.118 ± 0.262 versus mir-181a MJD: 0.669 ± 0.116; and mir-494 CTRL: 1.069 ± 0.181 versus mir-494 MJD: 0.816 ± 0.067), although not statistically significant given the small number of patients.
Figure 4.

Transcriptional Dysregulation of miRNAs Targeting ATXN3 3′ UTR and of RISC-Associated Genes
(A) Schematic representation of hATXN3 3′ UTR with respective indication of predicted miRNA binding sites for selected miRNAs (mir-9, mir-181a, and mir-494). (B) miRNA qPCR quantification of mir-9, mir-181a, and mir-494 performed in post-mortem human samples from the dentate nuclei of control (CTRL) or MJD patients (n = 6/5) displaying a general downregulation profile for all miRNAs. (C) miRNA qPCR quantification of mir-9, mir-181a, and mir-494 performed in RNA extracted from SH-SY5Y cells stably expressing WtATXN3 (WT) or mutATXN3 (MUT) (n = 6). The Mir-494 was found to be significantly reduced in SH-SY5Y stably expressing mutATXN3. (D) Immunostaining of differentiated cultures of human neurons derived from diseased (MJD) or control (CTRL) patient neurospheres. (E) miRNA qPCR quantification of mir-9, mir-181a, and mir-494 performed in neurons derived from human neurospheres (n = 4/5). (F) miRNA qPCR quantification of mir-9, mir-181a, and mir-494 performed in cerebellar tissue from MJD transgenic mice (TG) or littermate controls (WT) with 8 weeks of age (n = 8/14). (G and H) Expression profiling of genes involved in the biogenesis and function of miRNAs (DGCR8, FMR1, Drosha, Dicer, DDX6, Ago2, and TARBP2) in cerebellum samples from MJD transgenic mice (TG) or littermate controls (WT) with 8 weeks of age (n = 5). A general downregulation profile for most of the evaluated genes was observed. qPCR analysis for miRNA was normalized with endogenous control mir-103-3p (B and C) or snRNAU6 (E and F). qPCR analysis for mRNA was normalized with endogenous control (GAPDH). The statistical significance was evaluated with unpaired Student’s t test (*p < 0.05, **p < 0.01, and ***p < 0.001). The data are expressed as mean ± SEM. The scale bars represent 40 μm.
To corroborate these results, we screened miRNA levels in in vitro and in vivo models of MJD. Total RNA was isolated from SH-SY5Y human neuroblastoma cells stably expressing either wild-type ataxin-3 with a 27 glutamines polyQ stretch (wtATXN3) or mutATXN3. miRNA levels were evaluated by qPCR and, as observed in the human brain samples, a tendency for decreased mir-9 and mir-181a expression was observed in cells expressing mutATXN3 compared with wtATXN3 (Figure 4C; mir-9 WT: 1.256 ± 0.427 versus mir-9 Mut: 0.812 ± 0.051 and mir-181a WT: 1.122 ± 0.257 versus mir-181a Mut: 0.825 ± 0.158). Moreover, the levels of mir-494 were significantly downregulated in SH-SY5Y cells expressing mutATXN3 (Figure 4C; mir-494 WT: 1.056 ± 0.141 versus mir-494 Mut: 0.509 ± 0.183).
In order to screen another in vitro model more relatable to the human condition, we successfully obtained neurons differentiated from human induced pluripotent stem cells (hiPSCs) derived from both control (CTRL) and MJD patient fibroblasts. Following 18 days of neurosphere differentiation, both cell lines generated a high fraction of β-III tubulin positive neurons (Figure 4D). miRNA profiling from these cells revealed a significant downregulation in the levels of mir-181a and mir-494 (Figure 4E; mir-181a CTRL: 1.016 ± 0.086 versus mir-181a MJD: 0.705 ± 0.041 and mir-494 CTRL: 1.022 ± 0.112 versus mir-494 MJD: 0.599 ± 0.077), whereas no significant differences were obtained for mir-9 (Figure 4E; mir-9 CTRL: 1.057 ± 0.170 versus mir-9 MJD: 0.917 ± 0.198).
In order to investigate if the levels of mir-9, mir-181a, and mir-494 were also dysregulated in an in vivo model of MJD, we took advantage of a previously developed MJD transgenic mouse model.19 This model is characterized by early pronounced ataxic motor behavior since the third week of age, which is accompanied by general cerebellar atrophy. Therefore, this model may be correlated with a late stage disease scenario of MJD patients, where motor impairments and cerebellar atrophy are also severe. Analysis of the miRNA profile in samples from transgenic MJD mice cerebella showed a significant and robust decrease in the levels of all miRNAs (Figure 4F; mir-9 WT: 1.051 ± 0.125 versus mir-9 TG: 0.4566 ± 0.042; mir-181a WT: 1.077 ± 0.150 versus mir-181a TG: 0.2345 ± 0.031; and mir-494 WT: 1.039 ± 0.106 versus mir-494 TG: 0.3606 ± 0.044). Taken together, these results indicate a general downregulation profile for mir-9, mir-181a, and mir-494 in human samples and in MJD in vitro and in vivo models.
Given the observed miRNA expression changes, we hypothesized that this dysregulation could be associated with an impairment in the expression of genes coding for components of the miRNA biogenesis pathway. Gene expression analysis from samples of transgenic MJD mice revealed a significant downregulation of genes implicated in miRNA biogenesis, such as DGCR8, Dicer, and FMR1 (Figure 4G; DGCR8 WT: 1.013 ± 0.082 versus DGCR8 TG: 0.602 ± 0.040; Dicer WT: 1.037 ± 0.051 versus Dicer TG: 0.891 ± 0.020; and FMR1 WT: 1.003 ± 0.039 versus FMR1 TG: 0.716 ± 0.052), whereas no significant differences were observed for Drosha levels (Figure 4G; Drosha WT: 1.004 ± 0.042 versus Drosha TG: 0.943 ± 0.013). We further evaluated the expression levels of genes involved in miRNA silencing machinery and found a significant downregulation of mRNA levels for Ago2, TARBP2, and DDX6 (Figure 4H; Ago2 WT: 1.006 ± 0.053 versus Ago2 TG: 0.727 ± 0.016; TARBP2 WT: 1.010 ± 0.067 versus TARBP2 TG: 0.637 ± 0.038; and DDX6 WT: 1.003 ± 0.036 versus DDX6 TG: 0.735 ± 0.036). Importantly, mRNA downregulation was also corroborated at the protein level as was observed for DGCR8 (Figure S4A; WT: 1.000 ± 0.1333 versus TG 0.5509 ± 0.0861) and Ago2 (Figure S4B; WT: 1.000 ± 0.0498 versus TG: 0.6285 ± 0.1021).
All in all, these results suggest not only an impairment in the miRNA biogenesis pathway, which could be related with the observed miRNA dysregulation, but also an impairment in miRNA function, which might contribute for a disturbance in normal post-transcriptional regulatory events.
ATXN3 Expression Is Regulated by a Specific Set of miRNAs
To evaluate whether mir-9, mir-181a, and mir-494 regulate ATXN3 expression, we first developed a lentiviral vector system for the overexpression of these candidate miRNAs in vitro and in vivo (Figure 5A).
Figure 5.

Human ATXN3 Expression Is Regulated by mir-9, mir-181a, and mir-494
(A) Schematic representation of the lentiviral vector system used for overexpression of candidate miRNAs. The endogenous genomic sequences encoding for pre-miRNA sequences plus approximately 400 bp of flanking sequences were cloned downstream of an H1 promoter. The mir-Neg sequence was used as control. Simultaneously, the reporter gene expression was mediated by PGK controlled GFP expression. (B) Validation of miRNA overexpression. qPCR for mir-9, mir-181a, and mir-494 in transfected HEK293T cells confirming efficient overexpression for each miRNA (n = 4). (C) MutAtxn3 mRNA levels in HEK293T co-transfected with miRNA constructs and mutATXN3-3′ UTR (n = 4). Mir-181a and mir-494 significantly reduced the mutATXN3 mRNA levels. (D) Western blot analysis of HEK293T cells transfected with mutATXN3-3′ UTR and different miRNA constructs. The optical densitometry analysis of mutATXN3 protein levels was normalized with actin. The results are expressed as ratio ataxin-3/actin relative to mir-neg control (n = 3). All miRNAs significantly reduced mutATXN3 protein levels. (E) Validation of direct interaction between miRNAs and ATXN3 3′ UTR using a dual luciferase assay. Luminescence activity from HEK293T cells co-transfected with a dual luciferase construct containing ATXN3 3′ UTR or a control luciferase vector and miRNA/mir-neg encoding constructs is shown. The results are expressed as a ratio of firefly luminescence/Renilla luminescence (FL/RL) relative to control (LucCTRL) (n = 4). All miRNAs specifically reduced Luc-3′ UTR activity when compared to mir-neg. The qPCR analysis for mRNA was normalized with endogenous control (ACTB). The qPCR analysis for miRNA was normalized with endogenous control (snRNAU6). The statistical significance was evaluated with unpaired Student’s t test (B) or one-way ANOVA comparing all miRNAs to mir-neg (C–E) (*p < 0.05 and **p < 0.01). The data are expressed as mean ± SEM.
