

Abstract
Human studies demonstrate that sleep impairment is a concurrent comorbidity of autism spectrum disorders (ASD), but its etiology remains largely uncertain. One of the prominent theories of ASD suggests that an imbalance in synaptic excitation/inhibition may contribute to various aspects of ASD, including sleep impairments. Following the identification of Nlgn3R451C mutation in patients with ASD, its effects on synaptic transmission and social behaviours have been examined extensively in the mouse model. However, the contributory role of this mutation to sleep impairments in ASD remains unknown. In this study, we showed that Nlgn3R451C knock-in mice, an established genetic model for ASD, exhibited normal duration and distribution of sleep/wake states but significantly altered electroencephalography (EEG) power spectral profiles for wake and sleep.
Keywords: Nlgn3 R451C mouse model, Autism, EEG, NREM, REM, Sleep deficit
Introduction
Autism spectrum disorders (ASD) are a class of pervasive neurodevelopmental conditions, affecting 1 in 110 children [1, 2]. ASD are diagnosed based on impaired social interaction, impaired communication, and restricted behavioural stereotypies. In addition, sleep impairment is reported as one of the most concurrent symptoms in this population [3, 4]; 40–80% of children with ASD are reported to have some sleep abnormalities [3–11]. Currently, sleep abnormalities in patients with ASD are largely undertreated as other symptoms take precedence [3]. However, studies show that core symptoms of ASD as well as behavioural measures are worsened by poor sleep quality [3], highlighting the importance to understand and treat sleep abnormalities in ASD. For example, the severity of social and communication deficits are exacerbated following sleep deprivation [3, 12, 13]. In addition, sleep impairments were also linked to increased aggression, irritability, hyperactivity, and affective problems, that may further impair daytime functioning of patients with ASD [3, 13–16]. Despite the pervasiveness of sleep impairment in the ASD population, its neurochemical underpinnings remain unclear.
ASD are usually diagnosed between the ages of 2 to 4, a developmental period with extensive activity-dependent neuronal remodelling, but their symptoms persist throughout adulthood [2, 17]. A prominent neurochemical notion for ASD etiology is the excitation/inhibition (E/I) imbalance theory which suggests that altered neuronal network excitability may underline ASD [2, 18, 19]. For example, while an upregulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, the principal mediator of fast excitatory synaptic transmission, was found in post-mortem brain samples of patients with ASD, a concomitant downregulation of the inhibitory γ-aminobutyric acid (GABA) receptors was found [2]. Furthermore, abnormal cortical and subcortical excitability was also proposed to be a critical contributor to sleep impairment in ASD [4].
Neuroligins (Nlgns), a family of postsynaptic cell adhesion proteins, are crucial for neuronal E/I balance through their regulation of GABAergic and glutamatergic synaptic strength [18, 20–23]. To date, 5 Nlgn family members have been identified in humans, including Nlgn 1-3, which have close homologs in mice [24, 25]. Importantly, a missense mutation in Nlgn3 (Nlgn3R451C), which results in its reduced synaptic expression due to increased retention in the endoplasmic reticulum [24, 26], is linked with ASD in humans [24, 25, 27] as well as impaired social interaction and vocal communication in rodent models [24, 28–30]. Although Nlgn3 is ubiquitously expressed in the brain, its function in synaptic transmission appears to be region-specific, affecting the somatosensory cortex and the hippocampus differently [24, 30]. Recently, a number of studies have looked at the role of neuroligins in sleep. In one study, Nlgn1 knockout mice were unable to maintain wakefulness and spent more time in NREM sleep [31] while drosophila deficient in neuroligin 4 exhibited impaired night sleep in another study [32]. To date, although numerous studies have demonstrated that Nlgn3R451C mutation is linked to impaired social interaction and memory, whether it also contributes to sleep impairment is unknown. In this study, we employed electroencephalography (EEG)-electromyography (EMG) recordings to evaluate sleep properties in Nlgn3R451C knock-in (KI) mice. We demonstrated altered EEG spectral profiles in wakefulness and sleep in these mice.
