

Abstract
Systemic viroimmunotherapy activates endogenous innate and adaptive immune responses against both viral and tumor antigens. We have shown that therapy with vesicular stomatitis virus (VSV) engineered to express a tumor-associated antigen activates antigen-specific adoptively transferred T cells (adoptive cell therapy, ACT) in vivo to generate effective therapy. The overall goal of this study was to phenotypically characterize the immune response to VSV+ACT therapy and use the information gained to rationally improve combination therapy. We observed rapid expansion of blood CD8+ effector cells acutely following VSV therapy with markedly high expression of the immune checkpoint molecules PD-1 and TIM-3. Using these data, we tested a treatment schedule incorporating mAb immune checkpoint inhibitors with VSV+ACT treatment. Unlike clinical scenarios, we delivered therapy at early time points following tumor establishment and treatment. Our goal was to potentiate the immune response generated by VSV therapy to achieve durable control of metastatic disease. Despite the high frequency of endogenous PD-1+ TIM-3+ CD8+ T cells following virus administration, antibody blockade did not improve survival. These findings provide highly significant information about response kinetics to viroimmunotherapy and juxtapose the clinical use of checkpoint inhibitors against chronically dysfunctional T cells and the acute T cell response to oncolytic viruses.
Keywords: oncolytic virotherapy, checkpoint inhibitors, tumor therapy
Graphical Abstract
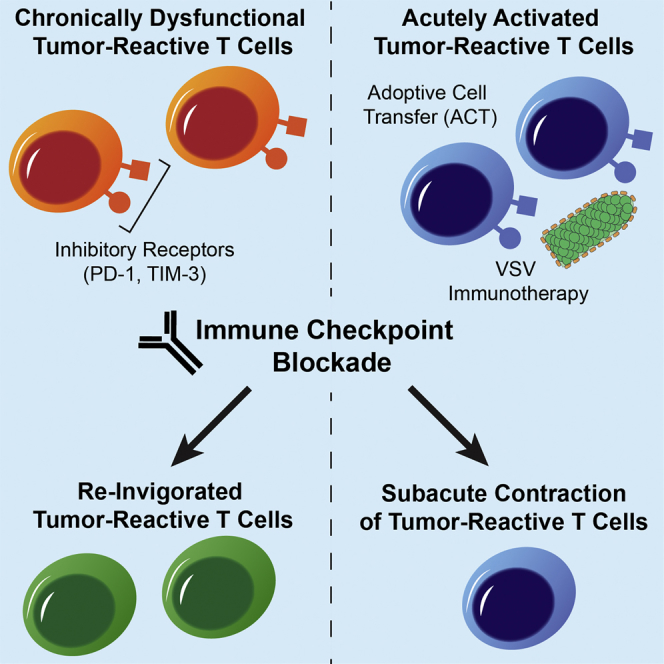
Shim et al. designed a treatment strategy combining VSV viroimmunotherapy and adoptive T cell transfer with immune checkpoint blockade. Viroimmunotherapy caused significant upregulation of the inhibitory receptors PD-1 and TIM-3 on effector T cells, but signaling through these receptors was not a dominant immunomodulatory axis of their therapeutic strategy.
Introduction
Driving the immune system to mount a response against malignancy has emerged as one of the primary goals of contemporary cancer therapies. In this respect, viroimmunotherapy seeks to harness the immunogenicity of viruses to initiate an anti-tumor response. Led by the first in class viral therapeutic T-VEC, a wealth of preclinical and clinical studies suggest that there is real capacity for these therapies to translate to clinically meaningful outcomes.1 There is now considerable evidence that the ability of oncolytic virotherapy to control local disease involves both direct viral oncolysis and multiple host-derived effectors of the innate and adaptive immune system.2, 3 Moreover, we, and others, have focused on understanding how systemically delivered viral therapies may be used to mount a systemic anti-tumor response against widely disseminated metastatic disease, a major challenge to effective cancer therapy.
We have engineered the Rhabdovirus, vesicular stomatitis virus (VSV) to express tumor-associated antigens (TAAs) to generate a host immune response against tumors. Previously, we demonstrated that intra-tumoral administration of VSV controlled B16 melanomas and generated an immunological recall response to specific tumor antigens.4 Additionally, the oncolytic activity of VSV engaged both adaptive and innate immune responses that contributed significantly to anti-tumor effects, which have been validated in a variety of models.3 We, and others, have expanded the concept of oncolytic virotherapy as an immune stimulant against disseminated malignancy by showing that systemic treatment with VSV expressing a defined tumor associated antigen (such as OVA or gp100) activated adoptively transferred naive antigen specific T cells in vivo and led to effective therapy.5 Taken together, these results demonstrate that oncolytic viruses can generate significant therapy against tumors by various mechanisms including direct oncolysis, stimulation of anti-tumor innate immunity, and activation of adaptive T cell responses against both the virus itself and tumor-associated antigens.6, 7, 8
The success of immune inhibitory receptor blocking therapies (immune checkpoint inhibitors) has also highlighted the capacity of the immune system to contribute to the therapy of even widely disseminated metastatic disease. These monoclonal antibody therapies block signaling through inhibitory receptors and are thought to re-invigorate tumor-reactive cells of the immune system that have become chronically dysfunctional (“exhausted”). These therapies, most notably those blocking programmed cell death protein-1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA4), have demonstrated exciting success in the clinic with roughly 60% of metastatic melanoma patients responding to combination therapy and 30% of patients achieving a complete response.9 Another potential therapeutic target is the inhibitory receptor T cell immunoglobulin and mucin-domain containing-3 (TIM-3) associated with T cell effector dysfunction in tumor infiltrating lymphocytes.10 Expression of both TIM-3 and PD-1 has been strongly linked to chronic immune dysfunction in both clinical and preclinical studies.11, 12
The immunogenic mechanism of both viroimmunotherapy and immune checkpoint inhibitors make them ideal candidates for combination therapy. Indeed, there have been several reports of success utilizing a variety of viral (or other pro-inflammatory) therapeutics in conjunction with immune checkpoint inhibitors.13, 14, 15 However, the therapeutic significance of PD-1 and TIM-3 expression is complex. These receptors are expressed both during acute infection (as occurs following oncolytic virus administration) and in chronic dysfunctional states (as occurs in patients with long term exposure to tumors). The continued expression of inhibitory receptors is associated with changes in transcriptional, phenotypic, epigenetic, and metabolic profiles defining a state of chronic dysfunction or exhaustion.16, 17, 18 Therefore, the similarities and differences between these physiological states are likely to have a profound impact on their suitability as targets for enhancing therapy against malignancy at an early versus late stage.
