

Abstract
Morganella morganii is a common but frequent neglected environmental opportunistic pathogen which can cause deadly nosocomial infections. The increased number of multidrug-resistant M. morganii isolates motivates the search for alternative and effective antibacterials. We have isolated two novel obligatorily lytic M. morganii bacteriophages (vB_MmoM_MP1, vB_MmoP_MP2) and characterized them with respect to specificity, morphology, genome organization and phylogenetic relationships. MP1’s dsDNA genome consists of 163,095 bp and encodes 271 proteins, exhibiting low DNA (<40%) and protein (<70%) homology to other members of the Tevenvirinae. Its unique property is a >10 kb chromosomal inversion that encompass the baseplate assembly and head outer capsid synthesis genes when compared to other T-even bacteriophages. MP2 has a dsDNA molecule with 39,394 bp and encodes 55 proteins, presenting significant genomic (70%) and proteomic identity (86%) but only to Morganella bacteriophage MmP1. MP1 and MP2 are then novel members of Tevenvirinae and Autographivirinae, respectively, but differ significantly from other tailed bacteriophages of these subfamilies to warrant proposing new genera. Both bacteriophages together could propagate in 23 of 27 M. morganii clinical isolates of different origin and antibiotic resistance profiles, making them suitable for further studies on a development of bacteriophage cocktail for potential therapeutic applications.
Bacteriophages (phages) are bacterial viruses which have received renewed interest as alternative or complementary antibacterial agents in the fight against multidrug-resistant pathogens1. Phages are also important genetic tools that have contributed significantly to the development of the fields of molecular biology and biotechnology2,3.
Phage taxonomy was originally based on phage morphology and genome type. Phages of complex virions which consist of a head that contain linear dsDNA and a tail have been classified to a separate order - Caudovirales (tailed phages)4. They evolved before the separation of bacteria and archaea. Their long evolutionary history, as well as, frequent genetic exchanges of their genes by horizontal gene transfer has led to the enormous diversification of this group of viruses and often the disappearance of sequence similarities between members of the same families, at the DNA or protein level. Thus, uniform rules for classifying tailed phages, analogous to those, based on the sequence of the 16S rRNA gene that are used for bacteria, do not exist. For example, the Salmonella virus P22 and the coliphage T7 have been placed in the same Podoviridae family on the basis of morphology despite the lack of DNA sequence relatedness between them5. With the advance of genomics, newer approaches based on the comparative analysis of terminase sequences6, structural genes7 or whole proteomes8 have been proposed to classify phages. More recently, a novel BLAST-based function prediction has emerged as a novel tool to distinguish phage subfamilies9. Therefore, the taxonomic positioning of phages is in constant evolution due to the continuous deposition of a large number of novel genomes in the GenBank, especially for phages targeting clinically relevant pathogens.
Morganella morganii is an opportunistic pathogen often associated with nosocomial infections10, with several clinical isolates being resistant to multiple antibiotics11. Several studies have demonstrated the frequency of spontaneous antibiotic resistance (e.g. β-lactams)12,13,14, with the ability to acquire new resistance mechanisms (e.g. horizontal transfer of resistance genes)15,16. Relative to other Gram-negative bacteria such as Escherichia coli and Pseudomonas aeruginosa, only a few phages have been characterized against M. morganii. Historically, several phage typing systems were developed for this bacterium17,18,19,20 and a generalized transducing phage, subsequently called phage M21,22,23, has also been reported. More recently, Morganella phage FSP1 was characterized as a member of the Myoviridae with a genome of approximately 47.7 kb24. To our knowledge, only one Morganella phage has been sequenced, but is lacking an associated microbiological characterization (phage MmP1)25.
This study reports the isolation and characterization of two novel phages that are able to propagate in multidrug resistant M. morganii isolates, and may potentially find a therapeutic use. They are named MP1 (vB_MmoM_MP1) and MP2 (vB_MmoP_MP2), according to recent nomenclature recommendations26. Morphological analyses and whole-genome comparisons reveal that MP1 and MP2 belong to Myoviridae and Podoviridae families and represent new clades within the Tevenvirinae and Autographivirinae subfamilies, respectively. This work provides a detailed characterization of MP1 and MP2 contributing to a better understanding of the biodiversity of phages infecting different hosts.
Materials and Methods
Bacterial strains and culture conditions
All bacterial strains used to evaluate the lytic spectra of the isolated phages have been listed in Table S1. The panel includes strains from the Salmonella Genetic Stock Centre, the American Type Culture Collection as well as several clinical isolates: 27 M. morganii and one representative of each of the following closely-related species: Citrobacter freundii and Citrobacter koseri, Proteus mirabilis, Proteus vulgaris, Providencia stuartii and E. coli. Clinical isolates have undergone typing and antimicrobial susceptibility tests with Vitek2 (BioMerieux) or Walk Away (Beckman Coulter), according to CLSI guidelines. Strains producing beta-lactamases (AmpC) were identified using an Etest (BioMerieux) with positive results for cefotetan/cefotetan + cloxacillin. Bacteria were grown at 37 °C in Tryptic Soy Broth (TSB, Merck) or in Tryptic Soy Agar (TSA, Merck).
Phage Isolation and propagation
Phages were isolated from wastewater treatment plant sewage samples located in Braga (Portugal) following the enrichment procedure described previously27. Briefly, 20 ml of 2x TSB were added to 20 ml of previously centrifuged (10 min, 9,000 × g, 4 °C) raw sewage. After that, a loop of each M. morganii bacterial strain was added, and the mixture was incubated for 16 h (37 °C, 120 rpm, ES-20/60). The suspension was centrifuged (10 min, 9,000 × g, 4 °C) and the presence of phages in the supernatant was confirmed by spotting 10 μl on each of the bacterial lawns, previously incorporated in 0.6% TSA. After a 16 h incubation period at 37 °C, clear and turbid areas on the lawns were considered positive for phages and single plaques were repeatedly purified until only one plaque morphology was achieved.
Phages were propagated in accordance with a previously described protocol27. Briefly, purified phages were incorporated into a 0.6% TSA layer with bacteria, and incubated at 37 °C for 16 h. SM buffer (5.8 g/L NaCl, 2 g/L MgSO4.7H2O, 50 mL 1 M Tris, pH 7.5) was added to the plates, which were then incubated at 4 °C (16 h, 120 rpm, ES-20/60). After that, all liquid was removed from the Petri dishes, and 0.58 g of NaCl was dissolved in each 10 mL of sample. The resultant suspension was incubated for 1 h at 4 °C, centrifuged for 10 min (9,000 × g, 4 °C), and the supernatant was collected. Phages were concentrated via PEG 6000 precipitation and purified with chloroform as described previously28. Briefly, PEG 6000 (10% (w/v)) was added to the samples and incubated for 16 h at 4 °C. The pellet (containing phages) was recovered by centrifugation (10 min, 9,000 × g, 4 °C), suspended in SM buffer and purified with chloroform (1:4 (v/v)). After centrifugation (15 min, 3,000 × g, 4 °C), the aqueous phase was collected, filtered through a 0.22 μm Whatman PES membrane and stored at 4 °C.
Lytic spectra and efficiency of plating (EOP)
The lytic spectra of phages were evaluated using the drop spot test. Phages were tested against all the bacterial strains listed in Table S1. The phage titers were adjusted to 108 PFU/mL, 10-fold serial diluted, and 10 μL of each dilution were spotted onto the lawns of each strain, before incubation for 16 h at 37 °C. The results were observed and a score attributed. The relative efficiency of plating (EOP) was calculated by dividing the titer of the phage (PFU/mL) of each isolate by the titer for the relevant propagating host.
One-Step-Growth curve
One-step growth curves were performed as described previously2. Briefly, 10 ml of a mid-exponential-phase culture were harvested by centrifugation (9,000 × g, 5 min, 4 °C) and suspended in 5 ml of fresh TSB to an OD620 of 1.0. An equal volume of phage was added in order to have a multiplicity of infection (MOI) of 0.001. This culture was then incubated at room temperature for 5 min to allow phage adsorption to the host. The mixture was centrifuged as described above, the pellet was suspended in 10 ml of fresh TSB medium and incubated at 37 °C. Samples were collected for plaque forming unit counts at 5, 10, 15, 20, 25, 30, 40, 50 and 60 min and plated immediately. Averages ± standard deviations for all experiments are given for n = 3 repeats.
