

Summary
The proneural transcription factor Ascl1 coordinates gene expression in both proliferating and differentiating progenitors along the neuronal lineage. Here, we used a cellular model of neurogenesis to investigate how Ascl1 interacts with the chromatin landscape to regulate gene expression when promoting neuronal differentiation. We find that Ascl1 binding occurs mostly at distal enhancers and is associated with activation of gene transcription. Surprisingly, the accessibility of Ascl1 to its binding sites in neural stem/progenitor cells remains largely unchanged throughout their differentiation, as Ascl1 targets regions of both readily accessible and closed chromatin in proliferating cells. Moreover, binding of Ascl1 often precedes an increase in chromatin accessibility and the appearance of new regions of open chromatin, associated with de novo gene expression during differentiation. Our results reveal a function of Ascl1 in promoting chromatin accessibility during neurogenesis, linking the chromatin landscape at Ascl1 target regions with the temporal progression of its transcriptional program.
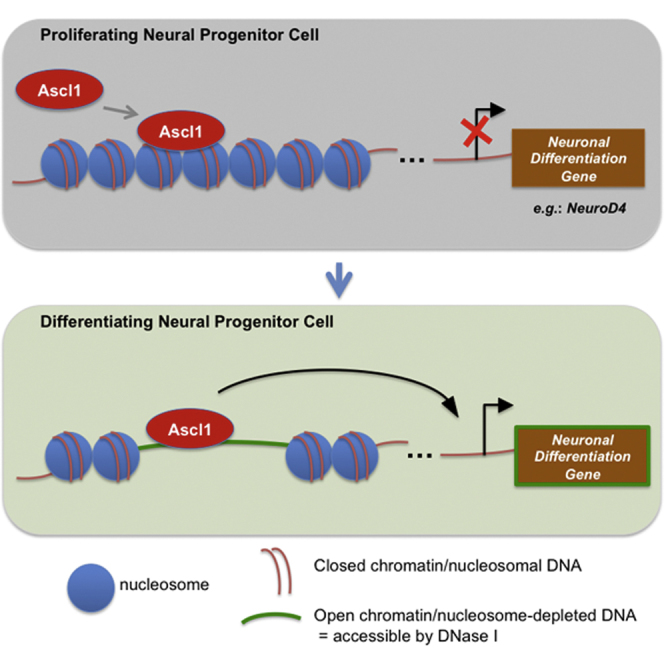
Graphical Abstract
Highlights
-
•
Genome-wide binding of Ascl1 correlates with transcription activation
-
•
Ascl1 can bind to both open and closed chromatin in proliferating cells
-
•
Ascl1 promotes local chromatin accessibility at its target sites
-
•
Chromatin dynamics at Ascl1 sites regulates temporal progression of its program
The proneural transcription factor Ascl1 sequentially activates target genes in proliferating and differentiating progenitors during neurogenesis. Here Raposo et al. show that Ascl1 binds closed and open chromatin in proliferating cells. Binding to closed chromatin promotes accessibility and activation of differentiation specific genes. Thus, dynamics of chromatin landscape at Ascl1 target regions regulate the temporal onset of Ascl1 targets.
Introduction
The generation of neurons in the developing central nervous system requires a number of precisely orchestrated steps, whereby proliferating neural progenitors become committed to the neuronal fate, exit cell cycle, and undergo a long and complex program of migration and differentiation (Kriegstein and Alvarez-Buylla, 2009). Proneural transcription factors (TFs) of the bHLH family, such as Ascl1/Mash1, are the main regulators of neurogenesis in the mammalian brain, and gain and loss-of-function analyses have shown that they are both required and sufficient to promote neurogenesis (Bertrand et al., 2002, Wilkinson et al., 2013). Accordingly, while genetic ablation of proneural genes in mice results in neural developmental defects associated with reduced neurogenesis, overexpression of proneural factors in neural progenitors induces a full neuronal differentiation program (Berninger et al., 2007b, Casarosa et al., 1999, Geoffroy et al., 2009). In addition to its pivotal role in development, Ascl1 has been extensively used in protocols to reprogram somatic cells, including fibroblasts, astrocytes, and pericytes, into induced neurons (Berninger et al., 2007a, Karow et al., 2012, Vierbuchen et al., 2010), renewing interest in understanding the neurogenic activity of this proneural factor.
Previously, we characterized the transcriptional program of Ascl1 in the ventral telencephalic region of the embryonic mouse brain by combining gene expression profiling with chromatin immunoprecipitation (ChIP), followed by hybridization to promoter oligonucleotide arrays (ChIP-chip). This work resulted in the identification of a set of Ascl1 target genes with various biological roles at distinct stages of the differentiation program, raising intriguing questions concerning the molecular basis for such temporal pattern (Castro et al., 2011, Vasconcelos and Castro, 2014). In addition, it led to the identification of a novel function for Ascl1 in maintaining cell proliferation, mediated by the direct activation of genes that promote cell cycle progression. This resulted in a model whereby this proneural factor sequentially promotes the proliferation and differentiation of progenitor cells along the neuronal lineage, reconciling the classical view of this proneural protein as a differentiation factor with the fact that it is mostly expressed in cycling progenitors. Moreover, a recent study has shown that these two opposing activities are associated with distinct modes of Ascl1 expression, with oscillating or sustained Ascl1 promoting proliferation or differentiation, respectively (Imayoshi et al., 2013).
In spite of the significant progress made on the characterization of its transcriptional targets, little is still known about how Ascl1 regulates gene expression. In particular, the relationship between Ascl1 binding, regulation of the chromatin landscape, and gene transcription is poorly understood. It was recently shown that during neuronal reprogramming, Ascl1 can access its cognate sites in nucleosomal-DNA when ectopically expressed in fibroblasts, defining it as a pioneer TF (Wapinski et al., 2013). However, it remains to be seen whether Ascl1 works as a pioneer factor in a neurogenic context and whether binding of Ascl1 results in alterations to the chromatin landscape at its target regions, as it has been shown for some, but not all, other pioneer TFs (Zaret and Carroll, 2011).
Mammalian neurogenesis is not a synchronized process at a cell population level and studies to investigate the mechanistic basis of Ascl1 function at a genome-wide scale are difficult to perform in the developing embryo or in the adult brain. An alternative is the use of adherent cultures of neural stem (NS) cell lines derived from embryonic stem cells or embryonic neural precursors (Conti et al., 2005, Pollard et al., 2006). These cultures provide us with reliable models to study neurogenesis in culture, without the confounding effects of cellular heterogeneity, characteristic of other cellular models such as neurospheres. In proliferating culture conditions, endogenous Ascl1 regulates a progenitor program that functions to maintain cell proliferation (Castro et al., 2011), whereas overexpression of Ascl1 leads to efficient cell cycle exit and neuronal differentiation.