Here, we used our successful strategy of anti-tumor viroimmunotherapy combining intravenous VSV-TAA in combination with antigen-specific T cells to analyze in detail the immune profiles of both endogenous and adoptively transferred T cell subsets. Our overall goal was to predict rationally when, and how, to improve therapy with immune checkpoint blockade. Despite the presence of a large fraction of targetable endogenous PD-1+ and TIM-3+ CD8+ T cells following oncolytic virus administration, antibody blockade of neither molecule added any significant survival benefit. This was in spite of the evidence that inhibitory receptor signaling during TCR activation or the acute effector differentiation in response to infection leads to dampening of the immune response.17, 19, 20 Our findings are significant because they characterize the kinetics of inhibitory receptor expression following the administration of oncolytic virotherapy and test a strategy to take advantage of this expression as a therapeutic target. Furthermore, they highlight the significant differences between inhibitory receptor expression on immune effectors in response to acute virotherapy administration and the current clinical use of inhibitory receptor blockade at late, chronic time points after the development of T cell exhaustion.
Results
VSV Viroimmunotherapy and Adoptive Cell Transfer Controls Metastatic Disease
Building upon our previous work, we utilized a combination of systemic VSV-TAA therapy and adoptive cell transfer (ACT) of naive CD8+ transgenic Pmel T cells in a model of metastatic disease.4, 5, 21 We generated two models of oligometastatic disease in which C57BL/6 immunocompetent mice were challenged both subcutaneously (s.c.) and intravenously (i.v.) with either B16 or B16-OVA tumors (Figure 1A).22 In these models, mice eventually developed tumors throughout the body at late time points when the s.c. tumor was controlled by therapy. In the less aggressive B16-OVA tumor model, combination treatment with VSV-hgp100 and adoptive cell transfer of Pmel T cells significantly improved both overall survival (OS) and median survival (MS) when compared to VSV-hgp100 alone (OS: p < 0.0001 and MS: 22 versus 42 days) (Figure 1B). As well as prolonging overall survival, the combination therapy was successful at delaying the growth of s.c. tumors (Figure 1C). Even at late time points (day 40 or greater), mice treated with combination therapy were free of gross metastases at the time of sacrifice. In the more aggressive B16 tumor model, as expected, Pmel therapy alone offered no improvement in overall survival and though VSV-hgp100 + Pmel therapy statistically prolonged overall survival, it had a modest effect on prolonging median survival (OS: p < 0.0001 and MS: 15 versus 20 days) (Figures 1D and 1E). These data extend our previous results by demonstrating that our combination therapy was successful at treating widely disseminated metastatic disease.
Figure 1.

VSV-hgp100+ACT Treatment Strategy Controls Disseminated Disease
(A) Mice were challenged first s.c. tumor and i.v. tumor. When the s.c. tumor was visible (day 6–8), CD8+ Pmel T cells were adoptively transferred into the mice. Beginning on day 7–9, and continuing three times per week, the mice received a total of six doses of VSV-hgp100 or PBS. (B) Mice challenged with s.c. and i.v. B16-OVA received the treatment schedule above, in addition to a second Pmel treatment on day 21 following tumor challenge. n = 10 mice/group. (C) Subcutaneous tumor volumes measured three times weekly with calipers. Each line represents an individual mouse, grouped by treatment. (D) Mice challenged s.c. and i.v. with B16 cells received the treatment schedule above. n = 11 mice/group. (E) Subcutaneous tumor volumes measured three times weekly with calipers. Each line represents an individual mouse grouped by treatment. The significance for overall survival was determined at p < 0.01.
VSV Immunotherapy Generates an Acute CD8+ Response Characterized by a High Proportion of Inhibitory Receptor Expressing Cells
Despite the improvements in overall survival, the combination therapy of VSV-TAA and ACT of Pmel T cells was unable to cure mice (Figure 1). Therefore, we reasoned that it might be possible to improve the efficacy of this viroimmunotherapy by understanding the systemic immune response to treatment. Hence, with the goal of developing rational strategies to enhance our systemic therapy, we performed detailed immune phenotyping of mice treated with VSV through serial sampling of the peripheral blood. Live CD4+ or CD8+ T cells that had an effector phenotype (CD44hi CD62Llo) were assessed for their expression of the inhibitory receptors PD-1 and TIM-3 at predetermined time points during the course of therapy (Figures 2A and 2B). Consistent with a T cell response against an invading virus, the frequency of CD8+ effector cells peaked rapidly at 5 days after initial VSV therapy, followed by an immediate contraction phase (Figure 2C). A majority of the CD8+ endogenous cells were double positive for both inhibitory receptors PD-1 and TIM-3, with 80% of all effector CD8+ cells expressing PD-1 and TIM-3 at day 14. The frequency of double positive effector CD8+ T cells returned to basal levels at a more gradual rate than the contraction of the overall CD8+ effector cell population (Figure 2D). In contrast, the frequency of CD4+ effector cells, and their expression of inhibitory receptors, remained stable (K.G.S. and R.G.V., unpublished data). The acute expansion and contraction of antigen experienced CD44+ CD8+ splenocytes mirrored the frequency of effector cells expressing both PD-1 and TIM-3 in the blood (Figures 2E and 2F). We observed that the increase in the frequency of PD-1+ TIM-3+ cells in the blood was due primarily to an increase in the fraction of effector cells expressing TIM-3. At baseline levels, tumor-bearing mice expressed PD-1 at a high frequency (>60%) of circulating effector cells, whereas TIM-3 was present on a much smaller fraction of cells (Figures 2G and 2H). Immediately after administration of VSV immunotherapy, the fraction of cells expressing TIM-3 increased dramatically, then gradually declined to a point where a TIM-3 single positive population was actually dominant in the circulation (Figure 2H). The biological significance of this shift from baseline dominance of PD-1 expression to TIM-3 expression on circulating effector cells is unknown, but suggested TIM-3 as a potential target to block. Furthermore, we validated the specificity of the above effector T cell population by utilizing tetramer staining. Data gathered from the blood (Figures 2I and 2J) and spleen (Figures 2K and 2L) demonstrate that between 20%–40% of CD8+ effector cells recognize the immunodominant VSV N protein peptide RGYVYQGL. An assessment of tumor infiltrating lymphocyte (TIL) populations was also conducted to determine the PD-1 and TIM-3 expression status of this population. Tumors were of similar weights at day 22 post challenge (Figure 3A). While treatment with VSV-hgp100 showed a trend toward an increase in the number and fraction of CD8+ and CD4+ cells in the tumor (Figures 3B–3E), there were no statistically significant changes. Mice treated with VSV-hgp100 did demonstrate a statistically significant increase in the fraction of CD8+ cells that were of the effector phenotype (Figure 3F), which expressed both inhibitory receptors PD-1 and TIM-3 at high levels (Figure 3G).