After co-transfection of these miRNA-encoding vectors together with mutATXN3-3’ UTR in HEK293T cells, we could successfully validate a robust overexpression of all miRNAs of interest when compared to control (Figure 5B). Moreover, qPCR analysis of mutATXN3 mRNA levels showed a significant downregulation in mutATXN3 expression for both mir-181a and mir-494 transfected cells when compared to mir-neg (Figure 5C; mir-neg: 1.001 ± 0.018 versus mir-181a: 0.626 ± 0.051 versus mir-494: 0.701 ± 0.023). However, no difference was observed in cells co-transfected with mutATXN3 and mir-9 when compared to control. Analysis of mutATXN3 at the protein level revealed similar results, but with a significant downregulation of mutATXN3 protein levels for all miRNAs including mir-9 (Figure 5D; mir-neg: 1.000 ± 0.005 versus mir-9: 0.674 ± 0.041 versus mir-181a: 0.741 ± 0.092 versus mir-494: 0.569 ± 0.008).
These results suggest that, in vitro, mir-181a and mir-494 downregulate mutATXN3 expression either by inducing the degradation of its mRNA or by inhibiting its translation, while mir-9 acts mostly through translational inhibition.
Nevertheless, it is known that miRNAs can modulate the expression of multiple genes at the same time. Therefore, a dual luciferase assay was performed to confirm that mir-9, mir-181a, and mir-494 directly bind to ATXN3 3′ UTR in order to downregulate its expression. All candidate miRNAs significantly reduced luciferase activity when co-transfected with a vector encoding for a luciferase gene bound to ATXN3 3′ UTR (Figure 5E; Luc-3′ UTR: mir-neg: 104.0 ± 2.400 versus mir-9: 80.00 ± 4.864, mir-181a: 83.56 ± 2.245, and mir-494: 79.98 ± 2.137), but not when a control vector was used (Figure 5E; Luc-CTRL: mir-neg: 97.908 ± 4.121 versus mir-9: 97.447 ± 4.437, mir-181a: 123.055 ± 3.733, and mir-494: 103.670 ± 1.040.
Altogether, these results indicate that ATXN3 3′ UTR is a validated target of mir-9, mir-181a, and mir-494, and genetic modulation of these particular miRNAs can be seen as a potential tool for the manipulation of mutATXN3 levels in the disease.
In Vivo Overexpression of mir-9, mir-181a, and mir-494 Reduces mutATXN3-3′ UTR Expression and Associated Neuropathology
In order to evaluate the therapeutic potential of mir-9, mir-181a, and mir-494 in vivo, we stereotaxically co-injected lentiviral vectors encoding for mutATXN3-3′ UTR with either mir-neg or each miRNA in the striatum of 5-week-old mice (Figure 6A).
Figure 6.

In Vivo Overexpression of mir-9, mir-181a, and mir-494 Reduce mutATXN3 Levels and Associated Neuropathology
(A) Schematic representation of the stereotaxic procedure. The lentiviruses encoding for mutATXN3-3′ UTR were injected bilaterally in the striatum of 5-week-old C57/BL6 mice. Simultaneously, lentiviruses encoding for mir-9, mir-181a, or mir-494 were co-injected with mutATXN3, while mir-Neg was co-injected as control. (B) Lentiviruses encoding for interest miRNAs efficiently mediate in vivo transduction of mouse striatum, as can be seen through reporter expression of GFP. (C) qPCR analysis confirmed miRNA overexpression levels for each miRNA in injected striatum tissue compared to control. (D, F, H, J, and L) Immunohistochemical peroxidase staining using anti-Ataxin3 antibody (1H9 ab), 5 weeks post injection. The control mutATXN3-3′ UTR/LV-mir-Neg injected animals displayed a large number of mutant ataxin-3 inclusions (D), which were significantly decreased after co-injection with any of the study miRNAs (F, H, and J) as quantified in (L) (n = 4–5). (E, G, I, K, and M) Immunohistochemical analysis using an anti-DARPP-32 antibody and lesion identification. The MutATXN3-3′ UTR/LV-mir-Neg injected hemisphere displayed a higher depletion in DARPP-32 volume when compared to the miRNA injected hemispheres (G, I, and K), as quantified in (M) (n = 5). (N) MutATXN3 mRNA levels quantification after miRNA co-injection. LV-mir-9, mir-181a, and mir-494, significantly reduced the levels of mutATXN3 in vivo (n = 4–5). (O) Evaluation of endogenous mouse ataxin-3 mRNA levels in injected mice. The LV-mir-9 and LV-mir-181a significantly reduced mouse ataxin-3 in vivo (n = 4–5). (P) Schematic representation of mouse ATXN3 3′ UTR displaying predicted mir-9, mir-181a, and mir-494 binding sites. The qPCR analysis for mRNA was normalized with endogenous control (18S). The qPCR analysis for miRNA was normalized with endogenous control (snRNAU6). The statistical significance was evaluated with one-way ANOVA comparing all miRNAs to mir-neg (L–O) (*p < 0.05 and **p < 0.01). The data are expressed as mean ± SEM. The scale bars represent 200 μm (B, E, G, I, and K) and 50 μm (D, F, H, and J).
Lentiviral vectors encoding for the miRNAs of interest and EGFP, efficiently transduced a large area of the striatum (Figure 6B). Analysis of miRNA expression levels from transduced striatal tissue shows effective overexpression of all miRNAs, although at different magnitudes, probably due to differences in pre-miRNA processing efficiencies49 and in the endogenous levels of each miRNA50 (Figure 6C; mir-9 LV-Neg: 1.001 ± 0.028 versus mir-9 LV-mir-9: 1.237 ± 0.124; mir-181a LV-Neg: 1.032 ± 0.138 versus mir-181a LV-181a: 1.547 ± 0.116; and mir-494 LV-Neg: 1.047 ± 0.178 versus mir-494 LV-494: 2.197 ± 0.176).
Immunohistochemical analysis of coronal sections obtained from injected mice showed a marked reduction in the total number of 1H9 reactive mutATXN3 inclusions in the hemispheres injected with miRNA-encoding lentivirus when compared to the control hemisphere. (Figures 6C, 6E, 6G, 6I, and 6K; LV-neg: 13,966 ± 1,122 aggregates versus LV-9: 8,899 ± 849.8 aggregates, LV-181a: 8,832 ± 784.1 aggregates, and LV-494: 9,662 ± 996.7 aggregates). Moreover, western blot analysis revealed similar results in aggregated mutATXN3 levels (Figure S5A). Furthermore, and in accordance with the previous results, total DARPP-32 depleted volume was also significantly reduced after overexpression of the studied miRNAs (Figures 6D, 6F, 6H, 6J, and 6L; LV-neg: 0.082 ± 0.012 mm3 versus LV-9: 0.043 ± 0.007 mm3, LV-181a: 0.034 ± 0.007 mm3, and LV-494: 0.037 ± 0.007 mm3).
Similarly, analysis of mutATXN3 mRNA levels showed a strong reduction upon miRNA overexpression (Figure 6N; LV-neg: 1.027 ± 0.136 versus LV-9: 0.427 ± 0.128, LV-181a: 0.526 ± 0.085, and LV-494: 0.440 ± 0.062). Altogether, these results clearly demonstrate the potential for in vivo silencing of mutATXN3 both at the mRNA and protein level using miRNAs. Moreover, to evaluate if these miRNAs would modulate endogenous mouse Atxn3 (mAtxn3), qPCR for mAtxn3 mRNA was performed in the same samples. We observed a significant reduction for mAtxn3 mRNA levels for both LV-9 and LV-181a (Figure 6O; LV-neg: 1.002 ± 0.029 versus LV-9: 0.818 ± 0.045 and LV-181a: 0.769 ± 0.008), but not for LV-494 (Figure 6O; LV-neg: 1.002 ± 0.028 versus LV-494: 0.969 ± 0.068). Similar results were observed on endogenous mAtxn3 protein levels (Figure S5B). Note that mAtxn3 3′ UTR is considerably different from the human sequence. Still, when in comparison with the human 3′ UTR, it contains an identical mir-9 binding site and other non-conserved binding sites for mir-181a and mir-494, which fit with the observed results (Figure 6P).