Here we investigate how Ascl1 activity is restricted by and impacts the chromatin landscape, when driving neuronal differentiation. We combined expression profiling with genome-wide mapping of Ascl1 binding sites (ChIP-seq) (Park, 2009), and DNase I hypersensitivity sites (DNase-seq) (Song and Crawford, 2010), in a cellular model of neurogenesis driven by overexpressed Ascl1. We identify a large number of genes directly regulated by Ascl1 and characterize widespread changes in chromatin accessibility during differentiation. Ascl1 binding correlates with activation of gene transcription, targeting not only regions of accessible but also of closed chromatin. In addition, binding of Ascl1 to DNA precedes a local increase in chromatin accessibility at the regulatory regions of its target genes, providing the first direct link between Ascl1 regulation of gene expression and local changes in chromatin landscape.
Results
A Cellular Model of Ascl1-Driven Neurogenesis
Overexpression of Ascl1 promotes cell cycle exit and neuronal differentiation of neural progenitor cells (Castro et al., 2006). To study this process in controlled conditions, we established a cellular model of Ascl1-driven neurogenesis by expressing an inducible version of Ascl1 in NS cells in culture (NS5 cell line) (Pollard et al., 2006). Fusion of full-length Ascl1 to the modified ligand binding domain of the estrogen receptor (ERT2) renders Ascl1 activity dependent on the presence of 4-hydroxytamoxifen (herein referred to as tamoxifen) (Bergstrom et al., 2002, Burk and Klempnauer, 1991, Littlewood et al., 1995). In a transcriptional assay in transfected NS cells, Ascl1-ERT2 induces the transcriptional activation of an enhancer of the Ascl1 target gene Dll1 (Castro et al., 2006) in an inducible manner to levels that are similar to its WT counterpart (Figure S1A). In order to test for the ability of the inducible Ascl1 protein to promote neuronal differentiation, we transduced NS cells in culture with a retrovirus vector co-expressing Ascl1-ERT2 and green fluorescent protein (GFP). In the vast majority of transduced cells, drastic morphological changes characterized by the extension of long processes and associated with the expression of the neuronal marker Tuj1 were observed only upon the addition of tamoxifen, confirming the ability of Ascl1-ERT2 to activate the neurogenic differentiation program in an inducible manner (Figure S1B). Although activation is associated with the nuclear translocation of a small fraction of Ascl1-ERT2, most of the protein is already nuclear prior to addition of tamoxifen (as previously reported with other cases of TFs fused to ERT2) (Burk and Klempnauer, 1991, Roemer and Friedmann, 1993), and additional mechanisms must therefore contribute to the inducibility of Ascl1-ERT2 (Figure 1A).
Figure 1.

A Cellular Model of Neurogenesis Driven by Ascl1
(A) Cellular localization of Ascl1-ERT2 in NS5 cells before and 30 min after the addition of tamoxifen, as assessed by immunostaining against HA-tag (green). White arrowheads mark cytoplasmic expression in the absence of tamoxifen. Cell nuclei are labeled with DAPI (blue). Scale bar represents 30 μm.
(B) Quantification of total Ascl1 protein levels in E14.4 ventral telencephalic progenitors (left) or NS5 Ascl1-ERT2 cells (right). Histograms show absolute fluorescence intensity after normalization (see Experimental Procedures for details).
(C) State of differentiation of NS5 Ascl1-ERT2 cells in the presence or absence of tamoxifen 3 days after induction of differentiation, as assessed by the expression of the neuronal marker TUJ1 (red). Cell nuclei are labeled with DAPI (blue). Scale bar represents 200 μm.
(D) Expression of Gad65/67 (red) in NS5 Ascl1-ERT2 cells 4 days after induction of differentiation. Cell nuclei are labeled with DAPI (blue). Scale bar represents 200 μm.
(E) Electrophysiological properties of GFP-labeled neurons generated from NS5 Ascl1-ERT2 cells 14 days upon induction of differentiation. Representative responses of two neurons to step-current injection at a holding potential of −70mV in current-clamp mode. Note the brief burst of action potentials on top of a prolonged calcium spike following depolarization (red traces) and the rebound burst following release from hyperpolarization (black trace). Scale bar represents 200 μm.
(F) Experiment design for the transcriptome analysis and the resulting number of deregulated genes determined at different time points after induction of differentiation (fold change > 1.2, p < 10−3).
(G) Enrichment of Gene Ontology biological process terms among genes deregulated during neuronal differentiation. Total number of genes associated with each term is in brackets.
See also Figure S1.
To obtain large numbers of synchronized differentiating neural progenitors, we used antibiotic selection following the retroviral delivery of the Ascl1-ERT2 transgene. Quantification of immunofluorescence levels upon immunohistochemistry estimated the total Ascl1 protein level in transduced cells to be 8- to 9-fold higher when compared with endogenous Ascl1 expressed in embryonic ventral telencephalic progenitors (Figure 1B). NS cells expressing Ascl1-ERT2 plated at low density differentiate with great efficiency upon addition of tamoxifen (Figure 1C). Four days after induction, immunocytochemical analysis shows expression of the rate limiting enzyme for GABA synthesis glutamic acid decarboxylase GAD65/67 (15.1% ± 0.3%) (Figure 1D). Whole-cell patch-clamp recordings of retrovirus-transduced cells 14 days after onset of tamoxifen treatment show that these cells consistently exhibit electrophysiological properties of neurons (Figure 1E). Moreover, their action potential discharges pattern in response to step current injections is highly reminiscent of low-threshold burst spiking interneurons and very similar to those generated from medial ganglionic eminence progenitors of the ventral telencephalon (Figure 1D) (Martínez-Cerdeño et al., 2010). To characterize the transcriptome changes in our model, we performed expression profiling at various time points −4, 12, 24, and 50 hr after the onset of differentiation. A large number of genes (1,508) are differentially regulated at least at one time point (fold change >1.5, p < 10−3; see Figure 1F for a breakdown of numbers), as expected upon induction of a cellular differentiation program (Table S1). Functional annotation of differentially expressed genes by Gene Ontology shows considerable enrichment in terms associated with distinct phases of neurogenesis, describing both early events (e.g., “Notch signaling,” “cell division”) and later steps of the differentiation program (e.g., “cell migration,” “axon guidance”) (Figure 1G). Overall, these results show Ascl1 drives an extended program of neuronal differentiation in our model.
Genome-wide Location Analysis of Ascl1 in Differentiating NS Cells
To understand how the observed changes in gene expression relate to Ascl1 function, we mapped Ascl1 binding sites genome-wide by ChIP followed by massive parallel sequencing (ChIP-seq) (Wapinski et al., 2013), using an antibody against the HA tag of Ascl1-ERT2. This was performed 18 hr after the onset of differentiation, at a time point when NS cells have already undergone large transcriptional changes. Using input chromatin as control, we defined an extended list comprising 21,582 Ascl1 binding sites, with FDR <0.5% and p < 10−10 (Table S2).