Figure 2.

VSV-hgp100 Therapy Induces Acute CD8+ Effector Cell Expansion and Marked Expression of PD-1 and TIM-3
(A) Mice were challenged first with B16-OVA s.c. tumor then with i.v. tumor. Beginning on day 9, and continuing every other day during the week, the mice received a total of six doses of VSV-hgp100 or PBS. The mice were sacrificed at the indicated time points. (B) Gating strategy showing representative CD8+ effector (CD44hiCD62Llo) cells and their expression of PD-1 and TIM-3. (C and D) Blood flow cytometry. The frequencies of effector cells gated on CD8+ cells and frequencies of PD-1+ TIM-3+ cells gated on CD8+ effectors are shown. Each of the points represents 3–5 mice/group. (E and F) Splenic frequencies of CD8+ CD44+ effector cells and PD-1 and TIM-3 double positive effector cells. Each of the points represents 1–5 mice/group. (G and H) Frequencies of PD-1 or TIM-3 double negative, single positive, or double positive cells gated on CD8+ effectors in the blood. The above results are representative of three independent experiments with n = 1–5 mice per group. (I and J) Blood tetramer staining for the frequency and number of CD8+ effector cells reactive to the immunodominant VSV N peptide. (K and L) Splenic staining for the frequency and number of VSV N Tetramer+ CD8+ effector cells. The median with interquartile range is shown. ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05. The significance was determined at p < 0.05.
Figure 3.

VSV Immunotherapy Causes Tumor Infiltration by Inhibitory Receptor Expressing Effector T Cells
Mice were challenged with B16-OVA and treated with VSV-hgp100 as described in Figure 2. The tumors were harvested at day 22 post-challenge and analyzed by flow cytometry. (A) Tumor weight in grams of subcutaneous tumors harvested from mice at the time of sacrifice. (B and C) The fraction of live cells that were CD8+ or CD4+ in the tumor. (D and E) The number of CD8+ or CD4+ cells in 50 mg of tumor. (F and G) The fraction of CD8+ effector cells and the fraction of those effector cells double positive for expression of the inhibitory receptors PD-1 and TIM-3 (G). The median and interquartile range are shown. *p < 0.05.
Targeting PD-1 or TIM-3 Induced by VSV Immunotherapy Does Not Improve Treatment Outcomes
The sequence of rapid effector cell expansion and contraction seen in response to VSV immunotherapy (Figure 2) led us to hypothesize that early inhibitory signaling through PD-1 and TIM-3 may hinder the capacity of our therapy to drive a more robust anti-tumor response. Inhibition may occur by causing rapid contraction and inhibitory receptor-mediated suppression of potentially tumor-reactive effector cells (either endogenous or adoptively transferred). Alternatively, inhibition may occur by dampening the response of VSV reactive host immune effectors in vivo that contribute to the immunogenic environment supporting Pmel activation. Thus, we hypothesized that early blockade of PD-1 or TIM-3 inhibitory receptor signaling in combination with VSV immunotherapy would improve treatment outcomes in tumor-bearing mice by potentiating the immune response against tumors generated by VSV immunotherapy (Figure 4A). This differs from clinical studies on the impact of PD-1 or TIM-3 blockade on late stage, chronically dysfunctional T cells. For these experiments, we challenged mice with i.v. tumors only to model widely disseminated metastatic disease. As in our previous tumor models, the VSV-hgp100+Pmel ACT combination therapy significantly improved overall and median survival compared to untreated mice (OS: p < 0.0001 and MS: 23 versus 53 days) (Figure 4B). The use of this model also allowed us to extend mouse survival for long-term study, which was prevented by the rapid growth of the s.c. tumors that eventually led to the sacrifice of all mice in the oligometastatic model of Figure 1. However, despite a high frequency of PD-1 expressing CD8+ effectors following virotherapy (Figure 2), the addition of early PD-1 blocking mAb therapy did not improve overall or median survival when compared to an isotype antibody control or no antibody (OS: ISO versus PD-1; p = 0.32 and MS: 56 versus 53 days) (Figure 4B). To ensure that this was not a function of the hgp100/Pmel model, we also tested the approach using VSV-OVA and adoptive transfer of OVA-specific transgenic OT-I T cells. Similar results were seen in a less aggressive model of B16-OVA (OS: ISO versus PD-1; p = 0.55 and MS: 118 days versus not reached) (Figure 4C). We have previously demonstrated the biological efficacy of the anti-PD-1 antibody utilized in a model of i.t. Reovirus administration into B16 tumors.23 Based on the increased frequency of effector cells expressing TIM-3 during the T cell expansion phase of our therapy (Figure 2), we tested a TIM-3 blocking mAb beginning at the same, early time point. As with antibody-mediated blockade of PD-1, anti-TIM-3 therapy had no effect on the overall or median survival of tumor bearing mice (OS: ISO versus TIM-3; p = 0.51 and MS: 66 versus 54 days) (Figure 4D).
Figure 4.

Addition of PD-1 or TIM-3 Blocking Antibody to VSV-hgp100+ACT Therapy Does Not Improve Treatment Outcomes
(A) Mice were challenged with i.v. tumor only. The mice were treated with Pmel ACT on day 7 or 8, a second treatment was delivered on day 21. Beginning on day 8 or 9, and continuing three times per week, the mice received a total of nine doses of VSV-hgp100 or PBS. The mice received six doses of isotype antibody control or immune checkpoint inhibitor beginning with the third dose of VSV (day 14–17). The antibody was delivered on the same day, after VSV administration. n = 10 mice/group. (B) Mice were challenged with i.v. B16 tumor and treated with PD-1 inhibitor. (C) Mice were challenged with i.v. B16-OVA tumor and treated with PD-1 inhibitor. (D) Mice were challenged as in (A), but treated with TIM-3 inhibitor. The significance for overall survival was determined at p < 0.008.