Overall, these data provide evidence that mir-9, mir-181a, and mir-494 efficiently reduce mutant ataxin-3 expression and associated neuropathology and may therefore be promising physiological tools to alleviate MJD.
Discussion
In the present study using both in vitro and in vivo models, we demonstrate that the 3′ UTR of the human ATXN3 mRNA is targeted by miRNAs, which mediate post-transcriptional negative regulation of ATXN3 itself. Moreover, we present compelling evidence of MJD-associated downregulation of the miRNA biogenesis and of miRNA machinery, which presumably contribute to the disease pathogenesis. Furthermore, we identified three specific endogenous miRNAs involved in the regulation of mutATXN3 in the brain and successfully incorporated these in gene therapy tools, which we validated for the treatment of MJD.
Despite the efforts that have been made in the development of different MJD animal models for the study of MJD pathogenesis, the vast majority of them have been based on the expression of the CDS of mutant forms of human or mouse ATXN3.15, 16, 17, 18, 20, 21, 22 Due to this fact, and regardless of having been effective in the development of promising strategies for the treatment of MJD, these studies lacked in the elucidation of the physiological regulation of ATXN3 expression and how this might be contributing to mutATXN3 toxicity.
Human ATXN3 mRNA possesses a long 3′ UTR, which might be a target of post-transcriptional regulators, such as miRNAs or RNA-binding proteins. In fact, our results are in accordance with recent reports suggestive of a role for miRNAs as regulators of ATXN3 mRNA.32, 33 Additionally, another study recently described two novel SNPs in ATXN3 3′ UTR found to modulate disease age-of-onset on MJD patients, highlighting the impact of 3′ UTR regulation on disease progression.51
On the other hand, our results gain further relevance when aligned with the findings of Ramani and colleagues.52 The authors observed a retention of intron 10 in mutATXN3 transcripts, generating an ATXN3-10e isoform which lacks ATXN3 3′ UTR. Furthermore, they report an upregulation of total ATXN3 transcripts in SCA3 knockin mice, also pointing for likely differences in 3′ UTR regulation.52 In light of our results, we believe this alternative splicing event leads to the expression of an isoform of mutATXN3 that, by lacking the endogenous 3′ UTR, will escape endogenous regulatory players such as miRNAs.
Considering previous evidence implicating miRNAs in neurodegenerative diseases including MJD,25, 26, 27, 28, 30, 53 we sought to investigate if they would be the mediators of ATXN3 3′ UTR regulation. Impairment of miRNA biogenesis led to an upregulation of mutATXN3 mRNA and protein levels. These results provide strong evidence that endogenous miRNAs play an important role in the regulation of mutATXN3 through its 3′ UTR. Moreover, the inclusion of 3′ UTR of ATXN3 in our previously characterized lentiviral mouse model of MJD resulted in a marked reduction of mutATXN3 aggregates and associated neuropathology, suggesting that the 3′ UTR of ATXN3 enables regulation by brain miRNAs in vivo. These results are in accordance with the report that the regulation of human alpha-synuclein at its 3′ UTR also decreases its levels and toxicity in vivo.54
Interestingly, overexpression of mutATXN3-3′ UTR in vivo resulted in remarkably low toxicity. Notably, this finding is in accordance with the previously mentioned MJD transgenic mouse models that include the 3′ UTR of ATXN3. YAC ATXN3Q84, knockin ATXN3Q82, and Ki91 models are all reportedly described with mild phenotypes, slow progression, and modest neuropathology.52, 55, 56 Being characterized by a moderate yet clear neuropathology, all in a considerably short time frame, we believe our modified mutATXN3-3UTR lentiviral mouse model of MJD expands the repertoire of existing MJD mouse models, presenting itself as a platform for the study of not only endogenous 3′ UTR regulators, which might be implicated in MJD pathogenesis, but also novel therapeutic strategies for the treatment of MJD.
Although recent studies have approached the topic of miRNA dysregulation in MJD, our study is the first to evaluate miRNA dysregulation in human samples isolated from the dentate nucleus, and in human neurons derived from hiPSCs, corroborating the results obtained in other models. Although the changes observed in the miRNA levels in vitro were modest, we observed a very significant downregulation in the levels of all miRNAs in MJD transgenic mice cerebellum. Such miRNA downregulation could be associated with a severely aggravated phenotype and correspondent marked cerebellar degeneration in MJD, suggesting that the extent of miRNA downregulation is associated with the progression of the disease and that the changes in miRNA expression in vitro may be representative of early pathogenic events mediated by mutATXN3. Importantly, the simultaneous downregulation of multiple miRNAs, even if modest, may additively contribute to the transcriptional dysregulation of target genes, such as ATXN3. Moreover, we should account that mir-9, mir-181a, and mir-494 participate in multiple molecular pathways, which will likely be affected by their downregulation. For example, mir-181a has been linked to the regulation of the production of pro-inflammatory cytokines57, 58 and mir-9, a highly conserved neuronal miRNA which is involved in the neuronal development and neuronal differentiation,59 is not only downregulated in HD and FTD, but is also dysregulated in other neurodegenerative diseases such as Alzheimer’s, Parkinson’s disease, and spinal muscular atrophy.60, 61, 62 Accordingly, we believe that in the future, miRNA dysregulation and its impact on their targets should be further assessed.
In accordance with the observed miRNA profiling data, we observed a general downregulation in the expression of genes controlling the biogenesis and function of miRNAs. Importantly, miRNA biogenesis appears to be impaired both at the pri- and pre-miRNA processing steps due to the decrease of both DGCR8 and Dicer. Nevertheless, we cannot exclude the existence of impairments at the transcription level of these miRNAs, particularly taking into account the transcriptionally dysregulated profile in MJD.18 Interestingly, it has been previously reported that Dicer deficiency in Purkinje cells leads to cerebellar degeneration and the development of ataxia.63 Therefore, our data strongly suggest that the dysregulation of both miRNAs and miRNA biogenesis and function components could be a novel pathogenic mechanism in the development of MJD, where mutATXN3 initiates a cycle of impaired miRNA biogenesis and processing, leading to decreased miRNA expression, which results in subsequent higher levels of mutATXN3. In any case, we believe individual modulation of key RNAi mediators should be performed in order to properly evaluate their role in MJD pathogenesis.
Other studies have identified miRNAs targeting ATXN3 mRNA. Mir-25 was the first miRNA validated to target ATXN3 3′ UTR. Interestingly, overexpression of this miRNA was only capable of reducing the levels of mutATXN3 at the protein level, but not at the mRNA level. Still concomitant reduction of apoptosis was observed in vitro.32 Nevertheless, no further attempts to pursue an in vivo application of mir-25 in MJD animal models were made.
In this study, we demonstrated that the overexpression of three distinct miRNAs was able to downregulate mutATXN3 in vitro and in vivo by directly targeting its 3′ UTR. Despite having already been employed in different neurodegenerative diseases,25, 26, 64 this was the first time that in vivo modulation of an endogenous miRNA was used as a therapeutic approach for the treatment of MJD.
Given the observed downregulated miRNA profile in MJD models, we anticipate that the reestablishment of normal levels of endogenous miRNAs targeting ATXN3 might be a promising approach for the reduction of mutATXN3 levels. In any case, whether the usage of endogenous miRNAs will be beneficial over the usage of artificial miRNAs, a strategy already employed by others,13, 14 will likely depend on the required silencing potency and off-targeting characteristics of each sequence.
In conclusion, our results support a 3′ UTR-based regulation of mutATXN3, whose dysfunction may be implicated in the pathogenesis of MJD, not only due to changes in mutATXN3 mRNA levels, but also due to defects in miRNA biogenesis and function. More importantly, we provide evidence that gene delivery of endogenous miRNAs targeting ATXN3 may be a promising strategy for the treatment of MJD.