In order to validate binding events (BEs) mediated by the Ascl1-ERT2 protein, we used ChIP-PCR against WT Ascl1, with chromatin extracted from embryonic E14.5 ventral telencephalic progenitors, or WT NS5 cells (Figure S2A). We reasoned that most BEs in differentiating NS cells should be identified in at least one of the chromatin samples. Indeed, this is the case for all ChIP-seq peaks associated with p < 10−18 (with the majority of BEs validated in both chromatins), indicating a good match between Ascl1-ERT2 and WT Ascl1. Most of the 11,782 Ascl1 BEs defined by this high-confidence list (p < 10−18) are located within intergenic regions (63%) and at long distances from the nearest identified transcription start site (TSS). Less than one third of Ascl1 binding occurs inside genes (30%) or their promoter regions (7%) (Figures 2A and 2B), suggesting that Ascl1 binds predominantly to distal enhancer regions. We next used a Hidden-Markov model to characterize the chromatin states at Ascl1-bound regions, using genome-wide profiles of histone modifications generated from proliferating and differentiating NS cells (before or 24 hr after addition of tamoxifen, respectively) (Ernst and Kellis, 2012). Ascl1 BEs fall mostly within regions of chromatin highly co-enriched for H3K4me1 and H3K27ac, characteristic of active enhancers (same result for p < 10−10; data not shown). Moreover, the same association with this chromatin state is found in proliferating NS cells, indicating that during differentiation Ascl1 binds predominantly to regions that are already marked as active enhancers in proliferating NS cells (Figures 2C, 2D, and S2B).
Figure 2.

Genome-wide Mapping of Ascl1 Binding Sites in Differentiating NS Cells
(A) Location of Ascl1 BEs respective to various genomic features.
(B) Location of Ascl1 BEs in relation to the closest annotated TSS.
(C) Chromatin state in differentiating NS cells, at Ascl1 bound regions in the vicinity of Fbxw7, Olig2, and Dll3 genes.
(D) Heat maps of chromatin states for H3K27ac and H3K4me1 within ±2 kb of Ascl1-ERT2 peak summits in proliferating or differentiating NS cells (before and 24 hr after the addition of tamoxifen, respectively).
(E) DNA motif enrichment for Ascl1 E-box within a 20-bp region centered at Ascl1 peak summits (R, enrichment over local genomic background; S, motif score).
See also Figure S2.
To determine the DNA sequence mediating Ascl1 binding, we searched for motifs enriched within the 20 nucleotides region centered under the Ascl1 peak summits. A strong preference is found for the hexamer sequence CAGCTG, corresponding to the E-box type sequence previously associated with Ascl1 binding (Castro et al., 2011) and in agreement with its role in mediating direct DNA binding (Figure 2E).
Ascl1 Functions Globally as a Transcriptional Activator
Although it is generally believed that proneural factors function by activating gene transcription, some reports have challenged this view (Alvarez-Rodríguez and Pons, 2009, Rheinbay et al., 2013). In order to investigate this issue, we started by performing an in depth cross-comparison between the location analysis and expression profiling results, annotating each Ascl1 BE to its nearest TSS. Direct comparison with the set of deregulated genes previously described shows that only a small fraction of BEs are considered to be regulatory using nearest gene annotation (Figure 3A). Nonetheless, we find a positive correlation between peak height (which is proportional to significance of binding, or p value) and its potential to upregulate gene expression (top peaks ∼20%). In contrast, the fraction of regulatory peaks that can be associated to repressed genes is invariant with changes in peak size, suggesting no direct relation between Ascl1 binding significance and downregulation of gene expression. Therefore, mere binding of Ascl1 to chromatin does not predict a regulatory event; however, the size of an Ascl1 peak may be correlated with its potential to activate gene transcription.
Figure 3.

Ascl1 Functions as a Transcriptional Activator at a Genome-wide Level
(A) Cumulative fraction of Ascl1 BEs considered regulatory by association with upregulated or downregulated genes following a nearest gene annotation. The dashed line marks the most stringent cutoff (p < 10−25) for peak enrichment and defines a list of BEs with the highest regulatory potential (n = 8,624, 2.5 times over control).
(B) Heat map displaying the cumulative fraction of deregulated genes 12 hr after induction of differentiation that is directly regulated by Ascl1 (up, top left panel; down, bottom left panel). Transcripts are divided in equal bins of decreasing expression fold change and plotted against Ascl1 BEs (bin = 166) with increasing p value. Control: 100 sets of random BEs (right, mean value shown).
(C) Heat maps displaying the cumulative fraction of upregulated genes at 4, 12, 24, and 50 hr after induction of differentiation, defined as in (B).
(D) Association of Ascl1 BEs to each gene cluster. The red (activation) or blue (repression) bar represents the number of high-confidence Ascl1 BEs (p < 10−25) annotated to genes belonging to each cluster and is juxtaposed to the distribution of Ascl1 binding annotations, which can be found in 1,000 different random clusters of genes. Test data represented as box with median of test and first and third quartiles; whiskers, ±1.5 × innerquartile range (IQR).
See also Figures S3–S5.
To have a global view of the association between the data sets of genes bound by Ascl1 and regulated during differentiation, we determined the frequency at which deregulated genes (grouped in bins of increasing thresholds of fold change of expression) are associated with Ascl1 BEs (grouped in bins of increasing p value cutoff) and therefore considered to be directly regulated by Ascl1 (Figure 3B, left). The statistical significance of the overlaps was assessed by a similar comparison against 100 randomly generated control ChIP-seq data sets of identical size (Figure 3B, right). The resulting heat maps indicate a strong association between Ascl1-bound genes and those upregulated during differentiation and a much less pronounced association with downregulated genes (Figure 3B, top left versus bottom left). Moreover, the fraction of downregulated genes that are considered Ascl1 targets is very similar to that expected by chance (Figure 3B, left versus right). Altogether, the above analysis indicates that Ascl1 functions globally as a transcriptional activator. In addition, this function is independent of the progress of differentiation during the first 50 hr, as shown with similar analyses for the different time points considered in the transcription profiling (Figures 3C and S3). Importantly, restricting the analysis to the Ascl1 BEs located within ±5 kb of a TSS, producing a more stringent gene annotation, results in a similar conclusion (Figure S4).
To integrate the transcriptional profiling data sets collected at distinct time points, we applied a fuzzy c-means clustering algorithm, resulting in the identification of seven distinct temporal clusters (clusters 1–4 comprised of upregulated genes, clusters 5–7 of downregulated genes) (Figure S5; Table S3). Clusters of genes that are upregulated during differentiation are all strongly associated with Ascl1 BEs (p ≤ 1.3 × 10−11, 8.0 × 10−126, 1.1 × 10−22, and 7.9 × 10−10 for clusters 1, 2, 3, and 4, respectively), whereas clusters of downregulated genes show no such association (Figure 3D), further indicating that Ascl1 binding correlates with transcriptional activation.
Characterization of Ascl1 Transcriptional Program
The relative size of each cluster (Figure S5B) and their enrichment for BEs indicate that most Ascl1 target genes fall within a cluster characterized by an early activation profile (cluster 2), while a smaller but significant number is upregulated at mid or late time points (clusters 3 and 4, respectively). We then compiled a high-confidence list of 272 Ascl1 direct targets comprising genes from clusters 2, 3, and 4 that are associated with at least one Ascl1 BE with high regulatory potential (p < 10−25) (Table S4). A search for terms describing “Biological Process,” “Molecular Function,” and “Pathways” (Panther Classification System) revealed the segregation in each temporal cluster of Ascl1 target genes encoding proteins of distinct classes and functions, suggesting the control of various subprograms at distinct developmental stages (Figure S5C). Genes with the earliest onset of activation (belonging to cluster 2) are associated with signal transduction and cell communication, while targets with a mid-onset of activation (cluster 3) encode mostly proteins with a function related to transcription or Notch signaling. Genes with latest onset (cluster 4) are enriched for generic terms associated with neural development and relate to late events in the differentiation program, from cell migration to axonal growth and guidance (Figure S5C). In summary and in agreement with our previous study performed on a smaller scale, the identification of Ascl1 target genes in our neurogenesis model demonstrates the direct control of distinct components of the neurogenic differentiation program by Ascl1.