PD-1 Blockade Does Not Alter the Kinetics of Endogenous Effector T Cells in Response to VSV Therapy
We used a serial phenotyping approach on the mice treated in Figure 4B to track the kinetics of inhibitory receptor expression on peripheral blood cells in each individual mouse in response to PD-1 therapy over a broad time window. CD4+ and CD8+ effector cells, and the frequency of these cells expressing PD-1 and TIM-3, were gated using the strategy shown in Figure 2. Additionally, endogenous cells (Thy1.1−) were distinguished from adoptively transferred Pmel T cells (Thy1.1+) (Figure 5A). Mice treated with PD-1 inhibitor showed very few subsequent phenotypic changes when compared with untreated or isotype treated mice. The frequency of circulating CD4+ and CD8+ endogenous effector cells demonstrated similar kinetics, irrespective of treatment with isotype control or PD-1 blocking antibody (Figures 5B and 5C). There was a modest elevation in the fraction of endogenous CD8+ effector cells between days 24 and 38 in mice treated with PD-1 blockade (Figure 5C), with a small, statistically significant increase in the fraction of CD8+ effector T cells in PD-1 blocking mAb-treated mice compared with no mAb or ISO treated mice 15 days after VSV therapy. This overall lack of difference challenged our hypothesis, which postulated that blockade of PD-1 signaling would disrupt the contraction phase of the immune response generated by VSV. Furthermore, the fraction of CD8+ endogenous effector cells double positive for PD-1 and TIM-3 remained unchanged when either isotype control or PD-1 blocking mAb was added to the treatment schedule (Figure 5D).
Figure 5.

Addition of PD-1 or TIM-3 Blocking Antibody Therapy to VSV-hgp100+ACT Therapy Minimally Alters Immune Kinetics
Mice from Figure 3B had serial submandibular vein blood samples taken at the time points indicated in (B)–(H). (A) Gating strategy shows representative plot to distinguish Pmel adoptively transferred Thy1.1+ from BL/6 endogenous Thy1.1− cells, gated on CD8+ cells. (B and C) Frequencies of effector cells (CD44hiCD62Llo) gated on CD4+ or CD8+ cells. (D) Frequencies of PD-1+ TIM-3+ cells gated on CD8+ effectors. (E and F) Frequency and number of Thy1.1+ Pmel T cells gated on CD8+ cells. (G) Frequency of effector (CD44hiCD62Llo) cells gated on CD8+ Thy1.1+ cells. (H) Frequencies of PD-1+ TIM-3+ cells gated on CD8+ Thy1.1+ effectors. Each of the points represents 1–5 mice/group. The median with interquartile range is shown. *p < 0.05. The significance was determined at p < 0.05.
Immune Kinetics of Adoptively Transferred T Cells following Virotherapy
Interestingly, the fraction, and number, of Thy1.1+ Pmel CD8+ cells that were adoptively transferred did not undergo the same expansion as endogenous cells in response to systemic VSV therapy (Figures 5E and 5F). Furthermore, there were variations in the frequency and numbers of detected Pmel T cells across all groups over time, with no distinct expansion/contraction phase or clear response associated with checkpoint blockade. However, adoptively transferred Pmel T cells demonstrated a striking shift from naive or non-activated (CD44lo CD62Lhi) phenotypes to an antigen experienced phenotype, with nearly 100% of Pmel effectors acquiring the CD44hi CD62Llo phenotype by 17 days following transfer (Figure 5G). A maximum of 25%–30% of adoptively transferred cells expressed both inhibitory receptors PD-1 and TIM-3 at 3 days following transfer and this fraction quickly diminished to ∼5% over a short period of 7 days (Figures 5G and 5H). This was in contrast to ∼80% of endogenous CD8+ T cells at 9 days following transfer (Figure 5D). Taken together, Figures 5E–5H demonstrate that there was no significant effect on the immune phenotype of adoptively transferred Pmel T cells when PD-1 inhibitor was added to the VSV-hgp100+Pmel treatment strategy. However, the data of Figure 4 emphasize that systemic treatment with oncolytic virus induces different kinetics of expansion and profiles of inhibitory receptor expression on endogenous versus adoptively transferred effector T cell populations.
Effects of PD-1 Blockade on Memory T Cell Generation
We also investigated whether PD-1 blockade at an acute time point after virus administration might impact the nature of the T cell memory generated when combined with VSV-TAA+ACT therapy. Using the B16-OVA/VSV-OVA/OT-I model, the peripheral blood of mice surviving tumor challenge, treated as described in Figure 4C, was analyzed just prior to, and 1 week after, re-challenge with intravenous B16-OVA tumor to model recurrent metastases (Figure 6A). Figures 6B and 6C demonstrate that re-challenge with tumor did not induce an increase in the frequency of CD8+ effector cells in circulation. Correspondingly, prior to re-challenge with tumor, there was no difference in the frequency of CD8+ central memory (CD44hi CD62Lhi) cells found in circulation when comparing across all treatment groups. Similarly, the fraction of central memory cells did not change following tumor re-challenge. To monitor antigen-specific immune response, we assessed the fraction of CD8+ T cells in circulation that recognized the SIINFEKL peptide and their effector phenotype (Figure 6D). We observed that treatment with PD-1 blocking antibody resulted in no change in the fraction of cells recognizing this specific antigen. We also did not observe a difference in the percent of SIINFEKL tetramer positive cells prior to, or following, re-challenge with intravenous B16-OVA tumor (Figures 6E and 6F). Splenocytes evaluated at the day 200 time point also showed no changes in the frequency of the populations assayed between treatment groups (K.G.S. and R.G.V., unpublished data). Overall, there were no statistically significant changes in the frequencies of the memory populations evaluated when comparing treatment groups at each time point.
Figure 6.