Materials and Methods
Vector Construction
Human ATXN3 3′ UTR (1–3,434 bp) was obtained by restriction digestion of the dual luciferase vector clone HmiT054676a-MT01 (GeneCopoeia) using EcoRI and XhoI enzymes (New England Biolabs). The insert was gel purified using a DNA gel extraction kit (MACHEREY-NAGEL), blunted using Klenow Polymerase (New England Biolabs), and later inserted into the previously described LTR-SIN-PGK-mutATXN3-LTR vector17 using a SmaI site downstream of the mutATXN3 coding sequence in order to generate the LTR-SIN-PGK-mutATXN3-3′ UTR-LTR vector (Figure S1).
Truncated C-terminal mutATXN3 with 72 glutamines (TmutATXN3) vectors were constructed using inverted PCR mutagenesis. Briefly, two 5′ phosphorylated oligonucleotides (forward: GCAGATCTCCGCAGGG and reverse: GTCAATTTCTTGGCGACTTAGTG) (Eurofins Genomics) were designed in order to amplify the parental vectors full sequence (LTR-SIN-PGK-mutATXN3-LTR or LTR-SIN-PGK-mutATXN3-3′ UTR-LTR), with the exception of the sequence corresponding to the first 252 amino acids (756 bp), using Phusion DNA Polymerase (Thermo Scientific) and a modified protocol for GC rich templates. Linear products were circularized using T4 DNA ligase (Thermo Scientific) and transformed into TOP10 chemically competent cells (Life Technologies) to generate LTR-SIN-PGK-TmutATXN3-LTR or LTR-SIN-PGK-TmutATXN3-3′ UTR-LTR vectors (Figure S2).
For the construction of the lentiviral plasmids encoding for miRNAs, a series of oligonucleotides (Life Technologies) was designed in order to amplify the corresponding genomic region encoding for the pre-miRNAs, plus the endogenous flanking sequences in the total range of approximately 400 bp (Table S2). Amplified inserts were then digested with BglII and HindIII enzymes (New England BioLabs) and ligated into pENTR/pSUPER+ (Addgene plasmid #17338). The H1-miRNA expression cassettes were then transferred into LTR-SIN-PGK-GFP-AttL1-ccdB-AttL2-LTR destination vector using a Gateway LR recombination reaction (Life Technologies), in order to generate LTR-SIN-PGK-GFP-H1-miRNA-LTR plasmids. The mir-Neg lentiviral vector was generated through PCR amplification of the mir-Neg cassette plus flanking sequences from the pcDNA6.2-GW/miR-neg vector (Life Technologies), ligated into pENTR/pSUPER+, and gateway recombined, in order to generate LTR-SIN-PGK-GFP-H1-mir-Neg-LTR.
The protocol for the construction of shRNA lentiviral vectors has been described in our previous study.65 Briefly, a shRNA negative (control) and a shRNA targeting either human DICER or DROSHA were created. For each one, a pair of oligonucleotides was designed, annealed, and ligated into linearized p-ENTR/pSUPER+ (Addgene 575-1) (Table S3). The H1-shRNA cassette was then transferred, into SIN-cPPT-PGK-EGFP-WHV-LTR or SIN-cPPT-PGK-lacZ-WHV-LTR, taking advantage of a LR Clonase recombination system (Life Technologies).
All constructs were verified by restriction analysis and sequencing. Oligonucleotide sequences are available in the Supplemental Materials and Methods (Tables S2 and S3).
Cell Line Culture, Transfection, and Transduction
HEK293T and Neuro2A cells were maintained in standard DMEM (Sigma) supplemented with 10% fetal bovine serum (Life Technologies) and 1% penicillin/streptomycin (Life Technologies). SH-SY5Y cells were maintained in DMEM/F12 (Gibco) medium supplemented with 10% fetal bovine serum (Life Technologies) and 1% penicillin/streptomycin.
For the transfection of HEK293T cell line, 2.75 × 104 cells were initially plated on 12-well cell culture treated multiwell plates (Fisher Scientific). After 24 hr, medium was changed and cells were transfected with a mixture of DNA/polethyleneimine (PEI) complexes (MW40000, Polysciences) diluted in complete DMEM. Complex formation was performed by combining 750 ng of plasmid DNA in 60 μL of serum free DMEM and 4.5 μL of PEI (1 mg/mL) per well. This mixture was vortexed for 10 s and incubated at room temperature for 10 min. After that, DNA/PEI complexes were diluted in complete DMEM and added to cell culture. Cell collection was performed 48 hr post-transfection.
For the transduction of N2A and SH-SY5Y cells, 24 hr after plating, the culture medium was replaced with fresh medium containing lentivirus (400 ng of P24 per 200,000 cells). At 12 hr later, the medium was replaced with regular complete medium and cells were cultured and expanded in standard conditions.
Dual Luciferase Assay
A dual luciferase reporter construct including the 3′ UTR of ATXN3 (FLuc-3′ UTR) and a control vector (FLuc-CTRL) clone pEZX-MT01 were purchased from GeneCopoeia.
To evaluate the direct targeting of miRNAs to ATXN3 3′ UTR, 2.75 × 104 HEK293T cells were seeded per well in 12-well plates (Fisher Scientific). In the next day, cells were transfected with 375 ng of the luciferase reporter vector and 375 ng of miRNA constructs per well (in triplicates). At 48 hr after transfection, cells were washed with 1× PBS and frozen at −80°C. Collected cells were lysed with 100 μL of FLAR-T buffer per well containing 20 mM tricine, 100 μM EDTA, 25 μM MgCl2, 2.67 mM MgSO4, 17 mM DTT, and 0.1% Triton in Milli-Q grade water pH 7.8. For the luminescent reaction, 30 μL of cell lysates were loaded on an opaque 96-well plate (Corning) and the firefly luminescence activity was measured on a LMax II 384 Luminometer (Molecular Devices) after automatic injection of 100 μL of FLAR buffer containing 20 mM tricine, 100 μM EDTA, 25 μM MgCl2, 2.67 mM MgSO4, 17 mM DTT, 250 μM ATP, and 250 μM D-luciferin (Synchem). Renilla luminescence activity was used as a normalization control and was measured after automatic injection of 100 μL of RAB buffer containing 1.1 M NaCl, 2.2 mM Na2EDTA, 0.22 M KxPO4, 0.44 mg/mL BSA, 1.3 mM NaN3, and 1.43 mM coelenterazine (Life Technologies) in Milli-Q grade water pH 5.0 to 30 μL of cell lysate per microplate well. Integration times were 10 s for firefly luciferase signal capture and 5 s for renilla luciferase signal capture. Unless stated, all reagents were purchased from Sigma-Aldrich.
RNA Stability Assay
Actinomycin D (10 mg/mL, Sigma-Aldrich) was used to inhibit de novo mRNA synthesis. Neuro2A cells stably expressing either mutATXN3 or mutATXN3-3′ UTR at 50%–60% confluence were treated with actinomycin D (10 mg/mL) for 0, 2, and 4 hr and collected for total mRNA extraction and analysis.
Lentivirus Production, Purification, and Titer Assessment
Lentiviral vectors encoding for human mutant ataxin 3 (mutATXN3 and mutATXN3-3′ UTR) or miRNAs (mir-9, mir-181a, mir-494, and mir-Neg) were produced in HEK293T cells with a four-plasmid system, as previously described.34 The lentiviral particles were resuspended in 1% BSA in PBS. The viral particle content of batches was determined by assessing HIV-1 p24 antigen levels (Retro Tek, GENTAUR). Viral stocks were stored at −80°C. For functional analysis of lentiviral transduction rates, genomic DNA from HEK293T was isolated 72 hr after cell transduction with equal amounts of p24 using a Genomic DNA Purification Kit (Thermo scientific). qPCR analysis was performed with WPRE and Albumin primers using SsoAdvanced SYBR Green Supermix (Bio-Rad) and following manufacturers recommended conditions (Table S4).
Animals
4-week-old C57BL/6J mice were obtained from Charles River. The animals were housed in a temperature-controlled room maintained on a 12 hr light/12 hr dark cycle. Food and water were provided ad libitum. The experiments were carried out in accordance with the European Union Directive 2010/63/EU covering the protection of animals used for scientific purposes. The researchers received adequate training (FELASA certified course) and certification to perform the experiments from Portuguese authorities (Direção Geral de Veterinária).