Methods
Animals, housing conditions, and genotyping
Adult Nlgn3R451C mice obtained from the Jackson Laboratory were housed and bred under a 12 h light-12 h dark cycle (light on 6:40 am, light off 6:40 pm) with constant ambient temperature. Food and water were provided ad libitum. All animal protocols were approved by the Animal Lab Services ethics committee of the Hospital for Sick Children. To determine the genotype (wild type or knock-in) of the mice, the DNA fragments were amplified using polymerase chain reaction (PCR) with primers 5’- TGTACCAGGAATGGGAAGCAG-3’ and 5’- GGTCAGAGCTGTCATTGTCAC-3’ using the conditions recommended by the Jackson Laboratory.
Stereotaxic surgery
Adult male mice [WT = 19.1 ± 1.0 (SEM) weeks, 33.4 ± 0.7 g, n = 7; Nlgn3R451C KI = 19.3 ± 1.1 (SEM) weeks, 30.6 ± 0.6 g, n = 7] were used for surgery. Three cortical electrodes for EEG recordings and two insulated stainless steel wires (0.011 mm diameter, Cooner Wire, Chatsworth, CA, USA) for EMG recordings were soldered to a multi-channel connector (Digi-Key Electronics, Thief River Falls, MN, USA) prior to implantation. Animals were anesthetized using ketamine (100–150 mg/kg as needed, intraperitoneal, i.p.) and xylazine (7–10 mg/kg as needed, i.p.) prior to surgery. The three electrodes were inserted through holes made in the skull and placed against the dura, over the cerebellum, left frontal lobe (1.7 mm lateral to midline and 1.5 mm anterior to bregma), and right parietal lobe (1.7 mm lateral to midline and 1.0 anterior to lambda). In addition, 2 stainless steel wires were sutured into the neck muscles. Dental acrylic (Lang Dental Manufacturing Co., Inc., Wheeling, IL, USA) was used to secure the multi-channel connector to the skull. After surgery, ketoprofen (5 mg/kg, subcutaneous, s.c.) was administered for post-operative pain control and 1 ml 0.9% saline (s.c.) was administered for fluid loading.
Experimental protocol and data acquisition
All mice were recovered for at least 8 days in their home cage and were habituated individually for another 3 days in the recording chamber prior to the 48 h EEG-EMG recording. Seven pairs of age-matched Nlgn3R451C and their wildtype (WT) littermate were used for EEG-EMG recordings. The light-dark cycle during recording was kept consistent with the housing room condition. EEG and EMG signals were recorded, amplified, and filtered with HFF at 1 Hz and LFF at 90 Hz using Grasslab (Natus Neurology Incorporated – Grass Products, Warwick, RI, USA) and sampled at 512 Hz.
Sleep wake states determination
The 48 h recordings were visually scored for sleep-wake states by independent investigators who were blind to the genotype of the mice. Sleep-wake states were identified by inspecting both EEG and EMG signals using 4-s epochs, and classified into wakefulness, NREM sleep, and REM sleep, as previously described [33]. Sample EEG-EMG traces of each sleep-wake state are shown in Fig. 1.
Fig. 1.