Early Treatment with PD-1 or TIM-3 Blocking Antibody Therapy Does Not Alter Late-Stage Memory Phenotypes
(A) Surviving mice from the experiment in Figure 3C were re-challenged with i.v. B16-OVA on day 193. A submandibular vein bleed was taken 1 day before re-challenge and the mice were sacrificed 1 week after re-challenge. (B and C) Frequencies of effector (CD44hiCD62Llo) or central memory (CD44hiCD62Lhi) cells gated on CD8+ cells. (D) Gating strategy to identify SIINFEKL tetramer+ CD8+ cells and effector or central memory phenotypes. (E and F) Frequency of SIINFEKL tetramer+ cells gated on CD8+ cells and effector cells gated on CD8+ tetramer+ cells. Each of the points represents 3–5 mice/group. The median and interquartile range are shown.
Pmel T Cells Are Ineffective Unless Adoptively Transferred to a New Host
Given the lack of a clearly defined VSV-induced expansion of Pmel T cells (Figure 5), we hypothesized that VSV-gp100+Pmel therapy of B16 tumors may be limited by the number of effector Pmel cells which are present following adoptive T cell transfer. To test this hypothesis, we established B16 tumors in Pmel transgenic mice with a potentially limitless number of Pmel T cells which could act as anti-tumor effectors after VSV-mediated activation in vivo (Figure 7A). In a model of the s.c. disease, treatment of Pmel mice with systemic VSV-hgp100 therapy statistically improved overall survival with a modest improvement in median survival (OS: p = 0.0005 and MS: 15 versus 24). When the i.v. tumor challenge was added with the s.c. tumor, no significant improvements in overall survival were observed along with a minimal prolongation of median survival (OS: 0.062 and MS: 15 versus 19 days) (Figures 7A and 7B). Circulating CD8+ T cells (with the transgenic Vα1/Vβ13 Pmel receptor) underwent only a modest differentiation to an effector (CD44hi CD62Llo) phenotype (Figure 7C), which was in stark contrast to the rapid and stable differentiation of adoptively transferred Pmel T cells in BL/6 mice (compare with Figure 5G). The effector cells in VSV treated Pmel mice expressed inhibitory receptors PD-1 and TIM-3 at roughly the same frequency that adoptively transferred Pmel T cells had (Figure 7D). Administration of PD-1 therapy in combination with VSV in a model of the s.c. tumor challenge resulted in no substantial changes in median or overall survival when compared to the administration of PD-1 alone (OS: p = 0.055 and MS: 14 versus 21 days) (Figure 7E).
Figure 7.

Pmel Therapy Is Dependent on the Environment of Adoptive Cell Transfer
(A) Pmel transgenic mice were challenged with s.c. B16 tumor and treated with a total of six doses of VSV-hgp100 or PBS. n = 8 mice/group. (B–D) Pmel mice were challenged both s.c. and i.v. with B16 tumor cells. The mice were treated as in (A). n = 10 mice/group. The significance for overall survival was determined at p < 0.05. The serial submandibular vein bleeds from mice treated in (B) show peripheral blood populations of effector (CD44hiCD62Llo) cells gated on CD8+ cells (C) and the frequency of PD-1+ TIM-3+ cells gated on CD8+ effector cells (D). Each of the columns represents 1–5 mice/group. (E) Mice were challenged with B16 tumor cells s.c. only. The mice were treated with three doses of i.v. PD-1 inhibitor beginning with the first dose of VSV (day 9). The antibody was administered on the same day following VSV therapy. n = 8 mice/group. The significance for overall survival was determined at p < 0.01.
Discussion
Our first goal was to conduct kinetic analysis of PD-1 and TIM-3 expression on lymphocytes in the context of oncolytic VSV administration. Our second goal was to use this information to design a rational strategy to improve our effective viroimmunotherapy regimen of VSV-hgp100+Pmel with inhibitory receptor blockade. Our treatment strategy was ideal for investigating the potential of adding checkpoint inhibitors because we could easily track the impact of therapy on both endogenous and exogenous adoptively transferred cell populations. We show here the immune response following treatment with oncolytic VSV is characterized by an acute expansion of endogenous CD8+ T cells, followed by contraction, as expected from an anti-viral immune response (Figure 2). A markedly high proportion of these expanding effector cells expressed both PD-1 and TIM-3. We show at the peak of acute response to VSV immunotherapy ∼35% of circulating and ∼25% of splenic CD8+ effector cells recognize the immunodominant VSV N protein peptide, alongside a proportion we have previously demonstrated to have anti-tumor activity.6
This acute profile of inhibitory receptor expression suggested that either PD-1 and/or TIM-3 might be therapeutic targets to enhance the efficacy of VSV-hgp100+Pmel in the B16 model. This strategy was supported by in vitro findings from others demonstrating that PD-1 signaling during early TCR activation can disrupt T cell function and in vivo studies utilizing LCMV, wherein a dysfunctional phenotype was imprinted on cells during acute effector differentiation in response to antigen.17, 19, 20 Studies characterizing the impact of blocking TIM-3 signaling also provided rationale for this strategy.24, 25 In particular, we hypothesized that antibody blockade of PD-1 and TIM-3 would enhance therapy in three ways: first, checkpoint blockade would directly enhance the activation (de-repression) of anti-tumor adoptively transferred PD-1+ TIM-3+ Pmel cells; second, blockade of checkpoint molecules on endogenously activated immune effectors would potentiate their activity directly against tumor; and finally, the immune checkpoint blockade may relieve inhibition of the endogenous anti-viral immune response, which provides a highly immunogenic environment to support the expansion of both Pmel and endogenous anti-tumor T cells.
Interestingly, despite the high fraction of endogenous PD-1+ TIM-3+ CD8+ T cells in the blood, spleens, and tumors (Figures 3 and 5D), as well as a smaller fraction of adoptively transferred PD-1+ TIM-3+ Pmel T cells following oncolytic virus administration (Figure 5H), antibody blockade of neither molecule added any significant survival benefit (Figure 4). Despite the known inhibitory signaling through PD-1 and TIM-3 during acute stimulation, the blockade of signaling through these receptors in our in vivo model did not significantly alter the kinetics of the immune response observed against systemic VSV treatment (Figure 5). This includes no difference in the late stage antigen-specific memory populations found in mice treated with PD-1 compared to those that were not treated or given isotype control antibody (Figure 6). Our data do not exclude the possibility that therapy with VSV-TAA+ACT would be further improved by the combination of multiple immune checkpoint inhibitors (such as anti-PD-1+TIM-3 or anti-CTLA4+PD-1 etc.). We have not undertaken these experiments here due to the lack of any improvement in therapy afforded by either anti-PD-1 or anti-TIM-3 therapy alone. These findings suggest that combination of these two agents may not have dramatic added efficacy. This is in contrast to the improvement in clinical outcomes that were observed when patients were treated with single agent CTLA-4 or PD-1 blocking therapy. In addition, we have also found that expression of CTLA-4 was not significantly upregulated on CD8+ cell populations or CD4+ FoxP3+ Treg populations by VSV-hgp100 therapy either with or without added Pmel therapy (K.G.S. and R.G.V., unpublished data).