Cytotoxicity assay
The cell line A549 (ATCC CCL-185) was grown in Dulbecco’s Modified Eagle’s Medium (DMEM with 3.7 g/L NaHCO3 and 4.5 g/L D-glucose, Biochrom) supplemented with 10% of fetal bovine serum (FBS, Biochrom) and 1% Penicillin/Streptomycin (Sigma-Aldrich), at 37 °C in a humidified incubator with 5% CO2. Cells were seeded into wells of a 96-well plate at 1 × 106 cells/mL and incubated for 24 h, to reach a cell confluence between 70 and 90%. Phage solution (in SM buffer) was added to the cells, so that a phage concentration of 108 PFU/mL and a final volume of 100 μL were achieved in the wells. Controls were performed by adding the same amount of SM buffer to the wells with cells grown in the same conditions. After 24 h, 20 μL of CellTiter 96 Aqueous One Solution Reagent (Promega) was added to each well and incubated for 1 h at 37 °C in a humidified 5% CO2 atmosphere, protected from light. The amount of soluble formazan produced by cellular reduction of MTS was measured by reading the absorbance at 490 nm. The experiment was performed in triplicate.
Mass spectrometry
A protein pellet from the phage virion proteins was obtained after a chloroform:methanol extraction (1:1:0.75, v/v/v) performed for both phage on high titre of purified samples (>1010 PFU/mL). Subsequently, this pellet was resuspended in loading buffer (1% SDS, 6% sucrose, 100 mM dithiothreitol, 10 mM Tris pH 6.8, 0.0625% w/v bromophenol blue) and heated to 95˚C (5 min). Samples were run on a 12% SDS-PAGE gel and stained using Gelcode™ Blue Safe Protein Stain (Thermo Scienitic, Waltham, Massachusetts, United States) to visualise virion proteins. Gel fragments were extracted and subjected to trypsinization for analysis using tandem electrospray ionization-mass spectrometry (ESI-MS/MS) exactly as described previously29.
Transmission electron microscopy
Phage particles were sedimented (20,000 × g, 1 h, 4 °C), using a Beckman J2-21 (Carlsbad, CA, USA) centrifuge, washed twice and resuspended in tap water. Phages were deposited on copper grids provided with carbon-coated Formvar films, stained with 2% uranyl acetate (pH 4.0), and examined using a Jeol JEM 1400 transmission electron microscope.
Genome sequencing and assembly
Phage genomic DNA was isolated using the proteinase K and sodium dodecyl sulphate, followed by phenol:chloroform extraction as described previously28. DNA was sequenced by the VIB Nucleomics Core (Leuven, Belgium) using a NGS Illumina MiSeq platform (Illumina Inc., San-Diego, USA) and a NEBNext® Ultra™ DNA Library Prep Kit to generate 500-bp fragments with 2 × 300 bp paired-end read length configuration. Quality controls of DNA before being sequenced on the MiSeq System were made by Agilent Bioanalyzer and Qubit measurements. Outputs were demultiplexed and de novo assembled into a single contig using CLC Bio Genomics Workbench v7.0 (Aarhus, Denmark) and manually inspected. To identify virion DNA ends the assembly patterns of subpopulations of random sequence reads representing the genome fragments of each phage (up to 100x coverage) were analysed using the SeqMan program of Lasergene 9.1 package (DNAStar, Madison, WI, USA).
Genome analysis
The phage’s genome was annotated using MyRAST30. The CDSs putative functions were assigned by BLASTP31 queried against non-redundant protein sequences databases. HHpred32 was used for the detection of homology and structure prediction. Genes coding tRNAs were predicted using tRNAscan-SE33 and ARAGORN34. Putative promoters were identified using 100 bp of CDS upstream DNA sequence and MEME35, followed by manual confirmation. The putative rho-independent terminators were identified using ARNold36 and the free energy of their secondary structures was calculated using Mfold37. The transmembrane domains and signal peptides were found using Phobius38, TMHMM39, HMMTOP40 and SignalP41. The complete genome sequence of phages MP1 and MP2 have been deposited in the NCBI database under accession no. KX078569 and KX078568, respectively.
Comparative analysis of the phage genomes
For MP1 the comparisons were made with Proteus phage vB_PmiM_Pm5461 (NC_028762), Escherichia phages T4 (NC_000866), R49 (NC_005066), RB69 (AY303349), CC31 (GU323318), JS98 (EF469154), Shigella phage SP18 (GQ981382), Salmonella phage S16 (NC_020416) and Vibrio phage KVP40 (NC_005083). MP2 was compared with Morganella phage MmP1 (NC_011085), Escherichia phage T7 (NC_001604), Salmonella phage SP6 (NC_004831), Pseudomonas phages φKMV (AJ505558) and Klebsiella phage KP34 (GQ413938). At the DNA level, homology comparisons between phages were conducted with progressiveMauve42. Dot plots of the whole-genomes were produced using Gepard under default parameters43. At the proteomic level, CoreGenes44 was used to analyze the proteome conservation between phages. Pairwise comparisons were also made using tbBLASTX plug-in in Easyfig45 to reveal intercluster relationships. The phylogenetic analysis of homologous phage large terminase subunit or DNA polymerase proteins was conducted in “one click” mode at phylogeny.fr (http://www.phylogeny.fr/)46. The trees were exported in Netwick format and visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Results and Discussion
Microbiological characterization of the Morganella phages
MP1 and MP2 were isolated from wastewater treatment plant sewage samples using Morganella strains from different human clinical specimens in the enrichment. The phages’ lytic spectra were assessed against a wide range of clinical isolates and six strains of other closely-related Enterobacteriaceae species (Table S1). MP1 and MP2 were able to lyse 67% and 56% of all the Morganella isolates tested, respectively. Therefore, in vitro assessments revealed their infection patterns to be promising for therapy. In addition, they have a complementary lytic range infecting a total of 85% of the strains, and showed to be specific, as they were unable to lyse the remaining Enterobacteriaceae species tested. Their ability to kill drug resistant M. morganii clinical isolates (AmpC strains, resistant up to 11 antibiotics) makes them especially promising antibacterials of potential use against hard-to-treat infections by M. morganii strains. The latent periods and burst sizes were also determined based on the results of one-step growth experiments. Phage MP2 bursts after 20 min releasing only 16 progeny viruses per cell, while MP1 has a longer (35-min) latent period and also higher burst size (41 phages) properties which it shares with Morganella phage FSP124.
To assess the potential safe application of these phages, cytotoxicity tests were performed. Since M. morganii is known to cause several nosocomial infections, including pneumonia and urinary tract infections, MP1 and MP2 cytotoxic effect was evaluated using the cell line A549 (ATCC CCL-185), derived from a human alveolar cell carcinoma47. Similar quantities of soluble formazan detected from A549 cells after 24 hours exposure to MP1 or MP2, compared with the SM buffer, are indicative of absence of toxicity (Figure S1). As such, no counter indications towards the application of MP1 and MP2 to treat bacterial infections in vivo have been observed.
MP1 and MP2 morphologies
MP1 produces uniformly small plaques (0.1-mm diameter), which are difficult to enumerate even in a bacterial lawn solidified with 0.4% agar. MP2 always forms small (0.5-mm diameter) clear and large (2-mm diameter) plaque variants, both surrounded by haloes for each Morganella strain tested. The presence of opaque halos suggests active production of exopolysaccharide depolymerase-like enzymes48, which have never been described for Morganella phages. Transmission electron micrographs revealed two morphotypes (Figs 1A and 2A). MP1 is a myovirus with an elongated icosahedral head (85 nm in length and 65 nm in width) and a contractile tail of 111 nm long and 17 nm wide. MP2 has a smaller isometric head between 48 and 51 nm in diameter and a short (7 nm) non-contractile tail, which is a feature common to members of the Podoviridae4. MP1 and MP2 are then morphologically similar to Morganella phages FSP1 and MmP1, respectively.
Figure 1. Morphology and genome overview of MP1.