Mouse Surgery
5-week-old C57BL/6J mice were anesthetized by intraperitoneal administration of Avertin (14 μL/g, 250 mg/Kg). Mice were stereotaxically injected into the striatum in the following coordinates calculated from bregma: anteroposterior: +0.6 mm; lateral: ±1.8 mm; ventral: −3.3 mm; and tooth bar: 0, by receiving a single injection of concentrated lentiviral vectors in a final volume of 2 μL containing 400 ng of p24 antigen for each vector encoding for the specific transgenes, at an infusion rate of 0.25 μL/min using a 10 μL Hamilton syringe. At 5 min after the infusion was completed, the needle was retracted 0.3 mm and allowed to remain in place for an additional 3 min prior to its complete removal from the mouse brain.
Immunohistochemical Analysis
Tissue Preparation
Mice were sacrificed with an overdose of Avertin (35 μL/g, 625 mg/kg, intraperitoneally), transcardially perfused with phosphate solution, and fixed with 4% paraformaldehyde. Brains were collected and post-fixed in 4% paraformaldehyde for 24 hr, cryoprotected by incubation in 25% sucrose/phosphate buffer, and frozen at −80°C. Coronal sections of 25 μm thickness were obtained using cryostat equipment (LEICA CM3050S, Leica Microsystems).
Bright-Field Immunohistochemistry
Free-floating brain coronal sections were treated with a phenylhydrazyne/phosphate solution for the blockage of endogenous peroxidases for 30 min at 37°C. After permeabilization in PBS/0.1% Triton X-100 with 10% normal goat serum (Gibco), sections were incubated overnight at 4°C in blocking solution with primary antibodies: mouse monoclonal anti-ataxin 3 antibody (1H9; 1:5,000; Merck Millipore), rabbit anti-dopamine, and cyclic AMP-regulated neuronal phosphoprotein 32 (DARPP-32) antibody (1:1,000; Merck Millipore); followed by incubation with respective biotinylated secondary goat anti-mouse or anti-rabbit antibodies (1:200; Vector Laboratories). Bound antibodies were visualized using the VECTASTAIN ABC kit, with 3′,3′-diaminobenzidine tetrahydrochloride (DAB metal concentrate; Pierce) as substrate. Dry sections were mounted in gelatin-coated slides, dehydrated with ethanol solutions and Xylene Substitute (Sigma-Aldrich), and mounted in Eukit quick-hardening mounting medium (Sigma-Aldrich).
Fluorescence Immunohistochemistry
Free-floating sections were incubated in PBS/0,1% Triton X-100 containing 10% normal goat serum (Sigma-Aldrich) and then incubated overnight at 4°C in blocking solution with primary antibody: rabbit anti-ionized calcium binding adaptor molecule 1 (Iba-1) antibody (1:1,000; Wako Chemicals USA). Sections were washed and incubated for 2 hr at room temperature with the secondary antibody: goat anti-mouse Alexa Fluor 594 (1:250; Molecular Probes-Invitrogen) diluted in blocking solution. The sections were washed and incubated during 10 min with DAPI (1:5,000; Sigma), washed, and mounted in mowiol (Sigma) on gelatin-coated slides.
Immunoreactivity of mouse sections was analyzed as previously described.43 Staining was visualized with Zeiss Axioskop 2 imaging microscope (Carl Zeiss MicroImaging) equipped with AxioCam HR color digital cameras (Carl Zeiss MicroImaging) and Plan-Neofluar 5×/0.15 Ph 1 (440321), Plan-Neofluar 20×/0.50 Ph 2 (1004-989), Plan-Neofluar 40×/0.75 Ph 2 (440351), and Plan-Neofluar 63×/1.25 Oil (440460-0000) objectives using the AxioVision 4.7 software package (Carl Zeiss Microimaging) or with a Zeiss Axio Imager Z2 imaging microscope (Carl Zeiss MicroImaging) equipped with EC Plan-Neofluar 5×/0.16 M27 (420330-9901), Plan-Apochromat 20×/0.8 M27 (420650-9901), Plan-Apochromat 40×/1.3 Oil DIC M27 (420762-9800), and Plan-Apochromat 63×/1.40 Oil DIC M27 (420782-9900) using ZEN software. Quantitative analysis of fluorescence was performed with a semiautomated image-analysis software package and images were taken under the same image acquisition conditions and uniform adjustments of brightness and contrast were made to all images (ImageJ).
Fluorescence Immunocytochemistry
Cell cultures were washed with PBS and fixed with PBS/4% PFA. After permeabilization in PBS/1% Triton X-100 solution for 5 min and blocking in PBS/3% BSA, cells were incubated overnight at 4°C with the primary antibodies: mouse anti-Myc tag clone 4A6 (1:1,000; Millipore), mouse anti-βIII tubulin clone 38F4 (1:1,000; Life Technologies), and rabbit anti-GFAP Z0334 (1:1,000; Dako) diluted in PBS/3% BSA. On the next day, cells were washed and incubated for 2 hr at room temperature with the secondary antibody: goat anti-mouse/rabbit Alexa Fluor 488/594 (1:250; Molecular Probes-Invitrogen). Cells were washed and incubated during 5 min with DAPI (1:5,000; Sigma), washed, and mounted in mowiol (Sigma) on gelatin-coated slides. Staining in HEK293T was visualized using a Zeiss LSM 510 Meta confocal microscope (Carl Zeiss MicroImaging), equipped with EC Plan-Neofluar 40×/1.30 Oil DIC M27 (420462-9900) and Plan-Apochromat 63×/1.40 Oil DIC M27 (420782-9900) objectives and LSM Image software. In order to quantify mutATXN3 aggregates in transfected HEK293T cells, five images were randomly acquired per condition and total aggregates and nuclei were manually counted using ImageJ software. The total number of cells counted ranged from 800 to 1,100 cells per experimental condition.
Quantification of mutATXN3 Inclusions and DARPP-32 Depleted Volume
Quantitative analysis of the total number of mutATXN3 inclusions and the extent of DARPP-32 loss in the striatum was performed by scanning 12 stained sections per animal that were distanced 200 μm from each other. Total number of inclusions were manually counted in all 12 sections and multiplied by 8 to account for the intermediate sections. To calculate the DARPP-32 loss, sections were imaged using a 5× objective. The quantifications were then performed using image-analysis software (ImageJ software). The DARPP-32-depleted volume was estimated using the following formula: Volume = d(a1 + a2 + a3 +…), where d is the distance between serial sections and a1, a2, and a3 are the areas for individual serial sections.
Isolation of Total RNA from Cells and Mouse Tissue
Total RNA was isolated with miRCURY RNA isolation kit (Exiqon) according to manufacturer’s instructions. Monolayer cell cultures were washed with 1× PBS and stored at −80°C before extraction. For the extraction from mouse tissue, mice were sacrificed with a lethal dose of Avertin. Striatum of mice were dissected and stored at −80°C until RNA isolation. All samples were submitted to on-column DNase I digestion (QIAGEN) during isolation. Total amount of RNA was quantified by optical density (OD) using a NanoDrop 2000 Spectrophotometer (Thermo Scientific) and RNA was stored at −80°C.
cDNA Synthesis and Real-Time qPCR
Specific cDNAs for miRNA quantification were synthetized using a TaqMan MicroRNA Reverse Transcription Kit combined with specific TaqMan MicroRNA Assays (Applied Biosystems) for each miRNA (mir-103-3p ID000439; mir-9-5p ID000583; mir-181a-5p ID000480; mir-494-3p ID002365; and U6 snRNA ID001973) according to manufacturer’s instructions. qPCR was performed using TaqMan Universal PCR Master Mix II with UNG (Applied Biosystems), combined with the TaqMan MicroRNA Assays, following manufacturer’s recommended cycling conditions, in a StepOnePlus Real-Time PCR System (Applied Biosystems).
cDNA for mRNA quantification was obtained by conversion of total RNA with iScript cDNA Synthesis kit (Bio-Rad) according to manufacturer’s. qPCR was performed in the StepOnePlus Real-Time PCR System (Applied Biosystems) and SsoAdvanced SYBR Green Supermix (Bio-Rad). Primers for mouse DICER, DROSHA, GAPDH, and human ACTB genes were pre-designed and validated by QIAGEN (QuantiTect Primers, QIAGEN). Primers for mouse TARBP2, 18S, DDX6, AGO2, DGCR8, FMR1, and human DICER and DROSHA were designed using PrimerBlast Software (Table S4). Appropriate negative controls were also prepared. All reactions were performed in duplicate and using the following cycling conditions: 95°C for 30 s, followed by 45 cycles at 95°C for 5 s, and 58°C for 30 s. The amplification rate for each target was evaluated from the cycle threshold (Ct) numbers obtained with cDNA dilutions. Differences between control and experimental samples were calculated using the 2−ΔΔCt method.