Our data also illustrate the importance of understanding the interactions of the host immune system with adoptively transferred T cells in the context of a strong immunogenic stimulus like VSV. Treatment of B16 tumors with Pmel ACT alone (in the absence of VSV-hgp100) was completely ineffective.5 Ongoing studies have shown that this is due to a rapid loss of adoptively transferred T cells in the absence of VSV-mediated antigen presentation, suggesting that further combination with checkpoint inhibition is unlikely to have additional therapeutic effects (K.G.S. and R.G.V., unpublished data). Our results here show that the immunological environment a T cell is transferred into shapes its ability to control tumor. Here, we observed that Pmel T cells in a Pmel mouse were completely ineffective against an established tumor even when treated with VSV-hgp100. In contrast, when a much smaller number of Pmel T cells were adoptively transferred into BL/6 mice, we observed rapid and stable effector differentiation of the Pmel T cells with effective anti-tumor therapy (Figures 5G and 7D). Understanding the basis of these effects has significant implications for the development of effective adoptive transfer therapies, including with chimeric antigen receptor T cells.
We believe that these data are highly significant in three principal ways. First, they re-affirm that not all inhibitory receptor expression is equal and that the simple presence of inhibitory receptors on immune effector cells following virotherapy is not a guarantee of improved efficacy through antibody-mediated blockade. Second, this conclusion highlights the importance of understanding exactly which immune subsets are relevant to tumor clearance during different forms of virotherapy. In our model here, therapy is principally mediated through in vivo activation of adoptively transferred, naive Pmel T cells. This treatment is enhanced by global immune activation by the anti-viral immune effector response. Although we observed the induction of large fractions of inhibitory receptor positive T cells upon VSV administration (both endogenous and adoptively transferred), the induction of PD-1 and TIM-3 expression on these cells represented markers of acute T cell activation and expansion rather than of chronic T cell dysfunction (exhaustion). In this context, checkpoint inhibitor blockade might have been ineffective at enhancing therapy because there was minimal T cell dysfunction to reverse. Furthermore, these data suggest that an acute immune response to VSV infection is not suppressed or limited by signaling through either of the inhibitory receptors expressed on the circulating or splenic cells assayed. Third, our data illustrate the important differences between the clinical contexts in which antibody blockade of checkpoint molecules has been successful and the acute T cell response to oncolytic viruses, especially in a model such as this where true T cell exhaustion against tumor antigens has not been induced.
Clinical data demonstrate that targeting checkpoint molecules such as PD-1 holds considerable promise to re-invigorate exhausted T cell responses against tumors. However, these data also suggest that checkpoint blockade is effective in patients in which an anti-tumor response exists, but which has become functionally exhausted over time. In many models of oncolytic virotherapy, including ours here, checkpoint inhibitor expression on immune effectors is not a reflection of this chronic T cell exhaustion, but rather a marker of acute T cell expansion. Though it is has been demonstrated that signaling through both PD-1 and TIM-3 diminish T cell responses by regulating transcription factor activity, it remains to been seen how this signaling is qualitatively different than the inhibitory signaling that occurs with T cells possessing a chronically dysfunctional phenotype.26, 27 We hope these results will stimulate further investigation into the differences between infection and tumor-induced expression of inhibitory receptors and serve as a potentially cautionary result for use of immune checkpoint inhibitors in contexts which may not reflect clinically analogous situations. Therefore, it will be important to appreciate these differences in future studies of combinations of oncolytic virotherapy with checkpoint inhibitor blockade. In particular, it will be informative to develop preclinical models in which genuine anti-tumor T cell exhaustion is induced before treatments with oncolytic viruses are attempted in combination with checkpoint inhibitor blockade.
Importantly, other studies, including from our own laboratory, have shown successful synergy of checkpoint blockade with oncolytic virotherapy.13, 23, 28 These studies indicate that checkpoint inhibitor expression on various immune effector cells at early time points can still be inhibitory to an anti-tumor response under certain circumstances. For example, we have shown that direct intra-tumoral Reovirus therapy of B16 tumors can be enhanced by combination with anti-PD-1 therapy. In that model, therapy was partly mediated by PD-1 inhibited NK cells that represented a productive target for inducing a more effective anti-tumor response. In addition, Fourcade et al. showed that blockade of TIM-3 combined with tumor peptide vaccination resulted in the expansion of tumor-specific T cells.29 These studies suggest that perhaps each immunogenic tumor therapy has a unique activation profile characterized by differential expression levels of inhibitory receptors on different populations of cells. Understanding these variables for each immunotherapy will be critical to identify if therapy is to be rationally improved. Our data suggest that the high TIM-3 expression noted on endogenous and exogenous adoptively transferred cells (Figure 2H) may simply be part of the immune signature of acute VSV infection. Thus, the success of the combination of oncolytic virotherapy with checkpoint inhibitor blockade will depend upon the virus used, the route of administration, the tumor model, the immune effectors which are responsible for tumor clearance induced by oncolytic virotherapy, and the amount of immune cell exhaustion which is induced by the tumor model.
In summary, our results here show that combination therapy between oncolytic virotherapy and checkpoint inhibitor antibody blockade is not automatically assured of success by the mere presence of checkpoint molecules on immune effectors induced by the virus. Instead, it will be important to monitor inhibitory receptor expression quantitatively, kinetically, and (perhaps most significantly) qualitatively, on different immune effector populations following virus administration. It will also be critical to correlate those data with an understanding of the immunological effector mechanisms by which anti-tumor therapy is operating in vivo. Finally, better models will need to be developed which reflect the clinical situation in which genuinely exhausted tumor-reactive T cells exist prior to treatment with oncolytic viruses and checkpoint blockade.