(A) Electron micrographs of Morganella-infecting phage MP1 negatively stained with 2% uranyl acetate. (B) Genome map with predicted 271 CDSs numbered and coloured (yellow, green, blue and gray) according to their predicted function. Protein identified by mass spectrometry are pointed with vertical arrows. Above genome, the nucleotide positions in kb are given.
Figure 2. Morphology and genome overview of MP2.

(A) Electron micrograph of Morganella-infecting phage MP2 negatively stained with 2% uranyl acetate. (B) Genome map with predicted 55 CDSs numbered and coloured (yellow, green, blue and gray) according to their predicted function. Protein identified by mass spectrometry are pointed with vertical arrows. Above genome, the nucleotide positions in kb are given.
Genome general characteristics
The MP1 and MP2 genomes are both linear dsDNA molecules of 163,095 bp and 39,394 bp, respectively. The MP1 genome has a GC content of 34.7% whereas MP2 genome GC content is 46.9%. These values fall below the GC content of 51% observed in sequenced M. morganii strains49,50. The assembly pattern of MP1 sequence reads indicated a terminal redundancy and a circular permutation of virion DNA molecules, as observed for coliphage T4, whose DNA is packed into capsids from concatamers, by a headful mechanism51. The assembly pattern of MP2 sequence reads is characteristic for phages with a sequence specific packaging site and indicated the presence of 200-bp terminal repeats at ends of its genome, similar to that of coliphage T752. Genes in both, MP1 and MP2, are densely packed and occupy 95% and 89% of DNA, respectively. The genes mostly belong to one of the three distinct functional modules: DNA packaging and structural proteins; DNA replication, recombination and modification; and cell lysis (Figs 1B and 2B). While in the case of MP2 the genes of particular modules occupy distinct genome regions, in MP1 they are dispersed throughout the genome.
MP1 DNA encodes 271 predicted proteins with coding sequences situated on both the positive and negative strands and 10 tRNAs. The translation of 263 of these proteins starts from an ATG codon, five from a TTG codon, and three from a GTG codon. For 109 of the MP1 proteins a specific function can be assigned, while the other 162 have no known function, including 86 which are unique to this phage (Table S2). Identified homologs of MP1 proteins are mostly encoded by Proteus phage vB_PmiM_Pm5461. Proteins of assigned function include several structural proteins such as the major capsid, tail and neck proteins and proteins related to DNA replication, packaging and regulation. Genes encoding a small and large subunit of aerobic ribonucleotide reductase are separated from each other by a gene encoding HNH-type homing endonuclease (gp233), similarly as it is in the case of Enterobacteria phages RB32 and T4. A few proteins show similarities to products of bacterial genes (e.g. M. morganii, Xenorhabdus griffiniae, Escherichia coli). One example is the endolysin gene (gp100), which encodes a protein with peptidase activity (VANY, PG02557) with similarity to an E. coli peptidoglycan hydrolase. Regarding regulatory elements, 12 promoters and 27 rho-independent terminators were identified.
MP2 has a modular genome containing 55 proteins coding genes, all transcribed in the Watson strand. The functions of 24 proteins can be predicted (Table S3). Most proteins are closely related to those of phage MmP1, with identities ranging from 53 to 100%. Only the products of two predicted genes are homologous to proteins from different phages, namely Citrobacter phage CR8 and Escherichia phage CICC. Genes of assigned function encode several structural proteins, such as the major capsid protein, the capsid assembly protein, several tail proteins (tail tubular protein A and B, tail fiber, head-to-tail joining protein) and DNA replication and transcription-associated proteins, DNA polymerase and RNA polymerase among them. It was possible to identify gp21 (endolysin), gp47 (holin) and gp49 (Rz-like spanin) encoding the lytic cassette. The functions of the remaining 31 protein coding genes are unknown, being 16 of them unique to this phage. Only two host-dependent promoters, and three rho-independent terminators were identified in MP2. All genes start with the ATG codon, with the exception of gp50 (DNA packaging protein), which starts with TTG.
Phage structural proteins
To determine the protein composition of MP1 and MP2 virions, the structural components of both phages were precipitated, separated by thin-layer SDS-PAGE, trypsinized and analysed by electrospray ionization-tandem mass spectrometry (Fig. 3, Tables 1 and 2). Although some proteins were clearly visualized with Simply Blue™ Safe Stain, the whole gel was extracted, fragmented and analysed as previously described53. The resulting peptides allowed the identification of 58 and 21 proteins scattered around the MP1 and MP2 genome, respectively.
Figure 3. SDS-PAGE analysis of the purified structural proteins of MP1 and MP2.
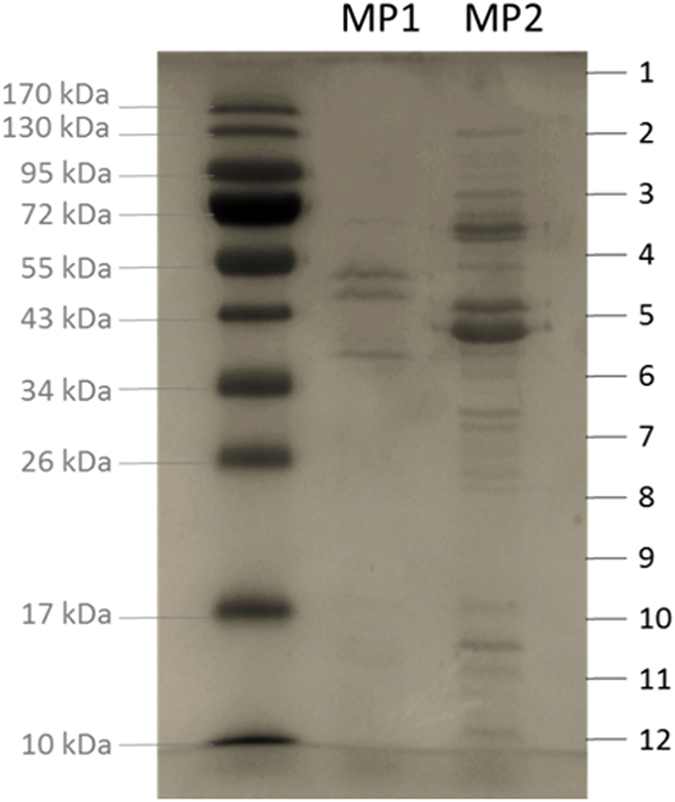
A 12% SDS-PAGE separation gel of phage proteins were made alongside with a PageRulerTM prestained protein ladder. The entire lane was cut into 12 slices for mass spectrometry analysis.
Table 1. Bacteriophage MP1 structural proteins identified by ESI-MS/MS.