Protein Extraction and Western Blotting
Monolayer cells were scraped in radioimmunoprecipitation lysis buffer (50 mM Tris HCl, pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) containing protease inhibitors (Roche) followed by sonication by two series of 4 s pulses. Cerebellar tissue from MJD transgenic mice was lysed in identical radioimmunoprecipitation lysis buffer followed by sonication by two series of 4 s pulses. Striatal tissue from mice injected with lentiviral vectors encoding for mutATXN3 and miRNAs was dissected from 12 sections per animal that were distanced 200 μm from each other in order to cover the full injection region. Tissue lysis was performed in 20 Mm Tris-HCl, pH 9, and 2% SDS for 2 hr at 80°C followed by sonication by two series of 4 s pulses. Protein lysates were stored at −20°C and protein concentration was determined with Bio-Rad Protein Assay (Bio-Rad). Protein samples were denatured with 6× sample buffer (0.375 M Tris pH 6.8 [Sigma-Aldrich], 12% SDS [Sigma-Aldrich], 60% glycerol [Sigma-Aldrich], 0.6 M DTT [Sigma-Aldrich], and 0.06% bromophenol blue [Sigma-Aldrich) and 40 μg of protein were resolved by electrophoresis on 12% SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes (GE Healthcare). Membranes were blocked by incubation in 5% non-fat milk powder in 0.1% Tween 20 in Tris buffered saline (TBS-T) and incubated overnight at 4°C with primary antibody: mouse monoclonal anti-ataxin-3 antibody (1H9; 1:5,000; Merck Millipore); mouse monoclonal antibody anti-myc tag (4A6; 1:1,000; Merck Millipore); rabbit DGCR8 polyclonal antibody (10996-1-AP; 1:500; Proteintech); rabbit Ago2 monoclonal antibody (C34C6; 1:1,000; Cell Signaling), and mouse anti-β-actin antibody (AC74; 1:10.000; Sigma-Aldrich), followed by the incubation with alkaline phosphatase-linked secondary goat anti-mouse/anti-rabbit antibody (1:10,000; Thermo Scientific Pierce). Bands were visualized with Enhanced Chemifluorescence substrate (ECF) (GE Healthcare) and chemifluorescence imaging (ChemiDoc Imaging System, Bio-Rad). Semiquantitative analysis was carried out based on the optical density of scanned membranes (ImageLab version 5.2.1; Bio-Rad). The specific optical density was then normalized with respect to the amount of β-actin loaded in the corresponding lane of the same gel.
Neurosphere Culture and Neuronal Differentiation
Human neurospheres derived from hIPSCs generated from MJD or CTRL fibroblasts66 were maintained in neural expansion media containing DMEM/F12, B27 supplement (1:50; Invitrogen), MEM non-essential amino acid solution (1:100; Sigma-Aldrich), 20 ng/mL EGF, 20 ng/mL bFGF (PeproTech), 1% penicillin/streptomycin (Gibco), and 5 μg/mL Heparin (Sigma-Aldrich).
To initiate differentiation, NSCs were manually dissociated with a pipette in small aggregates and plated in poly-ornithin/laminin (Sigma-Aldrich) coated plates as small droplets. Neuronal differentiation was performed during 18 days on DMEM/F12 and Neurobasal (mixed 1:1), B27 supplement (1:100; Invitrogen), N2 supplement (1:200; Invitrogen), L-glutamine (1:100; Life Technologies), 20 ng/mL BDNF (both PeproTech), 1% penicillin/streptomycin (Gibco), 0.1 μM retinoic acid (Sigma-Aldrich), 200 nM ascorbic acid (Sigma-Aldrich), 0.5 mM Dibutyryl-cAMP (Sigma-Aldrich), and 10 μM forskolin (Sigma-Aldrich). Medium was half-changed twice per week or as needed.67
Human Brain Tissue
Post-mortem human brain tissue from dentate nucleus was obtained from the Tissue Donation Program of the National Ataxia Foundation, Minneapolis, USA (3 MJD and 4 CTRL) and from the University of Groningen, the Netherlands (3 MJD and 1 CTRL).
MJD Transgenic Mice and Genotyping
C57BL/6-background transgenic mouse model expressing the C-terminal-truncated ataxin-3 with 69 glutamine repeats and an N-terminal haemagglutinin (HA) epitope driven by Purkinje-cell-specific L7 promoter were initially obtained from Gunma University Graduate School of Medicine. The mice used in this study were obtained from parallel breeding at the CNC animal facilities. All animals used in this study were age matched transgenic (Tg) and wild-type (WT) littermates at 8 weeks of age.
The experiments were carried out in accordance with the European Community directive (2010/63/EU) covering the protection of animals used for scientific purposes. The researchers received adequate training (FELASA-certified course) and certification to perform the experiments from the Portuguese authorities (Direcção Geral de Alimentação e Veterinária).
For mouse genotyping, genomic DNA was isolated from collected animal ear tissue. The presence of the transgene was validated by PCR amplification using DreamTaq (Thermo Scientific) using standard amplification conditions and the following primers forward: ATGTACCCATACGATGTTCC and reverse: CTAGCGAGGGAATGAAGAAT.
Author Contributions
V.C., J.C.-S., I.O., A.T.S., U.V., and L.P.d.A. contributed for the design of the experiments. B.L.D. contributed hypotheses and edited the manuscript. V.C., J.C.-S., and I.O. performed the experiments. V.C. and L.P.d.A. wrote the manuscript. All seven authors contributed for the discussion of the results.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
The authors wish to thank Professor Arnulf Koeppen and Professor Wilfred den Dunnen for providing the human samples. This work was supported by funds from FEDER, the Competitive Factors Operational Program - COMPETE, by national funds through the Portuguese Foundation for Science and Technology (FCT: PTDC/SAU-NMC/116512/2010, E-Rare4/0003/2012 and EU Joint Programme - Neurodegenerative Disease Research [JPND] projects SynSpread, ESMI, and ModelPolyQ), by the Richard Chin and Lily Lock Machado Joseph Disease Research Fund, and the National Ataxia Foundation to L.P.d.A. V.C., J.C.-S., and A.T.S. hold fellowships with FCT. U.V. was funded by the Marie Curie ITN - Treat PolyQ network. B.L.D. is supported by the National Ataxia Foundation, the NIH (UH2 N5094355), and the Research Institute of The Children’s Hospital of Philadelphia.
Footnotes
Supplemental Information includes five figures and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.ymthe.2017.01.021.