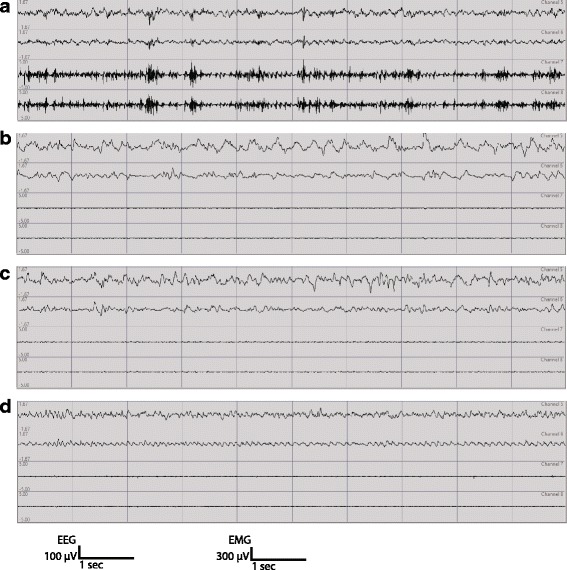
Representative traces of EEG-EMG recordings in a WT mouse. For each mouse, continuous recordings for 48 h were done (top 2 channels: EEG; bottom 2 channels: EMG). The top EEG channel is from left frontal electrode recording with reference to cerebellum. The second EEG channel is from right parietal electrode recording with reference to cerebellum. a Wake EEG is predominated by oscillations greater or equal to theta oscillations (≥5 Hz) with varying amplitude and is irregular in comparison to NREM and REM sleep. b, c NREM sleep is predominated by either high amplitude delta waves (<4 Hz) or mixed high amplitude theta/delta waves in the EEG channels and low muscle activity. The mixed high amplitude theta/delta waves in our mice often occur before transitioning into REM sleep. d REM sleep is characterized by regular low amplitude theta waves (≥7 Hz) in the EEG channels associated with muscle atonia. Every gridline marks 1 s in duration. The amplitude scale for EEG is 100 μV and the amplitude scale for EMG is 300 μV
EEG spectral analysis
The two EEG channels were subtracted to obtain a single EEG signal for Fast Fourier Transformation (FFT) using MatLab (MathWorks, Natick, MA, USA). These signals were further filtered to reduce noise prior to FFT and only frequency bands below 56 Hz were subjected to FFT. To achieve these, the filters were set as HFF = 0.55 Hz, band stop = 0.45 Hz, band stop = 60 Hz. Total EEG power (μV2) between 1 and 56 Hz for each 4 s epoch were obtained using MATLAB. To normalize the data, EEG power (μV2) for each frequency bin within each epoch was calculated as a percentage of the average of the total EEG powers (1–56 Hz) of all epochs.
Statistical analysis
The EEG-EMG signals of each mouse were recorded for 48 h. The data from the 2 days were averaged. As noise in our recordings was characterized by very high amplitude oscillations, we removed all epochs with any frequency band that has a power of greater than 300% after normalization (regardless of frequency bands and sleep state, but most of these occur during wake states). The data were then re-normalized after the removal of these epochs. Once again, epochs with any 1 Hz frequency bin that has a normalized power > 300% were removed. This accounted for 2.1% of WT data and 2.2% of KI data. This was done to reduce the effect of extremely high amplitudes outlier oscillations on the overall data by reducing skewing effect. All the averaged data were stated as mean ± SEM and statistically evaluated by Student’s t-test for comparisons of two groups, or ANOVA (one-way, two-way or repeated measures wherever appropriate) for comparisons of more than two groups followed by post-hoc Bonferroni t-tests.
Results
Nlgn3R451C mutant mice exhibit normal time-of-day distribution of sleep-wake states across time-of-day and sleep fragmentation
We first analyzed if the proportion of time spent in each state differed between the first day and second day of recordings. As day 1 and day 2 did not differ significantly for any of the state (all p >0.098), the two days’ data were averaged for subsequent analysis. As patients with ASD often have shortened sleep duration, longer waking duration during the night, and altered circadian rhythm for sleep [3, 10, 14, 34], we analyzed the proportion of time that the mice spent in each vigilance state and their distribution during the light (ZT1-12 h) and dark (ZT13-24 h) periods. During both the light and dark periods, the Nlgn3R451C mutant mice did not differ significantly from their WT littermates in the proportion of time that they spent in wakefulness, NREM sleep, and REM sleep (all p >0.14) (Fig. 2). To determine the effect of Nlgn3R451C mutation on the distribution of vigilance states across the time-of-day, we analyzed the proportion of time spent during each hour over the 24 h recording period, and found no significant difference between genotypes for all three states (genotype: all p >0.05) (Fig. 3). As patients with ASD are often reported to have more fragmented sleep or more frequent awakenings after sleep onset than non-ASD subjects [3, 34], we also examined if Nlgn3R451C mutant mice had alterations in the number of episodes and the duration of each individual sleep-wake episode. As shown in Fig. 4, there were no significant differences between Nlgn3R451C mutant mice and WT controls in the number (all p >0.45) or the duration of sleep/wake episodes (all p >0.13) (Fig. 4). Taken together, these results suggest that Nlgn3R451C mutation does not significantly affect the overall sleep/wake duration or contribute to fragmentation of sleep.