| Protein | Identified function | Band N° (most abundant) | Protein MW (Da) MS/MSa | Protein MW (Da)b | N° of unique peptides | Sequence coverage, % | HHPred significant match | E-valuec | Identitiy, %d |
|---|---|---|---|---|---|---|---|---|---|
| gp007 | — | 10 | 15,738 | 14,750 | 1 | 7.4 | — | — | — |
| gp012 | Exonuclease A | 7 | 28,702 | 26,030 | 2 | 9.9 | exonuclease A [Citrobacter phage Merlin] dCTP pyrophosphatase [Proteus phage vB_PmiM_Pm5461] | 1E-108 | 66% |
| gp016 | dCTP pyrophosphatase | 9 | 20,529 | 20,190 | 3 | 18.1 | 5E-91 | 72% | |
| gp018 | — | 7 | 33,158 | 32,490 | 5 | 22.9 | — | — | — |
| gp030 | RecA-like recombination | 5–7 (6) | 44,549 | 42,850 | 11 | 30.5 | RecA-like recombination protein [Serratia phage PS2] | 0.0 | 81% |
| gp038 | DNA polymerase accessory protein clamp loader subunit | 6–7 (6) | 36,767 | 36,470 | 3 | 10.8 | DNA polymerase accessory protein clamp loader subunit [Proteus phage vB_PmiM_Pm5461] | 1E-155 | 66% |
| gp039 | Sliding clamp | 7 | 26,260 | 25,380 | 5 | 21.3 | sliding clamp [Proteus phage vB_PmiM_Pm5461] | 4E-78 | 55% |
| gp042 | Hypothetical protein | 12 | 9,690 | 9,580 | 4 | 40.0 | hypothetical protein [Morganella morganii] | 2E-22 | 53% |
| gp049 | Hypothetical protein | 11 | 14,126 | 13,190 | 1 | 15.8 | hypothetical protein Pm5461_055 [Proteus phage vB_PmiM_Pm5461] | 7E-17 | 44% |
| gp051 | Sigma factor for late transcription | 9 | 21,122 | 20,500 | 4 | 26.4 | sigma factor for late transcription [Proteus phage vB_PmiM_Pm5461] | 7E-82 | 70% |
| gp059 | Hypothetical protein | 11 | 15,176 | 14,750 | 6 | 46.3 | hypothetical protein Aeh1p318 [Aeromonas phage Aeh1] | 3E-11 | 55% |
| gp069 | Thioredoxin | 7 | 40,600 | 39,370 | 2 | 6.6 | thioredoxin [Salmonella phage STML-198] | 0.0 | 80% |
| gp071 | Hypothetical protein | 10 | 19,622 | 17,150 | 4 | 29.8 | hypothetical protein Pm5461_072 [Proteus phage vB_PmiM_Pm5461] | 2E-24 | 39% |
| gp073 | — | 10–11 (10) | 20,635 | 19,200 | 8 | 44.3 | — | — | — |
| gp100 | Hypothetical protein | 11 | 17,714 | 16,480 | 3 | 17.0 | hypothetical protein Pm5461_100 [Proteus phage vB_PmiM_Pm5461] | 5E-42 | 60% |
| gp101 | Endolysin | 11 | 14,738 | 14,480 | 3 | 33.8 | peptidase M15 [Escherichia coli] | 3E-54 | 71% |
| gp113 | Hypothetical protein | 7-11 (8) | 23,179 | 22,310 | 6 | 35.3 | hypothetical protein Pm5461_106 [Proteus phage vB_PmiM_Pm5461] | 2E-42 | 43% |
| gp117 | Hypothetical protein | 10 | 16,842 | 16,160 | 4 | 28.9 | hypothetical protein VR7_gp147 [Enterobacteria phage vB_EcoM-VR7] | 9E-26 | 45% |
| gp119 | Hypothetical protein | 10 | 17,175 | 16,260 | 2 | 14.6 | hypothetical protein SP18_gp147 [Shigella phage SP18] | 5E-40 | 52% |
| gp136 | Hypothetical protein | 11 | 11,613 | 11,210 | 6 | 59.0 | hypothetical protein STP4a_129 [Salmonella phage STP4-a] | 2E-17 | 47% |
| gp137 | Hypothetical protein | 10 | 16,966 | 16,880 | 2 | 16.7 | gp57B conserved hypothetical protein [Enterobacteria phage T4] | 1E-79 | 77% |
| gp139 | dNMP kinase | 7 | 28,387 | 26,640 | 5 | 18.9 | dNMP kinase [Proteus phage vB_PmiM_Pm5461] | 1E-56 | 47% |
| gp140 | Tail completion and sheath stabilizer | 9 | 22,238 | 21,840 | 2 | 11.1 | gp3 tail completion and sheath stabilizer protein [Enterobacteria phage CC31] | 5E-80 | 58% |
| gp141 | DNA end protector | 7 | 31,464 | 30,950 | 3 | 9.2 | DNA end protector protein [Proteus phage vB_PmiM_Pm5461] | 4E-139 | 73% |
| gp147 | Baseplate wedge subunit | 2–3 | 72,921 | 72,370 | 18 | 37.9 | baseplate wedge subunit [Proteus phage vB_PmiM_Pm5461] | 0.0 | 72% |
| gp148 | Baseplate wedge subunit | 1 | 122,856 | 121,220 | 1 | 0,9 | baseplate wedge subunit [Proteus phage vB_PmiM_Pm5461] | 0.0 | 69% |
| gp149 | Baseplate wedge tail fiber connector | 5 | 39,290 | 38,630 | 2 | 7.4 | baseplate wedge tail fiber connector [Proteus phage vB_PmiM_Pm5461] | 0.0 | 77% |
| gp150 | Baseplate wedge subunit and tail pin | 7 | 31,824 | 31,340 | 2 | 6.3 | baseplate wedge subunit and tail pin [Proteus phage vB_PmiM_Pm5461] | 5E-135 | 67% |
| gp151 | Baseplate wedge subunit and tail pin | 2–3 | 67,569 | 67,110 | 14 | 33.9 | baseplate wedge subunit and tail pin [Citrobacter phage Moon] | 0.0 | 48% |
| gp152 | Baseplate wedge subunit and tail pin | 8 | 26,233 | 23,260 | 2 | 8,9 | baseplate wedge subunit and tail pin [Proteus phage vB_PmiM_Pm5461] | 3E-45 | 45% |
| gp153 | Short tail fibers | 5 | 48,108 | 47,610 | 9 | 27.8 | short tail fibers protein [Proteus phage vB_PmiM_Pm5461] | 3E-133 | 49% |
| gp158 | Tail sheath stabilization | 6 | 32,675 | 32,380 | 1 | 4.0 | tail sheath stabilization protein [Proteus phage vB_PmiM_Pm5461] | 8E-134 | 68% |
| gp161 | Tail sheath | 1–3,7 (3) | 73,206 | 72,620 | 26 | 51.3 | tail sheath protein [Proteus phage vB_PmiM_Pm5461] | 0.0 | 76% |
| gp162 | Tail tube | 9 | 20,855 | 18,570 | 4 | 21.3 | tail tube protein [Proteus phage vB_PmiM_Pm5461] | 3E-97 | 79% |
| gp163 | Portal vertex of head | 2–4 (3) | 61,044 | 60,300 | 16 | 41.0 | portal vertex of head [Proteus phage vB_PmiM_Pm5461] | 0.0 | 68% |
| gp165 | Prohead core | 11 | 17,074 | 16,190 | 1 | 6.9 | prohead core protein [Proteus phage vB_PmiM_Pm5461] | 3E-51 | 70% |
| gp166 | Prohead core scaffold and Protease | 10 | 24,248 | 23,130 | 3 | 16.1 | prohead core scaffold protein and protease [Proteus phage vB_PmiM_Pm5461] | 2E-111 | 79% |
| gp167 | Prohead core scaffold | 2–11 (6) | 32,335 | 30,360 | 17 | 77.9 | prohead core scaffold protein [Proteus phage vB_PmiM_Pm5461] | 3E-77 | 56% |
| gp168 | Major capsid | 1–11 (4) | 55,998 | 56,010 | 23 | 60.24 | major capsid protein [Proteus phage vB_PmiM_Pm5461] | 0.0 | 81% |
| gp169 | Head vertex | 4–6 (5) | 46,883 | 46,160 | 21 | 66.5 | head vertex protein [Proteus phage vB_PmiM_Pm5461] | 0.0 | 79% |
| gp187 | Baseplate tail tube initiator | 7 | 33,526 | 33,420 | 4 | 16.1 | baseplate tail tube initiator [Proteus phage vB_PmiM_Pm5461] | 7E-174 | 79% |
| gp189 | Baseplate hub subunit and tail length determinator | 2–3 | 63,213 | 62,730 | 2 | 4.4 | baseplate hub subunit and tail length determinator [Proteus phage vB_PmiM_Pm5461] | 3E-119 | 41% |
| gp190 | Baseplate hub distal subunit | 9 | 19,816 | 18,180 | 3 | 18.5 | baseplate hub distal subunit [Proteus phage vB_PmiM_Pm5461] | 3E-62 | 59% |
| gp191 | Baseplate hub subunit | 5 | 45,005 | 44,550 | 2 | 7.4 | baseplate hub subunit [Proteus phage vB_PmiM_Pm5461] | 0.0 | 68% |