Materials and Methods
Cells and Viruses
Murine B16 cells were maintained in DMEM with 10% (v/v) fetal calf serum. B16-OVA cells are B16 cells transfected with pcDNA3.1OVA and were maintained in 10% DMEM with 5 mg/mL G418 selection media. All cell lines were regularly shown to be free of Mycoplasma infection. All VSV used was generated as previously described.30 Briefly, VSV (Indiana serotype) expressing tumor-associated antigens was generated by cloning the respective antigen into the pVSV-XN2 plasmid by inserting between XhoI and NheI restriction sites between the VSV G and L proteins. All viruses were titered by standard plaque assay on BHK cells.
In Vivo Studies
Female C57BL/6 mice were obtained from The Jackson Laboratory at 6–8 weeks of age and maintained in a pathogen-free BSL2 biohazard certified housing facility. Mice were challenged with tumor cells in a total volume of 100 μL of PBS either s.c. in the right lower limb or i.v. through the tail vein. Mice were challenged with B16-OVA s.c. at a dose of 1–5 × 105 cells and i.v. at a dose of 4 × 104 cells. For studies with B16, mice were challenged with 2.5 × 105 cells s.c. and with 4 × 104 cells i.v. For i.v. B16 tumor re-challenge, 4 × 105 cells were delivered. In studies where mice were challenged with both a s.c. and i.v. tumor, the s.c. tumor was delivered first followed 2 or 3 days later with an i.v. tumor. All mice with the s.c. challenge had their tumors measured three times weekly with calipers. All mice with the i.v. tumor were checked for signs of distress (e.g., lethargy and labored breathing) daily. The presence of a systemic tumor was monitored at the time of death by conducting a necropsy, taking note of any gross metastatic disease. There were six or nine doses of VSV that were administered in 100 μL of PBS, i.v., three times weekly, at a dose of 5 × 106 PFU. ACT therapy was the delivery of 1 × 106 CD8+ cells isolated by a magnetic bead separation kit (Miltenyi Biotec) from transgenic OT-1 or Pmel combined spleens and lymph nodes.31, 32 ACT was delivered i.v. through the tail vein in 100 μL of PBS. Monoclonal blocking antibodies were administered as six doses of 250 μg each in 100 μL of PBS. Anti-PD1 antibody (RMP1-14) and anti-TIM3 antibody (RMT3-23) were delivered i.p. three times weekly (BioXCell). Rat IgG isotype control antibodies were delivered at the same dose and in the same manner (Jackson ImmunoResearch). All animal studies were conducted in accordance with the Mayo Clinic Institutional Animal Care and Use Committee guidelines.
Flow Cytometry
Flow cytometry was performed on freshly explanted spleens, blood, or tumors. Blood was taken either serially in a ∼200 μL submandibular vein bleed or from cardiac puncture at the time of sacrifice. Blood was collected in heparinized tubes, washed twice with ACK lysis buffer, and re-suspended in PBS for staining. Spleens were crushed through 100 μm filters and washed with PBS. Following one wash with ACK lysis buffer, splenocytes were re-suspended in PBS for flow cytometry. Tumors were weighed then crushed as the spleens were and washed twice with PBS. The equivalent of 50 mg of tumor, or the entire volume if 50 mg was not available, was suspended in PBS then analyzed by flow cytometry. There were 1 to 1.5 million events that were collected during flow cytometry analysis or until the entire sample was analyzed. All samples were fixed in 4% formalin and analyzed using a modified BD FACSCanto II flow cytometer. Antibody clones used include: CD8a (53-6.7), CD4 (RM4-5), CD44 (IM7), CD62L (MEL14), PD1 (RMP1-30), TIM3 (RMT3-23), H-2Kb restricted OVA peptide SIINFEKL tetramer (MBL International), Thy1.1 (H1551), and Zombie NIR Fixable Viability Dye (BioLegend). The following reagent was obtained through the NIH Tetramer Core Facility: H-2Kb restricted VSV N peptide tetramer (RGYVYQGL). All data were analyzed using FlowJo software (FlowJo). Biexponential transformation was carried out on figures showing gating strategies to improve visualization.
Statistical Analysis
All statistical analysis was conducted using GraphPad Prism software (GraphPad Software). All survival analysis was conducted using log rank tests. The threshold for significance was determined by using the Bonferroni correction for multiple comparisons. All quantitative flow cytometry data depict median values of a treatment group with error bars showing interquartile range of the sample sizes listed in the respective figure legends. To analyze quantitative flow cytometry data, one-way ANOVA testing was conducted with a Tukey post-test, p values reported from these analyses were corrected to account for multiple comparisons. For comparisons between two groups, an unpaired t test was conducted.
Author Contributions
Conceptualization, K.G.S. and R.G.V.; Investigation, K.G.S., S.Z., J.T., L.E., and T.K.; Writing - Original Draft, K.G.S. and R.G.V.; Writing - Review & Editing, K.G.S., R.G.V., L.E., S.Z., K.R.R., M.S., C.B.D., A.H., and J.S.P.; Supervision, R.G.V. and J.S.P.; and Funding Acquisition, R.G.V. and K.G.S.
Acknowledgments
The authors thank Toni L. Higgins for expert secretarial assistance. This work was funded in part by the European Research Council, the Richard M. Schulze Family Foundation, the Mayo Foundation, Cancer Research UK, and the NIH (R01CA175386 and R01CA108961). K.G.S. was supported by the NIH grants from the National Cancer Institute (F30 CA199962) and the National Institute of General Medical Sciences (T32 GM65841).