Fig. 2.
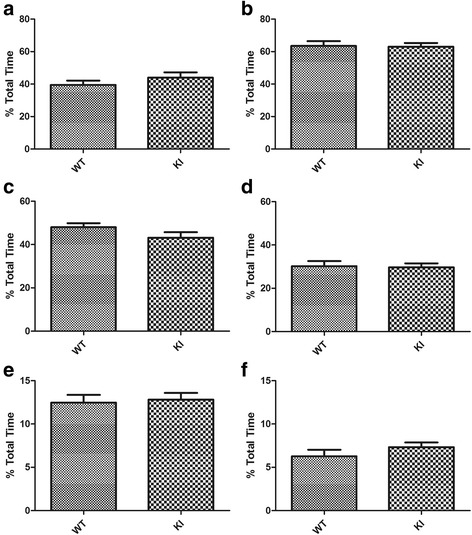
Total sleep-wake time in Nlgn3R451C mutant mice. Proportion of total time spent in each vigilance state during the 12-h light and 12-h dark periods in WT (n = 7) and Nlgn3R451C mice (KI, n = 7). Student t-test was used for this set of analysis. Both WT and Nlgn3R451C mice spent more time in NREM and REM sleep during the light period than they did during the dark period. Nlgn3R451C mice did not significantly alter the time that the mice spent in wake (a), NREM sleep (c), and REM sleep (e) during light period (all p >0.15. Nlgn3R451C mice did not significantly alter the time that the mice spent in wake (b), NREM sleep (d), and REM (f) sleep during dark period (all p >0.28)
Fig. 3.
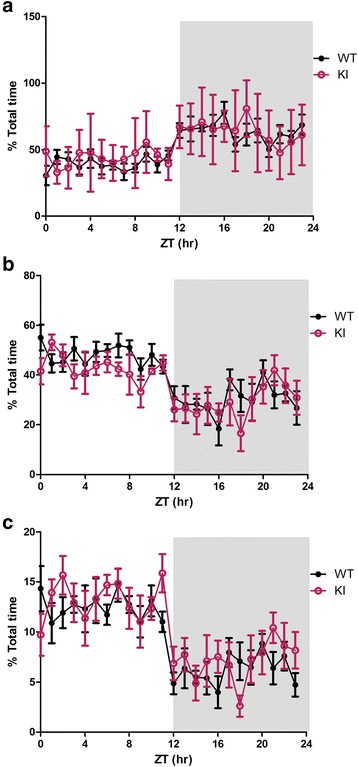
Time-of-day profile of vigilance states in WT and Nlgn3R451C mutant mice. The distribution profile of each vigilance state across the entire recording. Nlgn3R451C mutant mice exhibited a trend of less NREM sleep than WT (p = 0.051) (b), while the two groups did not differ from each other for wakefulness (a) and REM sleep (c) (genotype: both > 0.11)
Fig. 4.