| gp194 | Baseplate wedge subunit | 11 | 14,497 | 14,380 | 2 | 14.0 | baseplate wedge subunit [Proteus phage vB_PmiM_Pm5461] | 9E-56 | 66% |
| gp195 | recombination, repair and ssDNA binding protein | 10–11 | 17,680 | 15,740 | 3 | 22.4 | recombination, repair and ssDNA binding protein [Proteus phage vB_PmiM_Pm5461] | 3E-65 | 71% |
| gp201 | Head outer capsid | 5–6 (6) | 40,233 | 39,710 | 13 | 41.3 | head outer capsid protein [Proteus phage vB_PmiM_Pm5461] | 1E-176 | 68% |
| gp205 | RNA polymerase ADP-ribosylase | 1–3,5,7 (3) | 79,188 | 77,210 | 25 | 35.3 | RNA polymerase ADP-ribosylase [Proteus phage vB_PmiM_Pm5461] | 0.0 | 49% |
| gp206 | DNA ligase | 4,7 (7) | 55,737 | 55,740 | 10 | 27.4 | DNA ligase [Proteus phage vB_PmiM_Pm5461] | 0.0 | 61% |
| gp207 | Hypothetical protein | 8 | 24,524 | 24,410 | 4 | 21.6 | hypothetical protein Pm5461_192 [Proteus phage vB_PmiM_Pm5461] | 3E-126 | 84% |
| gp231 | RNA ligase 1 and tail fiber attachment catalyst | 5–7,10 (5) | 45,404 | 42,480 | 8 | 25.2 | RNA ligase 1 and tail fiber attachment catalyst [Proteus phage vB_PmiM_Pm5461] | 5E-147 | 58% |
| gp244 | Hypothetical protein | 10 | 16,419 | 16,420 | 2 | 15.7 | hypothetical protein Pm5461_225 [Proteus phage vB_PmiM_Pm5461] | 3E-11 | 31% |
| gp249 | dsDNA binding protein | 11 | 11,382 | 10,360 | 5 | 41.8 | dsDNA binding protein [Proteus phage vB_PmiM_Pm5461] | 8E-41 | 75% |
| gp251 | Long tail fiber proximal subunit | 1–3 (3) | 134,949 | 134,620 | 12 | 14.0 | long tail fiber proximal subunit [Proteus phage vB_PmiM_Pm5461] | 0.0 | 60% |
| gp253 | Long tail fiber distal connector | 10 | 19,223 | 17,300 | 3 | 30.7 | long tail fiber distal connector [Proteus phage vB_PmiM_Pm5461] | 2E-24 | 40% |
| gp254 | Long tail fiber distal subunit | 4 | 61,920 | 61,700 | 6 | 13.9 | long tail fiber distal subunit [Proteus phage vB_PmiM_Pm5461] | 2E-22 | 46% |
| gp266 | DNA topoisomerase | 4–5 (5) | 54,442 | 51,720 | 3 | 6.7 | DNA topoisomerase [Proteus phage vB_PmiM_Pm5461] | 0.0 | 63% |
| gp267 | Nucleoid disruption protein | 10 | 17,925 | 17,410 | 1 | 7.1 | nucleoid disruption protein [Proteus phage vB_PmiM_Pm5461] | 9E-25 | 44% |
aMolecular weight (MW) measured via mass spectrometry.
bMolecular weight (MW) predicted on the basis of amino acid sequence.
cHHpred E-value of the most significant matching protein model.
dHHpred percent sequence identity for a pairwise aa alignment with the template protein.
Identified phage proteins are listed below. Their mass and SDS-PAGE band have been indicated, as the number of identified peptides and the complete protein coverage %. It should be noted that Identifications with a low abundance of peptides (<4) could be the result of protein impurities remaining in the samples.
Table 2. Bacteriophage MP2 structural proteins identified by ESI-MS/MS.
| Protein | Identified function | Band N° (most abundant) | Protein MW (Da) MS/MSa | Protein MW (Da)b | N° of unique peptides | Sequence coverage, % | HHPred significant match | E-valuec | Identitiy, %d |
|---|---|---|---|---|---|---|---|---|---|
| gp06 | — | 12 | 12,086 | 11,550 | 2 | 25.2 | — | — | — |
| gp15 | Hypothetical protein | 7 | 22,670 | 21,720 | 1 | 4.5 | hypothetical protein MmP1_gp16 [Morganella phage MmP1] | 3E-48 | 53 |
| gp18 | Single-stranded DNA-binding protein | 6–8 (6) | 26,990 | 26,470 | 10 | 63.0 | single-stranded DNA-binding protein [Morganella phage MmP1] | 2E-133 | 87 |
| gp21 | Endolysin | 10 | 17,690 | 17,370 | 2 | 12.7 | lysozyme [Morganella phage MmP1] | 8E-102 | 93 |
| gp25 | Hypothetical protein | 11 | 8,294 | 7,830 | 1 | 21.6 | hypothetical protein MmP1_gp24 [Morganella phage MmP1] | 3E-24 | 93 |
| gp29 | — | 11–12 (11) | 13,080 | 10,520 | 4 | 38.5 | — | — | — |
| gp32 | Exonuclease | 5–6 | 36,568 | 35,060 | 5 | 19.4 | exonuclease [Morganella phage MmP1] | 0.0 | 90 |
| gp33 | Hypothetical protein | 12 | 9,793 | 9,360 | 1 | 15.1 | hypothetical protein MmP1_gp32 [Morganella phage MmP1] | 3E-17 | 74 |
| gp34 | Hypothetical protein | 10 | 8,919 | 8,440 | 4 | 59.0 | hypothetical protein MmP1_gp33 [Morganella phage MmP1] | 3E-23 | 70 |
| gp35 | Host Range Protein | 10 | 8,427 | 7,930 | 3 | 60.5 | hypothetical protein [Escherichia phage CICC 80001] | 2E-4 | 46 |
| gp36 | Head-to-tail joining protein | 1–5 (3) | 59,543 | 59,350 | 32 | 75.4 | head-to-tail joining protein [Morganella phage MmP1] | 0.0 | 92 |
| gp37 | Capsid assembly protein | 1–7 (5) | 38,204 | 35,430 | 15 | 56.9 | capsid assembly protein [Morganella phage MmP1] | 0.0 | 80 |
| gp38 | Major capsid protein | 1–12 (5) | 36,358 | 36,230 | 22 | 76.5 | major capsid protein [Morganella phage MmP1]; minor capsid protein [Morganella phage MmP1] | 0.0 | 89 |
| gp39 | Hypothetical protein | 1,4–7 (5) | 7,693 | 4,690 | 1 | 31.4 | hypothetical protein MmP1_gp37 [Morganella phage MmP1]; minor capsid protein [Morganella phage MmP1] | 7E-5 | 80 |
| gp40 | Tail tubular protein A | 7–8 (7) | 23,027 | 22,090 | 7 | 59.4 | tail tubular protein A [Morganella phage MmP1] | 6E-122 | 86 |
| gp41 | Tail tubular protein B | 1–5,7,10 (2) | 90,762 | 90,770 | 32 | 47.9 | tail tubular protein B [Morganella phage MmP1] | 0.0 | 88 |
| gp43 | Internal virio + n protein B | 8 | 21,669 | 20,410 | 10 | 50.5 | internal virion protein B [Morganella phage MmP1] | 1E-87 | 84 |
| gp44 | Internal virion protein C | 1–4,6–7,10 (3) | 82,793 | 82,570 | 38 | 54.4 | internal virion protein C [Morganella phage MmP1] | 0.0 | 76 |
| gp45 | Internal virion protein D | 1–7 (2) | 144,296 | 143,870 | 58 | 59.2 | internal virion protein D [Morganella phage MmP1] | 0.0 | 70 |
| gp46 | Tail fiber protein | 1–7,10,12 (3) | 63,591 | 63,260 | 31 | 64.5 | tail fiber protein [Morganella phage MmP1] | 0.0 | 72 |
| gp50 | DNA packaging protein B | 1,3 (3) | 68,739 | 66,550 | 7 | 11.9 | DNA maturase B [Morganella phage MmP1] | 0.0 | 92 |
aMolecular weight (MW) measured via mass spectrometry.
bMolecular weight (MW) predicted on the basis of amino acid sequence.
cHHpred E-value of the most significant matching protein model.
dHHpred percent sequence identity for a pairwise aa alignment with the template protein.
Identified phage proteins are listed below. Their mass and SDS-PAGE band have been indicated, as the number of identified peptides and the complete protein coverage %. It should be noted that Identifications with a low abundance of peptides (<4) could be the result of protein impurities remaining in the samples.