Ascl1 Access to Its Target Sites Does Not Depend on the Differentiation Stage of NS Cells
It was previously shown that Ascl1 target genes display different onsets of expression at distinct stages of the neuronal lineage (Borromeo et al., 2014, Castro et al., 2011). Such temporal patterning could be associated with distinct accessibilities of Ascl1 to its binding sites (e.g., due to differences in the chromatin landscape). To investigate this, we compared the binding profile of overexpressed Ascl1 at two distinct stages by extending the previous ChIP-seq analysis to a time point at the onset of differentiation (30 min upon addition of tamoxifen) (Table S9). Surprisingly, visual inspection of the genomic distribution of Ascl1 peaks finds a remarkable similarity between Ascl1 BEs at t = 30 min and t = 18 hr (Figure 4A). Comparison of the two Ascl1 binding profiles in a bin-by-bin approach within the confidence limits defined by the previous ChIP-PCR validation (Figure 4B, dashed line) shows an overlap of 86% between the two stages (Figure 4B). Furthermore, the presence of normalized sequencing signal across both samples in genomic regions centered at putative sample-specific BEs suggests that the overlap of occupancies may have been underestimated by peak calling conditions (Figure 4C). Overall, our results indicate that the accessibility of Ascl1 to the full complement of its binding sites remains largely identical throughout differentiation.
Figure 4.

Ascl1 Accessibility to Its Target Sites Remains Similar throughout Differentiation
(A) Visual representation of ChIP-seq profiles at t = 30 min (blue) and t = 18 hr (red) in a large genomic region centered on Dll3 gene.
(B) Comparison of Ascl1 BEs at t = 30 min and t = 18 hr (p < 10−10) by cumulative bins of increasing p value (bin = 1,007). The proportion of sites bound by Ascl1 in both conditions (overlapping peaks) is calculated in relation to the condition with the lowest number of BEs. Dashed lines represent higher confidence lists defined by p < 10−18, analyzed in (C).
(C) Density plot of Ascl1 ChIP-seq reads mapping to the genomic regions surrounding the summit of Ascl1 BEs. The signal intensity represents the Ascl1 ChIP-seq normalized tag count in the 4kb region surrounding the summit of each overlapping (top) and nonoverlapping peak at t = 30 min (blue) and t = 18 hr (red).
Characterization of Chromatin Landscape Changes during Differentiation
We next asked what impact Ascl1 may have on the chromatin landscape when it promotes neuronal differentiation of NS cells. We started by characterizing the chromatin landscape of proliferating NS cells and of NS cells undergoing differentiation by Ascl1, using a DNase I hypersensitivity assay coupled to massive parallel sequencing (DNase-seq). This technique identifies regions of decreased nucleosome occupancy (herein referred as “open chromatin”) on a genome-wide scale, which correspond mostly to active regulatory elements such as promoters, enhancers, insulators, and silencers (Boyle et al., 2008, Natarajan et al., 2012, Thurman et al., 2012). The density of mapped reads for each genome position was computed to generate a comprehensive list of DNase I hypersensitivity sites (DHSs). Using a constant threshold of p < 10−5, we identified ∼87,000 and ∼94,000 DHSs in proliferating and differentiating NS cells, respectively (Tables S5 and S6), numbers with a magnitude consistent with those obtained in other cell types (Song et al., 2011). Although the majority of these sites is shared by both experimental conditions, each cell state exhibits a specific set of ∼20,000 DHSs (Figure 5A).
Figure 5.

Characterization of Changes in Chromatin Accessibility during Differentiation of NS Cells
(A) Number of DHSs in proliferating NS cells and 24 hr upon Ascl1-mediated differentiation.
(B) FAIRE-PCR validation of differentiation-induced DHSs. Bars show fold enrichment of genomic DNA obtained from NS cells 24 hr after induction over cells prior to addition of tamoxifen. Data are represented as mean ± SD.
(C) Differentiation-induced DHSs are significantly associated with clusters of activated genes (left) and not with clusters of repressed genes (right). Red bars, total number of DHSs annotated to each set of genes; boxplots, distribution of DHSs associations with 1,000 random sets of genes. Test data are represented as box with median of test and first and third quartiles; whiskers, ±1.5 × IQR.
(D) Activated genes associated with differentiation-induced DHSs have low or no expression in proliferating cells. Gene expression levels in proliferating cells are significantly lower (p < 10−11, Wilcoxon test) for activated genes associated with induced DHSs (right) than for activated genes with no such association (left). Red bar, level of expression of NeuroD4 gene. Data distribution represented as box with median and first and third quartiles; whiskers, ±1.5 × IQR; notches, ±1.58 × IQR/n1/2).
(E) Frequency of motif occurrence at high occupancy sites found within differentiation-induced DHSs by Digital Genomic Footprinting (overrepresentation ratio indicated between brackets).
(F) Comparison of Ascl1 BEs at t = 30 min (left) or t = 18 hr (right) with the DNase-seq profile. Color shows the proportion of Ascl1 BEs by cumulative bins of increasing p value (bin = 1,842), which fall within regions of open chromatin (DHSs) in proliferating (top) or differentiating (bottom) NS cells.
Since Ascl1 functions as a transcriptional activator, we focused on the DHSs induced during differentiation (Table S7), as these are more likely to provide new insights into Ascl1 function. To validate these DHSs, we used formaldehyde-assisted isolation of regulatory elements (FAIRE), an alternative method to identify regions of open chromatin. Although distinct preferences have been described for each technique in the identification of distal or proximal regulatory regions, the two techniques have been shown to yield largely overlapping results on a genome-wide basis (Song et al., 2011). Indeed, 9 of 13 tested regions presented more than 2-fold enrichment by FAIRE-PCR across samples collected before and 24 hr after addition of tamoxifen, as opposed to control regions (Figure 5B).
Induced DHSs Are Associated with Genes Expressed De Novo during Differentiation
Next, we investigated how regions of chromatin newly opened during differentiation may be associated with the observed changes in gene expression. We find a statistically significant enrichment of differentiation-induced DHSs in the vicinity of upregulated genes (the sum of clusters 1–4; p < 1.3 × 10−29), in sharp contrast with downregulated genes (clusters 5–7) (Figure 5C). Moreover, a large fraction of all upregulated genes (413 of 760) is associated with at least one differentiation-induced DHS, suggesting the importance of these putative regulatory regions in activating gene expression during neurogenesis. Notably, upregulated genes associated with differentiation-induced DHSs are either not expressed in proliferating NS cells or expressed at a low level when compared with upregulated genes near constant DHSs (Figure 5D). This is well exemplified by the induction of NeuroD4, a pro-differentiation TF only expressed in post-mitotic precursors (Ohsawa et al., 2005), and which is associated with newly open DHSs (Figure 6B). Overall, our results indicate that activation of differentiation genes is strongly associated with the appearance of new DHSs.