References
- 1.Andtbacka R.H.I., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 2.Janelle V., Langlois M.-P., Lapierre P., Charpentier T., Poliquin L., Lamarre A. The strength of the T cell response against a surrogate tumor antigen induced by oncolytic VSV therapy does not correlate with tumor control. Mol. Ther. 2014;22:1198–1210. doi: 10.1038/mt.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aurelian L. Oncolytic viruses as immunotherapy: progress and remaining challenges. Onco. Targets Ther. 2016:2627–2637. doi: 10.2147/OTT.S63049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wongthida P., Diaz R.M., Pulido C., Rommelfanger D., Galivo F., Kaluza K., Kottke T., Thompson J., Melcher A., Vile R. Activating systemic T-cell immunity against self tumor antigens to support oncolytic virotherapy with vesicular stomatitis virus. Hum. Gene Ther. 2011;22:1343–1353. doi: 10.1089/hum.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rommelfanger D.M., Wongthida P., Diaz R.M., Kaluza K.M., Thompson J.M., Kottke T.J., Vile R.G. Systemic combination virotherapy for melanoma with tumor antigen-expressing vesicular stomatitis virus and adoptive T-cell transfer. Cancer Res. 2012;72:4753–4764. doi: 10.1158/0008-5472.CAN-12-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pulido J., Kottke T., Thompson J., Galivo F., Wongthida P., Diaz R.M., Rommelfanger D., Ilett E., Pease L., Pandha H. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat. Biotechnol. 2012;30:337–343. doi: 10.1038/nbt.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kottke T., Errington F., Pulido J., Galivo F., Thompson J., Wongthida P., Diaz R.M., Chong H., Ilett E., Chester J. Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors. Nat. Med. 2011;17:854–859. doi: 10.1038/nm.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boudreau J.E., Bridle B.W., Stephenson K.B., Jenkins K.M., Brunellière J., Bramson J.L., Lichty B.D., Wan Y. Recombinant vesicular stomatitis virus transduction of dendritic cells enhances their ability to prime innate and adaptive antitumor immunity. Mol. Ther. 2009;17:1465–1472. doi: 10.1038/mt.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larkin J., Chiarion-Sileni V., Gonzalez R., Grob J.-J., Cowey C.L., Lao C.D., Schadendorf D., Dummer R., Smylie M., Rutkowski P. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakuishi K., Apetoh L., Sullivan J.M., Blazar B.R., Kuchroo V.K., Anderson A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J.M., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fourcade J., Sun Z., Benallaoua M., Guillaume P., Luescher I.F., Sander C., Kirkwood J.M., Kuchroo V., Zarour H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010;207:2175–2186. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen W., Patnaik M.M., Ruiz A., Russell S.J., Peng K.-W. Immunovirotherapy with vesicular stomatitis virus and PD-L1 blockade enhances therapeutic outcome in murine acute myeloid leukemia. Blood. 2016;127:1449–1458. doi: 10.1182/blood-2015-06-652503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahrends T., Bąbała N., Xiao Y., Yagita H., van Eenennaam H., Borst J. CD27 agonism plus PD-1 blockade recapitulates CD4+ T-cell help in therapeutic anticancer vaccination. Cancer Res. 2016;76:2921–2931. doi: 10.1158/0008-5472.CAN-15-3130. [DOI] [PubMed] [Google Scholar]
- 15.Ali O.A., Lewin S.A., Dranoff G., Mooney D.J. Vaccines combined with immune checkpoint antibodies promote cytotoxic T-cell activity and tumor eradication. Cancer Immunol. Res. 2016;4:95–100. doi: 10.1158/2326-6066.CIR-14-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wherry E.J., Ha S.-J., Kaech S.M., Haining W.N., Sarkar S., Kalia V., Subramaniam S., Blattman J.N., Barber D.L., Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Youngblood B., Oestreich K.J., Ha S.-J., Duraiswamy J., Akondy R.S., West E.E., Wei Z., Lu P., Austin J.W., Riley J.L. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. 2011;35:400–412. doi: 10.1016/j.immuni.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patsoukis N., Bardhan K., Chatterjee P., Sari D., Liu B., Bell L.N., Karoly E.D., Freeman G.J., Petkova V., Seth P. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patsoukis N., Brown J., Petkova V., Liu F., Li L., Boussiotis V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012;5 doi: 10.1126/scisignal.2002796. ra46–ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schietinger A., Philip M., Krisnawan V.E., Chiu E.Y., Delrow J.J., Basom R.S., Lauer P., Brockstedt D.G., Knoblaugh S.E., Hämmerling G.J. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity. 2016;45:389–401. doi: 10.1016/j.immuni.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz R.M., Galivo F., Kottke T., Wongthida P., Qiao J., Thompson J., Valdes M., Barber G., Vile R.G. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 22.Blanchard M., Shim K.G., Grams M.P., Rajani K., Diaz R.M., Furutani K.M., Thompson J., Olivier K.R., Park S.S., Markovic S.N. Definitive management of oligometastatic melanoma in a murine model using combined ablative radiation therapy and viral immunotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2015;93:577–587. doi: 10.1016/j.ijrobp.2015.07.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajani K., Parrish C., Kottke T., Thompson J., Zaidi S., Ilett L., Shim K.G., Diaz R.M., Pandha H., Harrington K. Combination therapy with reovirus and anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol. Ther. 2016;24:166–174. doi: 10.1038/mt.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takamura S., Tsuji-Kawahara S., Yagita H., Akiba H., Sakamoto M., Chikaishi T., Kato M., Miyazawa M. Premature terminal exhaustion of Friend virus-specific effector CD8+ T cells by rapid induction of multiple inhibitory receptors. J. Immunol. 2010;184:4696–4707. doi: 10.4049/jimmunol.0903478. [DOI] [PubMed] [Google Scholar]
- 25.Ngiow S.F., von Scheidt B., Akiba H., Yagita H., Teng M.W., Smyth M.J. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71:3540–3551. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 26.Tomkowicz B., Walsh E., Cotty A., Verona R., Sabins N., Kaplan F., Santulli-Marotto S., Chin C.N., Mooney J., Lingham R.B. TIM-3 suppresses anti-CD3/CD28-induced TCR activation and IL-2 expression through the NFAT signaling pathway. PLoS ONE. 2015;10:e0140694. doi: 10.1371/journal.pone.0140694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boussiotis V.A., Chatterjee P., Li L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014;20:265–271. doi: 10.1097/PPO.0000000000000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zamarin D., Holmgaard R.B., Subudhi S.K., Park J.S., Mansour M., Palese P., Merghoub T., Wolchok J.D., Allison J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014;6:226ra32. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fourcade J., Sun Z., Pagliano O., Chauvin J.-M., Sander C., Janjic B., Tarhini A.A., Tawbi H.A., Kirkwood J.M., Moschos S. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8+ T cells induced by melanoma vaccines. Cancer Res. 2014;74:1045–1055. doi: 10.1158/0008-5472.CAN-13-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernandez M., Porosnicu M., Markovic D., Barber G.N. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J. Virol. 2002;76:895–904. doi: 10.1128/JVI.76.2.895-904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogquist K.A., Jameson S.C., Heath W.R., Howard J.L., Bevan M.J., Carbone F.R. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 32.Overwijk W.W., Theoret M.R., Finkelstein S.E., Surman D.R., de Jong L.A., Vyth-Dreese F.A., Dellemijn T.A., Antony P.A., Spiess P.J., Palmer D.C. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]