In MP1, 27 proteins had structural-related functions, being the baseplate wedge subunit (gp147), tail sheath (gp162), prohead core scaffold (gp167), major capsid (gp168) and the head vertex proteins, the most abundant (sequence coverage ranging from 37.9 and 79.9%). In several cases, however, the corresponding N- and C-terminal peptides were identified in several gel bands. This suggests posttranslational modifications, possibly due to proteolytic cleavage or degradation. Similar post-translational modifications have been previously observed in the case of T4 head proteins54. Furthermore, 20 proteins were identified with unknown functions, from which gp7, gp18 and gp73 that are located in the first halves of the MP1 genome, were unique. The remaining proteins identified specified unusual functions of DNA replication and cell lysis. Overall, for most identified proteins, relatively high homologies (>60% amino acid identity) were detected only to proteins of Proteus phage vB_PmiM_Pm546 (hereafter referred to as Pm5461).
In MP2, 12 proteins were previously predicted with structural function (gp35-gp41, gp43-gp46, gp50), including the internal virion protein, tail fiber and major capsid proteins. The product of gp45 is likely to facilitate the passage of phage DNA through the cell wall at infection by introducing a small gap in the peptidoglycan layer. It contains the amino acid sequence motif characteristic for soluble lytic transglycosylases, similarly to virion proteins of certain other phages that have this activity10,55. Three additional proteins of unassigned functions, encoded by genes located in a close vicinity to each other were also detected and presumably, are also virion components (gp33-gp34, gp39). BLASTP showed that most of these proteins had a relatively high homology only to Morganella phage MmP1 (>60% amino acid identity). Furthermore, mass spectrometry allowed the detection of two unique proteins, gp6 (22.6% coverage) and gp29 (38.5% coverage), whose genes are located upstream and downstream of the predicted DNA replication module, respectively.
Comparative genomics
The Bacterial and Archaeal Viruses Subcommittee of the International Committee on Taxonomy of Viruses (ICTV) has taken a holistic approach to the classification of phages, employing overall DNA and protein sequence identities coupled with phylogenetic analyses56,57. An initial BLASTN analysis of the complete genome sequence of MP1 revealed that Proteus phage Pm5461 is its closest relative, but these phages only share 39% overall sequence identity. Phage MP2, only exhibits significant overall DNA sequence identity of 70% with Morganella phage MmP1, as the sequence identity decreased below 40% when compared to other phages. Therefore, the creation of novel genus containing only MP1 on one hand and grouping MP2 together with MmP1 on the other hand seems appropriate at this stage. Nevertheless, owing to low nucleotide homology found with MP1 and MP2 against other viruses, we decided to perform multiple genome alignments with all prototype viruses currently classified within Tevenvirinae and Autographivirinae subfamilies, respectively, using progressiveMauve (Fig. 4). Within the T-even phages, despite being classified as a different genus, a remarkable synteny in three large conserved regions (>10 Kb) is observed. Interestingly, Mauve alignment also demonstrated that the second block of MP1, which is similar to Pm5461, is inverted when compared to the remaining phages, suggesting a local adaptive evolution due to genetic drift. As for Autographivirinae phages, a more extensive re-arrangement between the sequence-blocks was observed. Overall, MP2 genome was only collinear with an MmP1 and T7. These results were also further supported using sequence dot plots (Figure S2). In the case of MP1, an interrupted but straight diagonal lines against other Teven phages, being the phage Pm5461 the one with highest score. MP2 only presented homology to phages T7 and MmP1, being the latter with an evident higher score.
Figure 4. Multiple genome alignment of Morganella phage genomes against homologs.

(A) MP1 and (B) MP2 were compared to Tevenvirinae and Autographivirinae like phages, respectively. Genomes were compared using Mauve software, and similarity profiles were generated. Boxes with identical colors represent local colinear blocks (LCB) indicating homologous DNA regions shared by two or more genomes, indicated by a connected line. The placement of a block below the axis indicates inversion. Similarity inside the block is indicated by the height of the colored bars. For ease of interpretation, myovirus Vibro phage KVP40 with a 244,834 bp size was excluded from the Tevenvirinae comparisons.
While the taxonomic position of phages exclusively based on nucleotide similarity can give divergent interpretations due to the different algorithms available, phage relatedness should also be assessed with other factors such as the whole proteome and specific gene cluster organisations. Both Morganella phages have similar virion morphologies, genome size and number of CDSs when compared to the T4 (≈160 kb, 256–279 CDSs) and T7 archetypes (≈40 kb, 48–60 CDSs), respectively56. At the proteomic level, the complete set of MP1 proteins has a higher identity percentage with Pm5461 (67%), followed by T4, S16 and RB14 with proximally 50% homology. The remaining viruses fall below 50%. MP2 was compared with MmP1, presenting a high percentage of sequence identity (86%), followed by T7 (66%) and others (<54%). Taking the genomic and proteomic comparisons together, we conclude that MP1 and MP2 belong to the Tevenvirinae and Autographivirinae subfamilies, but with obvious distant relatedness to other subfamily members.
Phylogenetic relationships
To assess the exact taxonomic position of these two phages, the large subunit terminase proteins of MP1 and related phages, and the DNA polymerases of MP2 and related phages were analysed in “one click” mode at phylogeny.fr46. These genes have been previously used as marker in several phylogenetic studies of the T4- and T7-like phages58,59,60,61. The phylogenetic tree was constructed with all phages currently annotated as Tevenvirinae and Autographivirinae subfamilies along with MP1 and MP2. The resulting trees had a very good correlation with current taxonomy, meaning, every individual branch corresponded quite well with current ICTV recognized genus (Figs 5 and 6). In Tevenvirinae, which now has a total of eight genera, a novel branch containing the Morganella phage MP1 and Proteus phage Pm5461 was visualized. It is quite clear that MP1 and Pm5461 are phylogenetically related to each other, but significantly diverged from other tailed phages in evolution (Fig. 5). As for the Autographivirinae subfamily, the four genera (T7virus, Sp6virus, Phikmvvirus and Kp34virus) were also positioned on completely different branches in the resulting tree, as expected (Fig. 6). Phages MP2 and MmP1 form a distinct clade linked with the T7virus which is associated with a lower overall nucleotide (<10% BLASTN) and proteomic (<50% CoreGenes) identity. This suggests that both MP1 and MmP1 have diverged from a common T7-like ancestor due to genetic drift.
Figure 5. Phylogenetic analysis of MP1.

Phylogenetic tree from MP1 large subunit terminase and its most closely related proteins from other closely related and distant phages. The tree was generated using the “one click” mode at phylogeny.fr.
Figure 6. Phylogenetic analysis of MP2.

Phylogenetic tree from MP2 DNA polymerase and its most closely related proteins from other closely related and distant phages. The tree was generated using the “one click” mode at phylogeny.fr.
Similarities and distinctions features of MP1 and MP2 genomes
To individualize the similarities and differences of MP1 and MP2, we constructed comparative genome maps to show the detail diversion of the core gene organisation against phages with closer homology (Fig. 7). The starting gene and orientation of MP1 and MP2 followed the T4 and T7 convention, respectively.
Figure 7. Divergence of the core gene organization of Morganella phages genomes.

Pairwise comparisons of (A) MP1 and (B) MP2 genomes using tbBLASTX within EasyFig against the closest homolog (Pm5461 or MmP1) and the phage archetypes (T4 or T7), respectively. Arrows indicating CDSs are drawn to scale and colored in the reference genome (MP1 or MP2) according to their predicted function. Gene similarity profiles between phages are indicated in grayscale (and percentage). Major diversions are marked in roman numbers.
MP1 alignment demonstrates short conserved regions frequently interspersed by stretches with no homology (Fig. 7A). Notably, only 85 out of 271 CDSs share >50% amino acid sequence identity with Pm5461. The homology is even lower comparatively to the T4 proteome. The middle halves of the genome, which are closely related, include most of the structural and packaging proteins (e.g. baseplate wedge subunits, tail fibers, neck wiskers, tail sheath and major capsid), which have been identified by mass spectrometry. Interestingly, halves upstream (cluster I) and downstream (cluster II) of this region are the ones that display the most significant differences. Cluster I exclusively encode leftward-oriented hypothetical genes, with the exception of two endonucleases (gp96-97) and the endolysin (gp101). Some of these proteins with unknown functions might be related to MP1 adaptation to different host ranges as previously observed with other T4-like phages49,62. Interestingly, cluster II with >10 kb long (see also green box of Fig. 7A) coding for a second group of structural genes, mostly for distal host recognition devices (baseplate tail cap, wedge and hubs) and outer capsid related proteins, is shared with Pm5461 proteome but is completely inverted on T4 and all other T-even phages. Although it is known that chromosomal inversions can alter gene expression patterns and also result in major phenotypic alterations23,50, the specific biological meaning is unknown. Nevertheless, this example illustrates well that T-even evolution is not exclusively driven by exchanging, deleting and adding of genomic segments, but also by chromosomal inversions that may play an important role as well. This seems to be particularly true in MP1, fostering the ecological divergence in Tevenvirinae subfamily.