Figure 6.

Ascl1 Binds Closed Chromatin and Promotes Chromatin Accessibility
(A) Visual representation of ChIP-seq and DNase-seq enrichment profiles in the vicinity of various Ascl1 targets. Examples are shown of differentiation-induced and constant DHSs localized at Ascl1 binding sites (green and red arrows, respectively) and DHSs with no apparent association with Ascl1 binding (blue arrows).
(B) Induction of expression of genes upon Ascl1-induced differentiation as quantified by real-time PCR. Data are represented as mean ± SD.
(C) FAIRE-PCR validation of differentiation-induced DHSs at Ascl1 binding sites (green arrows in A). Bars show quantification of genomic DNA obtained from proliferating and differentiating NS cells (before and 24 hr after induction, respectively). Data are represented as mean ± SD.
(D) DNase-seq signal profile at genome-wide Ascl1 BEs at t = 30 min (top left) or restricted to BEs falling within differentiation-induced DHSs (top right). Profile determined as the median read count of DNase-seq reads mapped to the 4-kb regions centered at the peak summits in proliferating (blue) and differentiating cells (red). (Bottom) Corresponding fold change for the median profiles.
(E) Ascl1 BEs located at differentiation-induced DHSs are significantly associated with clusters of activated genes (left) and not with clusters of repressed genes (right). Red bars, total number of Ascl1 BEs annotated to each set of genes; boxplots, distribution of Ascl1 BEs associations with 1,000 random sets of genes. Test data represented as box with median of test and first and third quartiles; whiskers, ±1.5 × IQR. See also Figure S6.
(F) In vivo binding of Ascl1 to the NeuroD4 regulatory region assessed by ChIP-PCR using chromatin extracted from mouse ventral telencephalon. Data are represented as mean ± SD.
(G) Chromatin structure analysis by FAIRE-PCR of NeuroD4 and Dll1 regulatory regions bound by Ascl1, in mouse ventral telencephalon. Data are represented as mean ± SD.
Ascl1 Promotes Chromatin Accessibility
In order to investigate which DNA-binding TFs may be associated with the appearance of newly open chromatin regions, we took advantage of the high resolution potential of Digital Genomic Footprinting (DGF) applied to DNase-seq data to determine the position of sites occupied by TFs within differentiation-induced DHSs (Figure 5E) (Piper et al., 2013). DGF identified almost 17,000 genomic sites (Table S8) with significant occupancy levels (p < 10−10), and the corresponding sequences were then subject to a search for frequency of motif occurrence. The most abundant motif found corresponds to the consensus sequence of CTCF and is likely to reflect its frequent binding to insulator regions identified by DNase-seq (Figure 5E) (Thurman et al., 2012). Three additional motifs reveal binding by TFs of the bHLH or NFI families, with the most frequent one corresponding to the E-box consensus sequence, which mediates Ascl1 binding. This observation establishes Ascl1 as a prime candidate to promote the opening of chromatin structure and induce new DHSs during differentiation.
To understand how Ascl1 binding may relate to the observed changes in chromatin compaction, we compared the genomic distribution of Ascl1 BEs with that of DHSs before and after neuronal differentiation (Figure 5F). Ascl1 ChIP-seq and DNase-seq data sets show a high degree of overlap (e.g., 80.3% of Ascl1 BEs with p < 10−18 at t = 30 min fall within DHSs in proliferating cells), indicating that a large fraction of BEs, but not all, occur in regions of open chromatin in proliferating NS cells (Figure 5F). Strikingly, this overlap increases significantly under differentiation conditions (to 91.05% after differentiation, in the aforementioned case; Figure 5F). Altogether, a comparison between the Ascl1 ChIP-seq and DNase-seq suggests that (1) the Ascl1 binding profile remains constant at the two stages analyzed, (2) a large fraction of Ascl1 BEs occurs in regions of open chromatin in proliferating cells, with a subset falling within closed chromatin, and (3) binding of Ascl1 to closed chromatin precedes the appearance of new DHSs during differentiation.
Visualization of aligned Ascl1 ChIP-seq and DNase-seq data suggests that many differentiation-induced DHSs overlap with Ascl1 BEs (Figure 6A), as exemplified by those in the vicinity of genes activated during differentiation such as NeuroD4, Ap3b2, Mcf2l, and Nrxn3 (Figure 6B). Validation of changes in chromatin compaction at the selected Ascl1 binding sites was performed by FAIRE-PCR using chromatin extracted from NS5 cells expressing full-length Ascl1 under the regulation of a doxycycline inducible promoter, before and after Ascl1 induction (Figure 6C). Although not all DHSs that arise de novo during differentiation are associated with an Ascl1 BE, as not all Ascl1 BEs overlap with changes detected by DNase-seq, the vast majority of Ascl1 BEs fall within DHSs in differentiating cells (91.05% or 91.27% with p < 10−18, at t = 30 min or t = 18 hr, respectively; Figure 5F).
To investigate the correlation between Ascl1 binding and changes in chromatin compaction on a large scale, we quantified the DNase-seq signal from both time points centered on Ascl1 peak summits. Confirming previous observations, the overall DNase-seq profile at Ascl1 peak summits is consistent with many BEs occurring in regions of already open chromatin at t = 30 min (Figure 6D). Nevertheless, a significant increase is observed when comparing profiles before and after differentiation (2.33 increase of median read count) at Ascl1 peak summits genome-wide (Figure 6D). This increase is amplified when focusing our analysis on Ascl1 peaks that fall within differentiation-induced DHSs (4.37 increase of median read count; Figure 6D). Ascl1 BEs co-localizing with differentiation-induced DHSs are strongly associated with gene activation and not with gene repression (p < 6.8 × 10−32; Figure 6E), targeting a total number of 98 genes during differentiation. Overall, the integration of Ascl1 location analysis, DNase-seq results, and expression profiling demonstrate Ascl1 promotes genome-wide changes in chromatin compaction at its target sites during neuronal differentiation and the appearance of new DHSs associated with activation of gene expression.
Our results are in agreement with the observation that Ascl1 can bind to nucleosomal DNA when overexpressed in fibroblasts (Wapinski et al., 2013). To verify that Ascl1 can bind to closed chromatin in a physiological context, we focused on the Ascl1-bound regulatory region identified immediately downstream the NeuroD4 promoter (BS2 in Figure 6A), a target gene of Ascl1 in our cellular system that is not expressed in ventral telencephalon. We reasoned that binding to closed chromatin to NeuroD4 may still occur in this embryonic brain region at the neurogenic period, even in the absence of gene expression. Indeed, we detected strong binding of Ascl1, as compared with a negative control region within the NeuroD4 locus by ChIP-PCR in chromatin extracted from mouse ventral telencephalon (Figure 6F). However, in contrast to our previous findings with differentiating NS cells in culture, a lack of enrichment for non-nucleosomal DNA assessed by FAIRE-PCR shows this Ascl1 target site is found within closed chromatin in ventral telencephalon (Figure 6G, compare with Ascl1 site in Dll1). Thus, our results with the NeuroD4 regulatory region demonstrate that endogenous Ascl1 can bind to closed chromatin in an in vivo context.