Supplemental Information
References
- 1.Sudarsky L., Coutinho P. Machado-Joseph disease. Clin. Neurosci. 1995;3:17–22. [PubMed] [Google Scholar]
- 2.Dürr A., Stevanin G., Cancel G., Duyckaerts C., Abbas N., Didierjean O., Chneiweiss H., Benomar A., Lyon-Caen O., Julien J. Spinocerebellar ataxia 3 and Machado-Joseph disease: clinical, molecular, and neuropathological features. Ann. Neurol. 1996;39:490–499. doi: 10.1002/ana.410390411. [DOI] [PubMed] [Google Scholar]
- 3.Kawaguchi Y., Okamoto T., Taniwaki M., Aizawa M., Inoue M., Katayama S., Kawakami H., Nakamura S., Nishimura M., Akiguchi I. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994;8:221–228. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- 4.Seidel K., Siswanto S., Brunt E.R., den Dunnen W., Korf H.W., Rüb U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012;124:1–21. doi: 10.1007/s00401-012-1000-x. [DOI] [PubMed] [Google Scholar]
- 5.Paulson H.L., Perez M.K., Trottier Y., Trojanowski J.Q., Subramony S.H., Das S.S., Vig P., Mandel J.L., Fischbeck K.H., Pittman R.N. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 6.Simões A.T., Gonçalves N., Koeppen A., Déglon N., Kügler S., Duarte C.B., Pereira de Almeida L. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain. 2012;135:2428–2439. doi: 10.1093/brain/aws177. [DOI] [PubMed] [Google Scholar]
- 7.Nascimento-Ferreira I., Santos-Ferreira T., Sousa-Ferreira L., Auregan G., Onofre I., Alves S., Dufour N., Colomer Gould V.F., Koeppen A., Déglon N., Pereira de Almeida L. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain. 2011;134:1400–1415. doi: 10.1093/brain/awr047. [DOI] [PubMed] [Google Scholar]
- 8.Nóbrega C., Carmo-Silva S., Albuquerque D., Vasconcelos-Ferreira A., Vijayakumar U.G., Mendonça L., Hirai H., de Almeida L.P. Re-establishing ataxin-2 downregulates translation of mutant ataxin-3 and alleviates Machado-Joseph disease. Brain. 2015;138:3537–3554. doi: 10.1093/brain/awv298. [DOI] [PubMed] [Google Scholar]
- 9.Conceição M., Mendonça L., Nóbrega C., Gomes C., Costa P., Hirai H., Moreira J.N., Lima M.C., Manjunath N., Pereira de Almeida L. Intravenous administration of brain-targeted stable nucleic acid lipid particles alleviates Machado-Joseph disease neurological phenotype. Biomaterials. 2016;82:124–137. doi: 10.1016/j.biomaterials.2015.12.021. [DOI] [PubMed] [Google Scholar]
- 10.Alves S., Nascimento-Ferreira I., Auregan G., Hassig R., Dufour N., Brouillet E., Pedroso de Lima M.C., Hantraye P., Pereira de Almeida L., Déglon N. Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease. PLoS ONE. 2008;3:e3341. doi: 10.1371/journal.pone.0003341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nóbrega C., Nascimento-Ferreira I., Onofre I., Albuquerque D., Déglon N., de Almeida L.P. RNA interference mitigates motor and neuropathological deficits in a cerebellar mouse model of Machado-Joseph disease. PLoS ONE. 2014;9:e100086. doi: 10.1371/journal.pone.0100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nóbrega C., Nascimento-Ferreira I., Onofre I., Albuquerque D., Hirai H., Déglon N., de Almeida L.P. Silencing mutant ataxin-3 rescues motor deficits and neuropathology in Machado-Joseph disease transgenic mice. PLoS ONE. 2013;8:e52396. doi: 10.1371/journal.pone.0052396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Costa Mdo C., Luna-Cancalon K., Fischer S., Ashraf N.S., Ouyang M., Dharia R.M., Martin-Fishman L., Yang Y., Shakkottai V.G., Davidson B.L. Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease. Mol. Ther. 2013;21:1898–1908. doi: 10.1038/mt.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez-Lebron E., Costa M., Luna-Cancalon K., Peron T.M., Fischer S., Boudreau R.L., Davidson B.L., Paulson H. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol. Ther. 2013;21:1909–1918. doi: 10.1038/mt.2013.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goti D., Katzen S.M., Mez J., Kurtis N., Kiluk J., Ben-Haïem L., Jenkins N.A., Copeland N.G., Kakizuka A., Sharp A.H. A mutant ataxin-3 putative-cleavage fragment in brains of Machado-Joseph disease patients and transgenic mice is cytotoxic above a critical concentration. J. Neurosci. 2004;24:10266–10279. doi: 10.1523/JNEUROSCI.2734-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bichelmeier U., Schmidt T., Hübener J., Boy J., Rüttiger L., Häbig K., Poths S., Bonin M., Knipper M., Schmidt W.J. Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: in vivo evidence. J. Neurosci. 2007;27:7418–7428. doi: 10.1523/JNEUROSCI.4540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alves S., Régulier E., Nascimento-Ferreira I., Hassig R., Dufour N., Koeppen A., Carvalho A.L., Simões S., de Lima M.C., Brouillet E. Striatal and nigral pathology in a lentiviral rat model of Machado-Joseph disease. Hum. Mol. Genet. 2008;17:2071–2083. doi: 10.1093/hmg/ddn106. [DOI] [PubMed] [Google Scholar]
- 18.Chou A.H., Yeh T.H., Ouyang P., Chen Y.L., Chen S.Y., Wang H.L. Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol. Dis. 2008;31:89–101. doi: 10.1016/j.nbd.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 19.Torashima T., Koyama C., Iizuka A., Mitsumura K., Takayama K., Yanagi S., Oue M., Yamaguchi H., Hirai H. Lentivector-mediated rescue from cerebellar ataxia in a mouse model of spinocerebellar ataxia. EMBO Rep. 2008;9:393–399. doi: 10.1038/embor.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boy J., Schmidt T., Wolburg H., Mack A., Nuber S., Böttcher M., Schmitt I., Holzmann C., Zimmermann F., Servadio A., Riess O. Reversibility of symptoms in a conditional mouse model of spinocerebellar ataxia type 3. Hum. Mol. Genet. 2009;18:4282–4295. doi: 10.1093/hmg/ddp381. [DOI] [PubMed] [Google Scholar]
- 21.Boy J., Schmidt T., Schumann U., Grasshoff U., Unser S., Holzmann C., Schmitt I., Karl T., Laccone F., Wolburg H. A transgenic mouse model of spinocerebellar ataxia type 3 resembling late disease onset and gender-specific instability of CAG repeats. Neurobiol. Dis. 2010;37:284–293. doi: 10.1016/j.nbd.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 22.Silva-Fernandes A., Costa Mdo.C., Duarte-Silva S., Oliveira P., Botelho C.M., Martins L., Mariz J.A., Ferreira T., Ribeiro F., Correia-Neves M. Motor uncoordination and neuropathology in a transgenic mouse model of Machado-Joseph disease lacking intranuclear inclusions and ataxin-3 cleavage products. Neurobiol. Dis. 2010;40:163–176. doi: 10.1016/j.nbd.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 23.Conne B., Stutz A., Vassalli J.D. The 3′ untranslated region of messenger RNA: A molecular ‘hotspot’ for pathology? Nat. Med. 2000;6:637–641. doi: 10.1038/76211. [DOI] [PubMed] [Google Scholar]
- 24.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 25.Miyazaki Y., Adachi H., Katsuno M., Minamiyama M., Jiang Y.M., Huang Z., Doi H., Matsumoto S., Kondo N., Iida M. Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat. Med. 2012;18:1136–1141. doi: 10.1038/nm.2791. [DOI] [PubMed] [Google Scholar]
- 26.Cheng P.H., Li C.L., Chang Y.F., Tsai S.J., Lai Y.Y., Chan A.W., Chen C.M., Yang S.H. miR-196a ameliorates phenotypes of Huntington disease in cell, transgenic mouse, and induced pluripotent stem cell models. Am. J. Hum. Genet. 2013;93:306–312. doi: 10.1016/j.ajhg.2013.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee Y., Samaco R.C., Gatchel J.R., Thaller C., Orr H.T., Zoghbi H.Y. miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat. Neurosci. 2008;11:1137–1139. doi: 10.1038/nn.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Packer A.N., Xing Y., Harper S.Q., Jones L., Davidson B.L. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 2008;28:14341–14346. doi: 10.1523/JNEUROSCI.2390-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pourshafie N., Lee P.R., Chen K.L., Harmison G.G., Bott L.C., Katsuno M., Sobue G., Burnett B.G., Fischbeck K.H., Rinaldi C. MiR-298 counteracts mutant androgen receptor toxicity in spinal and bulbar muscular atrophy. Mol. Ther. 2016;24:937–945. doi: 10.1038/mt.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bilen J., Liu N., Burnett B.G., Pittman R.N., Bonini N.M. MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol. Cell. 2006;24:157–163. doi: 10.1016/j.molcel.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 31.Shi Y., Huang F., Tang B., Li J., Wang J., Shen L., Xia K., Jiang H. MicroRNA profiling in the serums of SCA3/MJD patients. Int. J. Neurosci. 2014;124:97–101. doi: 10.3109/00207454.2013.827679. [DOI] [PubMed] [Google Scholar]