In MP2, the major differences are located in the early genes cluster (cluster I), beginning of the DNA replication and recombination module (cluster II) and in the lysis cassette (cluster III) (Fig. 7B) that helped to provide phylogenetic distance between MP2 and T7. The high proteomic homology between MP2 to MmP1 is evident, but drastically decreases when compared to T7. From the early genes found (gp1 to gp9), only two proteins (gp1 and gp3) had homology with phages, namely Citrobacter phage CR8 and MmP1, with relatively low homology (<64% amino acid identity). In the remaining CDSs, no BLASP hits were found. This genomic region has been implicated with mechanisms that block the host macromolecular synthesis in T7-like phages, which in MP2 appears to be novel60. The RNAP, which is located in the DNA replication and recombination module, is a distinctive feature of the T7-like phages63,64, although located three CDSs ahead of T7. The MP2 RNAP has a relatively high homology to MmP1 gene (92% amino acid identity), dropping below 72% against the remaining phage proteins in the database. Three hypothetical CDSs located downwards of the DNA ligase (gp13) are novel. In the DNA packaging and structural protein cluster, a relatively strong homology was found, particularly against MmP1 (>70% coverage). However, mass spectrometry also allowed the identification of three unique proteins, which are probably also part of the mature virion (gp33-gp34, gp39). Notably, it seems that both MP2 and MmP1 have acquired complete novel genes coding for holin, DNA packaging and Rz-like genes (gp47-49), as lack of relatedness is observed to T7. This evolutionary adaptation makes sense, as while the similar endolysin found acts against a conserved peptidoglycan molecule (A1ϒ chemotype) shared by Gram-negative bacteria65, the acquisition of novel holins and spanins are necessary to deal with a diversified bacterial membrane structures, in this case of M. morganii cells. Overall, the genetic gains, losses, or replacements of MP1 and MP2 provide novel insights into the particular steps of genome divergence.
Conclusion
We have isolated and characterized two novel representatives of phages (MP1 and MP2) that infect drug-resistant M. morganii clinical isolates and feature unique characteristics. The obligatorily lytic nature of MP1 and MP2, their wide host range within M. morganii clinical isolates and the lack of toxicity of their virions for mammalian cells makes both these phages suitable for further studies in the development of phage cocktail against drug-resistant M. morganii strains. Additionally, based on limited DNA sequence identity (39% as shown using BLASTN) between Morganella phage vB_MmoM_MP1 and Proteus phage vB_PmiM_Pm5461, we cannot yet recommend that ICTV create a new genus within the Tevenvirinae. On the other hand, the similarity between Morganella phages vB_MmoP_MP2 and MmP1 (70% at DNA and 86% at protein levels) strongly indicate that these two phages should be considered as species within a new genus in the Autographivirinae. We propose that this be called “Mmp1virus” after the first characterized isolate.
Additional Information
How to cite this article: Oliveira, H. et al. Characterization and genomic analyses of two newly isolated Morganella phages define distant members among Tevenvirinae and Autographivirinae subfamilies. Sci. Rep. 7, 46157; doi: 10.1038/srep46157 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This study was supported by the Portuguese Foundation for Science and Technology (FCT) under the scope of the strategic funding of UID/BIO/04469/2013 unit, COMPETE 2020 (POCI-01-0145-FEDER-006684) and the Project PTDC/BBB-BSS/6471/2014 (POCI-01-0145-FEDER-016678). RL contributed to the genome sequencing analysis, supported by the KU Leuven GOA Grant ‘Phage Biosystems’. JP acknowledges the project R-3986 of the Herculesstichting.
Footnotes
The authors declare no competing financial interests.
Author Contributions H.O. and G.P. isolated, sequenced, annotated and analysed the two genomes and drafted the manuscript in equal manner. A.O. helped with the isolation and revised the manuscript. A.K. performed, wrote the phylogenetic studies and revised the manuscript. M.L. and A.K. helped with the phage annotation, analysis of the genome ends and revised the manuscript. J.N., H.H. and R.L. performed, analysed and wrote the proteomic related work. J.A. devised the study, assisted with experimental design and interpretation of data and edited the manuscript. All authors have read and approved the final manuscript.
References
- Summers W. C. Bacteriophage therapy. Annual review of microbiology 55, 437–451 (2001). [DOI] [PubMed] [Google Scholar]
- Oliveira H. et al. Characterization and genome sequencing of a Citrobacter freundii phage CfP1 harboring a lysin active against multidrug-resistant isolates. Applied microbiology and biotechnology (2016). [DOI] [PubMed] [Google Scholar]
- Thomas R. Regulation of gene expression in bacteriophage lambda. Current topics in microbiology and immunology 56, 13–42 (1971). [DOI] [PubMed] [Google Scholar]
- Ackermann H. W. Tailed bacteriophages: the order caudovirales. Advances in virus research 51, 135–201 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D. & Herskowitz I. Properties of hybrids between Salmonella phage P22 and coliphage lambda. Nature 251, 584–589 (1974). [DOI] [PubMed] [Google Scholar]
- Serwer P. et al. Improved isolation of undersampled bacteriophages: finding of distant terminase genes. Virology 329, 412–424 (2004). [DOI] [PubMed] [Google Scholar]
- Chibani-Chennoufi S., Dillmann M. L., Marvin-Guy L., Rami-Shojaei S. & Brussow H. Lactobacillus plantarum bacteriophage LP65: a new member of the SPO1-like genus of the family Myoviridae. Journal of bacteriology 186, 7069–7083 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwer F. & Edwards R. The Phage Proteomic Tree: a genome-based taxonomy for phage. Journal of bacteriology 184, 4529–4535 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavigne R. et al. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC microbiology 9, 224 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moak M. & Molineux I. J. Role of the Gp16 lytic transglycosylase motif in bacteriophage T7 virions at the initiation of infection. Molecular microbiology 37, 345–355 (2000). [DOI] [PubMed] [Google Scholar]
- Liu H., Zhu J., Hu Q. & Rao X. Morganella morganii, a non-negligent opportunistic pathogen. International journal of infectious diseases: IJID: official publication of the International Society for Infectious Diseases 50, 10–17 (2016). [DOI] [PubMed] [Google Scholar]
- Rhomberg P. R., Fritsche T. R., Sader H. S & Jones R. N. Clonal occurrences of multidrug-resistant Gram-negative bacilli: report from the Meropenem Yearly Susceptibility Test Information Collection Surveillance Program in the United States (2004). Diagnostic microbiology and infectious disease 54, 249–257 (2006). [DOI] [PubMed] [Google Scholar]
- Endimiani A. et al. Proteus mirabilis bloodstream infections: risk factors and treatment outcome related to the expression of extended-spectrum beta-lactamases. Antimicrobial agents and chemotherapy 49, 2598–2605 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J. C. et al. Detection of KPC-2 and qnrS1 in clinical isolates of Morganella morganii from China. Diagnostic microbiology and infectious disease 73, 207–209 (2012). [DOI] [PubMed] [Google Scholar]
- Philippon A., Arlet G. & Jacoby G. A. Plasmid-determined AmpC-type beta-lactamases. Antimicrobial agents and chemotherapy 46, 1–11 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery E. L., Mody L. & Mobley H. L. Identification of a modular pathogenicity island that is widespread among urease-producing uropathogens and shares features with a diverse group of mobile elements. Infection and immunity 77, 4887–4894 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- France D. R. & Markham N. P. Epidemiological aspects of Proteus infections with particular reference to phage typing. Journal of clinical pathology 21, 97–102 (1968). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovici M. [Research on the phago-bacterial systems in the Proteus group. I. Manifestations of lysosensitivity and lysogeny in bacteria of the Proteus group]. Archives roumaines de pathologie experimentales et de microbiologie 21, 307–314 (1962). [PubMed] [Google Scholar]