Finally, we wished to analyze the accessibility of chromatin at Ascl1 target sites in the absence of Ascl1 expression. For this, we used chromatin extracted from ventral telencephalon of Ascl1-null embryos and probed Ascl1-bound regions associated with previously validated Ascl1 target genes (Dll1, Fbxw7, and Stk33) in this embryonic structure (Castro et al., 2006). Ascl1 null embryos show a very significant reduction of FAIRE-PCR signal relative to WT chromatin at all tested Ascl1 binding sites, as compared with pairwise control regions, which is consistent with the results above and in support of a role for Ascl1 in regulating chromatin compaction in vivo (Figure S6).
Discussion
In spite of its pivotal role during neurogenesis, little is known on how Ascl1 functions to regulate gene expression. Here we investigated the reciprocal interactions between Ascl1 and the chromatin landscape when promoting the neuronal differentiation of NS cells in culture. We found that Ascl1 can bind to and promote the opening of closed chromatin regions in its native context. Below we discuss our findings and their implications to the mechanisms that govern the temporal progression of the Ascl1 transcriptional program.
Our work resulted in a fine-grained characterization of a differentiation program driven by Ascl1. In spite of displaying a wide range of biological roles, Ascl1 target genes are remarkably enriched in two categories. The first consists of genes encoding transcriptional regulators, an observation consistent with the master regulator function of Ascl1 in this developmental process. Importantly, the activation of some TFs may define feedback loops that modulate Ascl1 activity. This is the case of Id1, an inhibitor of bHLH activity that functions by sequestering E-proteins (Ruzinova and Benezra, 2003), and Cbfa2t2/MTGR1, a zinc-finger protein that has been shown to function as a co-repressor of proneural proteins, including Ascl1 (Aaker et al., 2010). Other examples, suggesting positive feedback mechanisms, are TFs previously linked to a relief of Notch inhibition by counteracting the activity of Hes1 (Hes6), repressing Notch1 (Prox1), or by as yet unknown mechanisms (MyT1) (Bellefroid et al., 1996, Gratton et al., 2003, Kaltezioti et al., 2010). These regulatory events define a less well-understood role of Ascl1 in modulating Notch signaling cell autonomously, of putative importance to the progression of differentiation. The second most prominent group of Ascl1 target genes encodes cytoskeleton-related proteins. The abundance of this group of genes is likely to result from its recognized role in many cellular functions, from signal transduction (where it may serve as a scaffold for components of signaling pathways) to cell shape change and locomotion, activities that are essential to the neurogenic differentiation program (Forgacs et al., 2004).
It was recently shown that Ascl1 promotes sequentially the proliferation and differentiation along the neuronal lineage, with the concomitant activation of distinct target genes (Castro et al., 2011). Our study focused on the transcriptional program activated by Ascl1 when promoting neuronal differentiation. Comparing the identity and molecular determinants for target gene specificity associated with both cellular functions will likely require the characterization of Ascl1 binding profile in an oscillatory versus sustained mode of expression. Nevertheless, the observation that overexpressed Ascl1 can readily access the full complement of its differentiation sites at the onset of differentiation suggests these may already be accessible to this TF in proliferating cells, an important observation in the context of the dynamics of Ascl1 function along the neuronal lineage.
It was previously shown that overexpressed Ascl1 can bind to closed chromatin in fibroblasts (as assessed by FAIRE-PCR), defining it as a pioneer TF (Wapinski et al., 2013), results that the present study validates for the first time in a neural context. Moreover, our analysis of binding of Ascl1 to the NeuroD4 locus in ventral telencephalic progenitors that do not express this gene allowed for the dissociation between binding and opening of chromatin, revealing binding of endogenous Ascl1 to closed chromatin in vivo. Altogether, our study supports the idea that Ascl1 displays its pioneer activity when regulating gene expression in its native context.
Our analysis of chromatin states suggests that most Ascl1 binding in our differentiation model occurs within enhancer regions that are already active in proliferating NS cells as shown by their enrichment for both H3K4me1 and H3K27ac. This may be due to the fact that many genes upregulated by Ascl1 during differentiation are already expressed in proliferating NS cells. Alternatively, it may reflect the trivalent mark (H3K4me1, H3K27ac, and H3K9m3) identified at Ascl1 binding sites in various cell types permissive to Ascl1-mediated reprogramming (Wapinski et al., 2013). Most importantly, Ascl1 promotes DNase I accessibility to regions of closed chromatin at a smaller but significant number of its target sites associated with the activation of a differentiation-specific component of its transcriptional program. Our results cast light into a previously unknown function of Ascl1 and establish the first link between the chromatin landscape at Ascl1-bound regulatory regions and the temporal pattern of the Ascl1 program along the neuronal lineage. Moreover, they suggest that the hierarchical model recently proposed for the reprograming paradigm (Wapinski et al., 2013) may also be representative of the neurogenesis process and that opening of chromatin regions at Ascl1 target sites may be important to allow subsequent binding of other TFs in differentiating cells. Finally, it is tempting to speculate that the broad effect of Ascl1 in promoting chromatin accessibility described in this study will be important for the reprogramming capacity of this TF upon its ectopic expression in various cell types.
Experimental Procedures
Plasmids and Virus Production
Ascl1-ERT2 encodes full-length mouse Ascl1 in frame with the modified ligand binding domain of the human estrogen receptor (ERT2) and an N-terminal Flag/HA tag sequence. For virus production, Ascl1-ERT2 was subcloned into pMX-IRES-GFP or pBABE-IRES-GFP-puro. Replication-incompetent retroviruses were produced from HEK293T cells transiently transfected with retroviral, viral envelope-, and VSVG-pseudotyping plasmids. Retroviral particles were concentrated from supernatant by ultracentrifugation at 90,000 g for 90 min. Viral titers were typically 105–106 infectious particles/ml.
Culture and Infection of NS5 Cells
NS5 cells were cultured as previously described (Conti et al., 2005) using laminin as media supplement (20 μg/ml). NS5 Ascl1-ERT2 cells were generated upon infection with pBABE-Ascl1-ERT2-IRES-GFP-puro, followed by selection in complete medium containing 1-μg/ml puromycin (Calbiochem). For differentiation, NS5 Ascl1-ERT2 cells were plated at 28,000 per cm2 of density and induced with 50-nM 4-Hydroxytamoxifen, while reducing EGF concentration to 5 ng/ml.
Quantification of Ascl1 Protein Levels
All experiments were carried out upon approval and following the guidelines of the ethics committee of Instituto Gulbenkian de Ciência.
Quantification of Ascl1 (endogenous + Ascl1-ERT2) done by parallel immunostaining with anti-Ascl1 antibody in acutely cultured primary ventral telencephalon progenitors from E14.5 mouse embryos and NS5 Ascl1-ERT2 cells (three replicates each). A total of 939 and 751 cells for embryonic and NS5 progenitors, respectively, was imaged with identical exposure and images thresholded and normalized—mean fluorescence per cell x (mean fluorescence of all cells in picture/mean fluorescence of all cells per sample)—to define positive cells. Fluorescence intensity was measured with ImageJ.