- 32.Huang F., Zhang L., Long Z., Chen Z., Hou X., Wang C., Peng H., Wang J., Li J., Duan R. miR-25 alleviates polyQ-mediated cytotoxicity by silencing ATXN3. FEBS Lett. 2014;588:4791–4798. doi: 10.1016/j.febslet.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koscianska E., Krzyzosiak W.J. Current understanding of the role of microRNAs in spinocerebellar ataxias. Cerebellum Ataxias. 2014;1:7. doi: 10.1186/2053-8871-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Almeida L.P., Ross C.A., Zala D., Aebischer P., Déglon N. Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J. Neurosci. 2002;22:3473–3483. doi: 10.1523/JNEUROSCI.22-09-03473.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikeda H., Yamaguchi M., Sugai S., Aze Y., Narumiya S., Kakizuka A. Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat. Genet. 1996;13:196–202. doi: 10.1038/ng0696-196. [DOI] [PubMed] [Google Scholar]
- 36.Yoshizawa T., Yoshida H., Shoji S. Differential susceptibility of cultured cell lines to aggregate formation and cell death produced by the truncated Machado-Joseph disease gene product with an expanded polyglutamine stretch. Brain Res. Bull. 2001;56:349–352. doi: 10.1016/s0361-9230(01)00578-0. [DOI] [PubMed] [Google Scholar]
- 37.Haacke A., Broadley S.A., Boteva R., Tzvetkov N., Hartl F.U., Breuer P. Proteolytic cleavage of polyglutamine-expanded ataxin-3 is critical for aggregation and sequestration of non-expanded ataxin-3. Hum. Mol. Genet. 2006;15:555–568. doi: 10.1093/hmg/ddi472. [DOI] [PubMed] [Google Scholar]
- 38.Antony P.M., Mäntele S., Mollenkopf P., Boy J., Kehlenbach R.H., Riess O., Schmidt T. Identification and functional dissection of localization signals within ataxin-3. Neurobiol. Dis. 2009;36:280–292. doi: 10.1016/j.nbd.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 39.Breuer P., Haacke A., Evert B.O., Wüllner U. Nuclear aggregation of polyglutamine-expanded ataxin-3: fragments escape the cytoplasmic quality control. J. Biol. Chem. 2010;285:6532–6537. doi: 10.1074/jbc.M109.036335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt M.F. Drug target miRNAs: chances and challenges. Trends Biotechnol. 2014;32:578–585. doi: 10.1016/j.tibtech.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Davies B.P., Arenz C. A fluorescence probe for assaying micro RNA maturation. Bioorg. Med. Chem. 2008;16:49–55. doi: 10.1016/j.bmc.2007.04.055. [DOI] [PubMed] [Google Scholar]
- 42.Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 43.Gonçalves N., Simões A.T., Cunha R.A., de Almeida L.P. Caffeine and adenosine A(2A) receptor inactivation decrease striatal neuropathology in a lentiviral-based model of Machado-Joseph disease. Ann. Neurol. 2013;73:655–666. doi: 10.1002/ana.23866. [DOI] [PubMed] [Google Scholar]
- 44.Agarwal V., Bell G.W., Nam J.W., Bartel D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:4. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paraskevopoulou M.D., Georgakilas G., Kostoulas N., Reczko M., Maragkakis M., Dalamagas T.M., Hatzigeorgiou A.G. DIANA-LncBase: experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Res. 2013;41:D239–D245. doi: 10.1093/nar/gks1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Betel D., Koppal A., Agius P., Sander C., Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krek A., Grün D., Poy M.N., Wolf R., Rosenberg L., Epstein E.J., MacMenamin P., da Piedade I., Gunsalus K.C., Stoffel M., Rajewsky N. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 48.Boudreau R.L., Jiang P., Gilmore B.L., Spengler R.M., Tirabassi R., Nelson J.A., Ross C.A., Xing Y., Davidson B.L. Transcriptome-wide discovery of microRNA binding sites in human brain. Neuron. 2014;81:294–305. doi: 10.1016/j.neuron.2013.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krol J., Sobczak K., Wilczynska U., Drath M., Jasinska A., Kaczynska D., Krzyzosiak W.J. Structural features of microRNA (miRNA) precursors and their relevance to miRNA biogenesis and small interfering RNA/short hairpin RNA design. J. Biol. Chem. 2004;279:42230–42239. doi: 10.1074/jbc.M404931200. [DOI] [PubMed] [Google Scholar]
- 50.Bak M., Silahtaroglu A., Møller M., Christensen M., Rath M.F., Skryabin B., Tommerup N., Kauppinen S. MicroRNA expression in the adult mouse central nervous system. RNA. 2008;14:432–444. doi: 10.1261/rna.783108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Long Z., Chen Z., Wang C., Huang F., Peng H., Hou X., Ding D., Ye W., Wang J., Pan Q. Two novel SNPs in ATXN3 3′ UTR may decrease age at onset of SCA3/MJD in Chinese patients. PLoS ONE. 2015;10:e0117488. doi: 10.1371/journal.pone.0117488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramani B., Harris G.M., Huang R., Seki T., Murphy G.G., Costa Mdo.C., Fischer S., Saunders T.L., Xia G., McEachin R.C., Paulson H.L. A knockin mouse model of spinocerebellar ataxia type 3 exhibits prominent aggregate pathology and aberrant splicing of the disease gene transcript. Hum. Mol. Genet. 2015;24:1211–1224. doi: 10.1093/hmg/ddu532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodriguez-Lebron E., Liu G., Keiser M., Behlke M.A., Davidson B.L. Altered Purkinje cell miRNA expression and SCA1 pathogenesis. Neurobiol. Dis. 2013;54:456–463. doi: 10.1016/j.nbd.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khodr C.E., Pedapati J., Han Y., Bohn M.C. Inclusion of a portion of the native SNCA 3′UTR reduces toxicity of human S129A SNCA on striatal-projecting dopamine neurons in rat substantia nigra. Dev. Neurobiol. 2012;72:906–917. doi: 10.1002/dneu.20989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cemal C.K., Carroll C.J., Lawrence L., Lowrie M.B., Ruddle P., Al-Mahdawi S., King R.H., Pook M.A., Huxley C., Chamberlain S. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum. Mol. Genet. 2002;11:1075–1094. doi: 10.1093/hmg/11.9.1075. [DOI] [PubMed] [Google Scholar]
- 56.Switonski P.M., Szlachcic W.J., Krzyzosiak W.J., Figiel M. A new humanized ataxin-3 knock-in mouse model combines the genetic features, pathogenesis of neurons and glia and late disease onset of SCA3/MJD. Neurobiol. Dis. 2015;73:174–188. doi: 10.1016/j.nbd.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 57.Hutchison E.R., Kawamoto E.M., Taub D.D., Lal A., Abdelmohsen K., Zhang Y., Wood W.H., 3rd, Lehrmann E., Camandola S., Becker K.G. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia. 2013;61:1018–1028. doi: 10.1002/glia.22483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie W., Li M., Xu N., Lv Q., Huang N., He J., Zhang Y. MiR-181a regulates inflammation responses in monocytes and macrophages. PLoS ONE. 2013;8:e58639. doi: 10.1371/journal.pone.0058639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coolen M., Katz S., Bally-Cuif L. miR-9: a versatile regulator of neurogenesis. Front. Cell. Neurosci. 2013;7:220. doi: 10.3389/fncel.2013.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cogswell J.P., Ward J., Taylor I.A., Waters M., Shi Y., Cannon B., Kelnar K., Kemppainen J., Brown D., Chen C. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 2008;14:27–41. doi: 10.3233/jad-2008-14103. [DOI] [PubMed] [Google Scholar]
- 61.Kim J., Inoue K., Ishii J., Vanti W.B., Voronov S.V., Murchison E., Hannon G., Abeliovich A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317:1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Catapano F., Zaharieva I., Scoto M., Marrosu E., Morgan J., Muntoni F., Zhou H. Altered levels of microRNA-9, −206, and −132 in spinal muscular atrophy and their response to antisense oligonucleotide therapy. Mol. Ther. Nucleic Acids. 2016;5:e331. doi: 10.1038/mtna.2016.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schaefer A., O’Carroll D., Tan C.L., Hillman D., Sugimori M., Llinas R., Greengard P. Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med. 2007;204:1553–1558. doi: 10.1084/jem.20070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miyazaki Y., Du X., Muramatsu S., Gomez C.M. An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNA1A second cistron. Sci. Transl. Med. 2016;8:347ra94. doi: 10.1126/scitranslmed.aaf5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cunha-Santos J., Duarte-Neves J., Carmona V., Guarente L., Pereira de Almeida L., Cavadas C. Caloric restriction blocks neuropathology and motor deficits in Machado-Joseph disease mouse models through SIRT1 pathway. Nat. Commun. 2016;7:11445. doi: 10.1038/ncomms11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Onofre I., Mendonça N., Lopes S., Nobre R., de Melo J.B., Carreira I.M., Januário C., Gonçalves A.F., de Almeida L.P. Fibroblasts of Machado Joseph disease patients reveal autophagy impairment. Sci. Rep. 2016;6:28220. doi: 10.1038/srep28220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Onofre I. University of Coimbra; 2016. Dissecting the Pathogenesis of Machado-Joseph Disease in a New Human Disease Model Derived from Induced Pluripotent Stem Cells. Faculty of Pharmacy, vol. PhD. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.