- Pavlatou M. & Vlassaki I. Preliminary research on the lysotyping of the Proteus genus. Archives de l’Institut Pasteur hellenique 7, 137–140 (1961). [PubMed] [Google Scholar]
- Schmidt W. C. & Jeffries C. D. Bacteriophage typing of Proteus mirabilis, Proteus vulgaris, and Proteus morganii. Applied microbiology 27, 47–53 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee J. N. Transduction in Proteus morganii. Nature 210, 220 (1966). [DOI] [PubMed] [Google Scholar]
- Coetzee J. N., Smit J. A. & Prozesky O. W. Properties of Providence and Proteus morganii transducing phages. Journal of general microbiology 44, 167–176 (1966). [DOI] [PubMed] [Google Scholar]
- Prozesky O. W., De Klerk H. C. & Coetzee J. N. The morphology of Proteus bacteriophages. Journal of general microbiology 41, 29–36 (1965). [DOI] [PubMed] [Google Scholar]
- Yamaki S., Omachi T., Kawai Y. & Yamazaki K. Characterization of a novel Morganella morganii bacteriophage FSP1 isolated from river water. FEMS microbiology letters 359, 166–172 (2014). [DOI] [PubMed] [Google Scholar]
- Zhu J. et al. Identification of lytic bacteriophage MmP1, assigned to a new member of T7-like phages infecting Morganella morganii. Genomics 96, 167–172 (2010). [DOI] [PubMed] [Google Scholar]
- Kropinski A. M., Prangishvili D. & Lavigne R. Position paper: the creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environmental microbiology 11, 2775–2777 (2009). [DOI] [PubMed] [Google Scholar]
- Sillankorva S., Neubauer P. & Azeredo J. Isolation and characterization of a T7-like lytic phage for Pseudomonas fluorescens. BMC biotechnology 8, 80 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. F. & Maniatis T. Molecular cloning: a laboratory manual, Edn. 3rd. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 2001). [Google Scholar]
- Van den Bossche A. et al. Systematic identification of hypothetical bacteriophage proteins targeting key protein complexes of Pseudomonas aeruginosa. Journal of proteome research 13, 4446–4456 (2014). [DOI] [PubMed] [Google Scholar]
- Aziz R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC genomics 9, 75 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F., Gish W., Miller W., Myers E. W. & Lipman D. J. Basic Local Alignment Search Tool. Journal of molecular biology 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- Soding J., Biegert A. & Lupas A. N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic acids research 33, W244–248 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattner P., Brooks A. N. & Lowe T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic acids research 33, W686–689 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett D. & Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic acids research 32, 11–16 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic acids research 37, W202–208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naville M., Ghuillot-Gaudeffroy A., Marchais A & Gautheret D. ARNold: a web tool for the prediction of Rho-independent transcription terminators. RNA biology 8, 11–13 (2011). [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic acids research 31, 3406–3415 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L., Krogh A. & Sonnhammer E. L. A combined transmembrane topology and signal peptide prediction method. Journal of molecular biology 338, 1027–1036 (2004). [DOI] [PubMed] [Google Scholar]
- Kall L. & Sonnhammer E. L. Reliability of transmembrane predictions in whole-genome data. FEBS letters 532, 415–418 (2002). [DOI] [PubMed] [Google Scholar]
- Tusnady G. E. & Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics 17, 849–850 (2001). [DOI] [PubMed] [Google Scholar]
- Bendtsen J. D., Nielsen H., von Heijne G. & Brunak S. Improved prediction of signal peptides: SignalP 3.0. Journal of molecular biology 340, 783–795 (2004). [DOI] [PubMed] [Google Scholar]
- Darling A. E., Mau B. & Perna N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PloS one 5, e11147 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumsiek J., Arnold R. & Rattei T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23, 1026–1028 (2007). [DOI] [PubMed] [Google Scholar]
- Zafar N., Mazumder R. & Seto D. CoreGenes: a computational tool for identifying and cataloging “core” genes in a set of small genomes. BMC bioinformatics 3, 12 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan M. J., Petty N. K. & Beatson S. A. Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A. et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic acids research 36, W465–469 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber M., Smith B., Szakal A., Nelson-Rees W. & Todaro G. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. International journal of cancer 17, 62–70 (1976). [DOI] [PubMed] [Google Scholar]
- Hughes K. A., Sutherland I. W. & Jones M. V. Biofilm susceptibility to bacteriophage attack: the role of phage-borne polysaccharide depolymerase. Microbiology 144 (Pt 11), 3039–3047 (1998). [DOI] [PubMed] [Google Scholar]
- Chen Y. T. et al. Whole-genome sequencing and identification of Morganella morganii KT pathogenicity-related genes. BMC genomics 13 Suppl 7, S4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash J. H. & Young N. M. Draft Whole-Genome Sequence of Morganella morganii Serotype O:1ab. Genome announcements 3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao V. B. B. L. In Viral Genome Packaging Machines:.Genetics, Structure, and Mechanisms (ed. C. C. S.) 40–58 (Springer, 2005). [Google Scholar]
- Serwer P. In Viral Genome Packaging Machines: Genetics, Structure, and Mechanisms (ed. C. C. S.) 59–79 (Springer, 2005). [Google Scholar]
- Lavigne R. et al. The structural proteome of Pseudomonas aeruginosa bacteriophage phiKMV. Microbiology 152, 529–534 (2006). [DOI] [PubMed] [Google Scholar]
- Baschong W. et al. Mass analysis of bacteriophage T4 proheads and mature heads by scanning transmission electron microscopy and hydrodynamic measurements. Journal of structural biology 106, 93–101 (1991). [DOI] [PubMed] [Google Scholar]
- Lehnherr H., Hansen A. M. & Ilyina T. Penetration of the bacterial cell wall: a family of lytic transglycosylases in bacteriophages and conjugative plasmids. Molecular microbiology 30, 454–457 (1998). [DOI] [PubMed] [Google Scholar]
- Adriaenssens E. M. et al. Taxonomy of prokaryotic viruses: 2016 update from the ICTV bacterial and archaeal viruses subcommittee. Archives of virology (2016). [DOI] [PubMed] [Google Scholar]
- Krupovic M. et al. Taxonomy of prokaryotic viruses: update from the ICTV bacterial and archaeal viruses subcommittee. Archives of virology 161, 1095–1099 (2016). [DOI] [PubMed] [Google Scholar]
- Chen Z. & Schneider T. D. Information theory based T7-like promoter models: classification of bacteriophages and differential evolution of promoters and their polymerases. Nucleic acids research 33, 6172–6187 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adriaenssens E. M. et al. Bacteriophages LIMElight and LIMEzero of Pantoea agglomerans, belonging to the “phiKMV-like viruses”. Applied and environmental microbiology 77, 3443–3450 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann H. W., Krisch H. M. & Comeau A. M. Morphology and genome sequence of phage varphi1402: A dwarf myovirus of the predatory bacterium Bdellovibrio bacteriovorus. Bacteriophage 1, 138–142 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheepudom J., Lee C. C., Cai B. & Meng M. Isolation, characterization, and complete genome analysis of P1312, a thermostable bacteriophage that infects Thermobifida fusca. Frontiers in microbiology 6, 959 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeau A. M., Bertrand C., Letarov A., Tetart F. & Krisch H. M. Modular architecture of the T4 phage superfamily: a conserved core genome and a plastic periphery. Virology 362, 384–396 (2007). [DOI] [PubMed] [Google Scholar]
- Lavigne R. et al. The genome of bacteriophage phiKMV, a T7-like virus infecting Pseudomonas aeruginosa. Virology 312, 49–59 (2003). [DOI] [PubMed] [Google Scholar]
- Ceyssens P. J. et al. Genomic analysis of Pseudomonas aeruginosa phages LKD16 and LKA1: establishment of the phiKMV subgroup within the T7 supergroup. Journal of bacteriology 188, 6924–6931 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleifer K. H. & Kandler O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 36, 407–477 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.