Expression Analysis of NS5 Ascl1-ERT2 Cells
Extraction of mRNA and cDNA synthesis performed as previously described (Castro et al., 2011). Quantitative real-time PCR (qRT-PCR) was performed according to the manufacturer using SYBR Green super-mix (Quanta BioSciences) on an ABI7500 machine (Applied Biosystems). For microarray analysis, quality of total RNA from biological triplicates was assessed using Agilent Bioanalyser, and samples were processed according to the Illumina Whole-genome Gene Expression Direct Hybridization Assay Guide, using Illumina TotalPrep-96 RNA Amplification kit (Life Technologies). Quality control was performed on labeled cRNA, and 750 ng of labeled cRNA was hybridized to Mouse Ref-8v2 arrays and scanned by BeedStudio v.3.1.3 (quantile normalization with background correction). Normalization among arrays and significant analysis were performed using GeneSpring X (Agilent). Further details of data processing, including gene clustering, are described in Supplemental Information.
ChIP-seq
For Ascl1 ChIP-seq, NS5 Ascl1-ERT2 cells were fixed sequentially with 2 mM di(N-succimidyl) glutarate and 1% formaldehyde in PBS and then lysed, sonicated, and immunoprecipitated with anti-HA antibody, as previously described (Castro et al., 2011). DNA libraries were prepared from 10 ng of immunoprecipitated DNA according to the standard Illumina ChIP-seq protocol and sequenced with Illumina GAIIx. Raw reads were mapped to the mouse genome (NCBI37/mm9) with Bowtie 0.12.7 (Langmead et al., 2009). Uniquely mapped reads data (∼11.5 million for both Ascl1 ChIP and input chromatin samples) were then subsampled before peak calling with MACS 1.4.1 (Zhang et al., 2008). For histone marks ChIP-seq, Ascl1-ERT2 cells were fixed in 1% formaldehyde in PBS, prepared as described above and immunoprecipitated with anti-H3K27ac and anti-H3K4me1 antibodies. Libraries were sequenced with Illumina HiSeq 2000. Raw reads were mapped as above (25 million uniquely mapped reads for all samples) and data processed with MACS 2.1.0. For chromatin state characterization, we used a Hidden Markov modeling of chromatin enrichment software—ChromHMM (Ernst and Kellis, 2012). Further details of data processing are described in Supplemental Information.
FAIRE-PCR
Chromatin preparation was done with a single fixation with 1% formaldehyde, from NS5 Ascl1-ERT2 cells, or when indicated with NS5 cells infected with a doxycycline inducible lentivirus expressing WT Ascl1 (Wapinski et al., 2013). Three rounds of phenol/chloroform extraction were followed by isopropanol precipitation of the DNA. Quantification of genomic regions was done using a standard curve generated with decross-linked input chromatin by qRT-PCR as above.
DNase-seq
DNase-seq samples from differentiating NS5 Ascl1-ERT2 cells or proliferating NS5 cells were prepared as previously described (Song et al., 2011). Libraries were generated as previously described (Boyle et al., 2008, Song and Crawford, 2010) with a slight modification to the linkers to increase ligation efficiency (Song et al., 2011) and then sequenced with Illumina GAIIx. Mapping was done as for ChIP-seq, resulting in 165.9 and 168.7 millions of unique reads for proliferating and differentiating conditions, respectively. Genomic location of DHSs was identified with MACS version 1.4.1, following a protocol described in Supplemental Information.
For oligonucleotides used in this study, see also Table S10.
Author Contributions
A.A.S.F.R. did the bioinformatics analysis. F.F.V. and D.D. designed research and performed experimental work, except electrophysiology tests performed by B.B. D.S.C. and A.A.S.F.R. wrote the manuscript, which was read and approved by all others. All other authors contributed with reagents and analytical tools.
Acknowledgments
We thank Daniel Sobral and the IGC Bioinformatics Unit for expert assistance and Abdul Sesay and the NIMR high-throughput sequencing facility for ChIP-seq library preparation and sequencing. This study was funded by FCT grants (PTDC/SAU-NEU/100208/2008 and PTDC/NEU-NMC/0315/2012 to D.S.C.), Marie Curie CIG (303644 to D.S.C.), Wellcome Trust (WT095908 and WT098051 to P.F.), the European Community (FP7-223210 to P.F, L.E. and F.G.), and an FCT doctoral fellowship to F.F.V. DSC is supported by the FCT investigator program.
Published: March 5, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information includes Supplemental Experimental Procedures, six figures, and ten tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.02.025.
Accession Numbers
The ChIP-seq dataset for Ascl1-ERT2 (t=30’) was submitted to the ArrayExpress database (www.ebi.ac.uk/arrayexpress) and is publicly available under the accession number E-MTAB-2384.
The DNase-seq datasets were submitted to the ArrayExpress database (www.ebi.ac.uk/arrayexpress) and are publicly available under the accession number E-MTAB-2270.
The ChIP-seq datasets for H3K4me1 and H3K27ac were submitted to the ArrayExpress database (www.ebi.ac.uk/arrayexpress) and are publicly available under the accession number E-MTAB-3104.
Supplemental Information
References
- Aaker J.D., Patineau A.L., Yang H.-J., Ewart D.T., Nakagawa Y., McLoon S.C., Koyano-Nakagawa N. Interaction of MTG family proteins with NEUROG2 and ASCL1 in the developing nervous system. Neurosci. Lett. 2010;474:46–51. doi: 10.1016/j.neulet.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Rodríguez R., Pons S. Expression of the proneural gene encoding Mash1 suppresses MYCN mitotic activity. J. Cell Sci. 2009;122:595–599. doi: 10.1242/jcs.037556. [DOI] [PubMed] [Google Scholar]
- Bellefroid E.J., Bourguignon C., Hollemann T., Ma Q., Anderson D.J., Kintner C., Pieler T. X-MyT1, a Xenopus C2HC-type zinc finger protein with a regulatory function in neuronal differentiation. Cell. 1996;87:1191–1202. doi: 10.1016/s0092-8674(00)81815-2. [DOI] [PubMed] [Google Scholar]
- Bergstrom D.A., Penn B.H., Strand A., Perry R.L., Rudnicki M.A., Tapscott S.J. Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol. Cell. 2002;9:587–600. doi: 10.1016/s1097-2765(02)00481-1. [DOI] [PubMed] [Google Scholar]
- Berninger B., Costa M.R., Koch U., Schroeder T., Sutor B., Grothe B., Götz M. Functional properties of neurons derived from in vitro reprogrammed postnatal astroglia. The Journal of neuroscience, 2007;27(32):8654–8664. doi: 10.1523/JNEUROSCI.1615-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berninger B., Guillemot F., Götz M. Directing neurotransmitter identity of neurones derived from expanded adult neural stem cells. Eur. J. Neurosci. 2007;25:2581–2590. doi: 10.1111/j.1460-9568.2007.05509.x. [DOI] [PubMed] [Google Scholar]
- Bertrand N., Castro D.S., Guillemot F. Proneural genes and the specification of neural cell types. Nat. Rev. Neurosci. 2002;3:517–530. doi: 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- Borromeo M.D., Meredith D.M., Castro D.S., Chang J.C., Tung K.-C., Guillemot F., Johnson J.E. A transcription factor network specifying inhibitory versus excitatory neurons in the dorsal spinal cord. Development. 2014;141:2803–2812. doi: 10.1242/dev.105866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle A.P., Davis S., Shulha H.P., Meltzer P., Margulies E.H., Weng Z., Furey T.S., Crawford G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk O., Klempnauer K.H. Estrogen-dependent alterations in differentiation state of myeloid cells caused by a v-myb/estrogen receptor fusion protein. EMBO J. 1991;10:3713–3719. doi: 10.1002/j.1460-2075.1991.tb04939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarosa S., Fode C., Guillemot F. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999;126:525–534. doi: 10.1242/dev.126.3.525. [DOI] [PubMed] [Google Scholar]
- Castro D.S., Skowronska-Krawczyk D., Armant O., Donaldson I.J., Parras C., Hunt C., Critchley J.A., Nguyen L., Gossler A., Göttgens B. Proneural bHLH and Brn proteins coregulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev. Cell. 2006;11:831–844. doi: 10.1016/j.devcel.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Castro D.S., Martynoga B., Parras C., Ramesh V., Pacary E., Johnston C., Drechsel D., Lebel-Potter M., Garcia L.G., Hunt C. A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Genes Dev. 2011;25:930–945. doi: 10.1101/gad.627811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti L., Pollard S.M., Gorba T., Reitano E., Toselli M., Biella G., Sun Y., Sanzone S., Ying Q.L., Cattaneo E., Smith A. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 2005;3:e283. doi: 10.1371/journal.pbio.0030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgacs G., Yook S.H., Janmey P.A., Jeong H., Burd C.G. Role of the cytoskeleton in signaling networks. J. Cell Sci. 2004;117:2769–2775. doi: 10.1242/jcs.01122. [DOI] [PubMed] [Google Scholar]
- Geoffroy C.G., Critchley J.A., Castro D.S., Ramelli S., Barraclough C., Descombes P., Guillemot F., Raineteau O. Engineering of dominant active basic helix-loop-helix proteins that are resistant to negative regulation by postnatal central nervous system antineurogenic cues. Stem Cells. 2009;27:847–856. doi: 10.1002/stem.17. [DOI] [PubMed] [Google Scholar]
- Gratton M.O., Torban E., Jasmin S.B., Theriault F.M., German M.S., Stifani S., Stifani S. Hes6 promotes cortical neurogenesis and inhibits Hes1 transcription repression activity by multiple mechanisms. Mol. Cell. Biol. 2003;23:6922–6935. doi: 10.1128/MCB.23.19.6922-6935.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I., Isomura A., Harima Y., Kawaguchi K., Kori H., Miyachi H., Fujiwara T., Ishidate F., Kageyama R. Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science. 2013;342:1203–1208. doi: 10.1126/science.1242366. [DOI] [PubMed] [Google Scholar]
- Kaltezioti V., Kouroupi G., Oikonomaki M., Mantouvalou E., Stergiopoulos A., Charonis A., Rohrer H., Matsas R., Politis P.K. Prox1 regulates the notch1-mediated inhibition of neurogenesis. PLoS Biol. 2010;8:e1000565. doi: 10.1371/journal.pbio.1000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karow M., Sánchez R., Schichor C., Masserdotti G., Ortega F., Heinrich C., Gascón S., Khan M.A., Lie D.C., Dellavalle A. Reprogramming of pericyte-derived cells of the adult human brain into induced neuronal cells. Cell Stem Cell. 2012;11:471–476. doi: 10.1016/j.stem.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Kriegstein A., Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood T.D., Hancock D.C., Danielian P.S., Parker M.G., Evan G.I. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Cerdeño V., Noctor S.C., Espinosa A., Ariza J., Parker P., Orasji S., Daadi M.M., Bankiewicz K., Alvarez-Buylla A., Kriegstein A.R. Embryonic MGE precursor cells grafted into adult rat striatum integrate and ameliorate motor symptoms in 6-OHDA-lesioned rats. Cell Stem Cell. 2010;6:238–250. doi: 10.1016/j.stem.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan A., Yardimci G.G., Sheffield N.C., Crawford G.E., Ohler U. Predicting cell-type-specific gene expression from regions of open chromatin. Genome Res. 2012;22:1711–1722. doi: 10.1101/gr.135129.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa R., Ohtsuka T., Kageyama R. Mash1 and Math3 are required for development of branchiomotor neurons and maintenance of neural progenitors. J. Neurosci. 2005;25:5857–5865. doi: 10.1523/JNEUROSCI.4621-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P.J. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper J., Elze M.C., Cauchy P., Cockerill P.N., Bonifer C., Ott S. Wellington: a novel method for the accurate identification of digital genomic footprints from DNase-seq data. Nucleic Acids Res. 2013;41:e201. doi: 10.1093/nar/gkt850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard S.M., Conti L., Sun Y., Goffredo D., Smith A. Adherent neural stem (NS) cells from fetal and adult forebrain. Cereb. Cortex. 2006;16(Suppl 1):i112–i120. doi: 10.1093/cercor/bhj167. [DOI] [PubMed] [Google Scholar]
- Rheinbay E., Suvà M.L., Gillespie S.M., Wakimoto H., Patel A.P., Shahid M., Oksuz O., Rabkin S.D., Martuza R.L., Rivera M.N. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013;3:1567–1579. doi: 10.1016/j.celrep.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer K., Friedmann T. Modulation of cell proliferation and gene expression by a p53-estrogen receptor hybrid protein. Proc. Natl. Acad. Sci. USA. 1993;90:9252–9256. doi: 10.1073/pnas.90.20.9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzinova M.B., Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003;13:410–418. doi: 10.1016/s0962-8924(03)00147-8. [DOI] [PubMed] [Google Scholar]
- Song L., Crawford G.E. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor Protoc. 2010;2010 doi: 10.1101/pdb.prot5384. pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L., Zhang Z., Grasfeder L.L., Boyle A.P., Giresi P.G., Lee B.K., Sheffield N.C., Gräf S., Huss M., Keefe D. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21:1757–1767. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasconcelos F.F., Castro D.S. Transcriptional control of vertebrate neurogenesis by the proneural factor Ascl1. Front. Cell. Neurosci. 2014;8:412. doi: 10.3389/fncel.2014.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T., Ostermeier A., Pang Z.P., Kokubu Y., Südhof T.C., Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski O.L., Vierbuchen T., Qu K., Lee Q.Y., Chanda S., Fuentes D.R., Giresi P.G., Ng Y.H., Marro S., Neff N.F. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell. 2013;155:621–635. doi: 10.1016/j.cell.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson G., Dennis D., Schuurmans C. Proneural genes in neocortical development. Neuroscience. 2013;253:256–273. doi: 10.1016/j.neuroscience.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Zaret K.S., Carroll J.S. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.