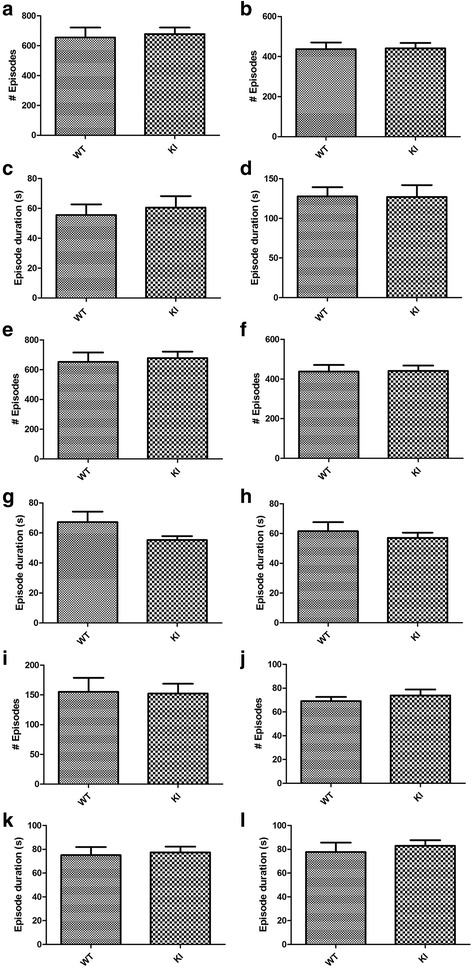
Sleep-wake episodes in Nlgn3R451C mutant mice. Total number of episodes and the mean episode duration for each vigilance state were compared between WT (n = 7) and Nlgn3R451C mice (KI, n = 7) using student t-test. Episode number of each state and their mean episode duration were separately assessed in the light (a, c, e, g, i, and k) or dark phase (b, d, f, h, j, and l). Nlgn3R451C mutation did not result in significant alteration in the number of episodes (all p >0.45) nor the mean episode duration (all p >0.13) of wake (a to d), NREM (e to h), and REM sleep (i to l) in the light period or the dark period
Nlgn3R451C mutant mice exhibit altered power spectral profiles for wakefulness, NREM, and REM sleep
In order to examine the role of Nlgn3 in sleep quality, power spectrum of each state was compared between genotypes. Firstly, the mean spectral powers of 1 Hz bands in each vigilance state were normalized (to the average of the total power of all 1 Hz bins from 1 to 56 Hz across all epochs) and expressed as percentage of the total power. Significant genotype-frequency interaction was found for NREM state (F = 2.856, p <0.001) (Fig. 5). More specifically, post-hoc Bonferroni test showed significantly reduced EEG power between 2-8 Hz in Nlgn3R451C mice (p <0.05) (Fig. 5). On the other hand, no significant “genotype” or “genotype-frequency” interactions were found for wake or REM sleep (both p >0.11) (Fig. 5). In addition, the normalized powers of delta (1–4 Hz), theta (5–8 Hz), alpha (9–12 Hz), sigma (13–15 Hz), and beta (15–30 Hz) frequency bands were examined in each sleep/wake state over time as these are indicators of cortical arousal and sleep quality [35–44]. These bands were normalized to the total power of 1–56 Hz as described above. These analyses revealed significantly altered wake/sleep EEG spectral profile in Nlgn3R451C mutant mice. Specifically, the EEG sigma (F = 7.363, p <0.01) and beta (F = 9.024, p <0.01) powers were all significantly higher in Nlgn3R451C mutant mice during wakefulness in comparison to WT mice (Table. 1, Fig. 6). Delta power during NREM sleep, which is an indicator of sleep depth [36], was reduced in Nlgn3R451C mutants (F = 20.981, p <0.001) (Table 1, Fig. 6). Theta (F = 30.622, p <0.001) and alpha (F = 6.162, p <0.05) powers during NREM sleep were also reduced in Nlgn3R451C mice (Table 1, Fig. 6). During REM sleep, while alpha power (F = 12.069, p <0.001) was reduced in Nlgn3R451C mice, beta power was increased in Nlgn3R451C mice (F = 4.829, p < 0.05) (Table 1, Fig. 6). During wakefulness, time had a significant main effect on beta power (F = 2.226, p <0.05) (Table 1). During NREM sleep, time had a significant main effect on delta power (F = 2.612, p < 0.05) (Table 1). Frequency bands with no main genotype effect (p >0.05) are not included in Fig. 6. In another set of analysis for time effect, we normalized the data of each frequency band by expressing it as a % of mean 24 h activity of each mouse for the targeted band within each arousal state. These analysis revealed significant time effect for all frequency bands (i.e. delta, theta, alpha, sigma, and beta; all p <0.001) in both genotypes. More specifically, EEG power for all of the stated bands were higher during the dark period for wakefulness, whereas EEG power for all of these bands were higher during the light period for NREM and REM sleep. Furthermore, we found no significant genotype-time interaction (all p >0.3), which is consistent with what we showed in Fig. 6 and in Table 1. This further supports that the effect of genotype on frequency bands is not time-dependent. The difference found on time effect between this analysis and the analysis done in Fig. 6 is likely due the fact that by normalizing the power of frequency band within each epoch to the total activity of the targeted band in the entire recording is more sensitive in revealing the time effect.
Fig. 5.
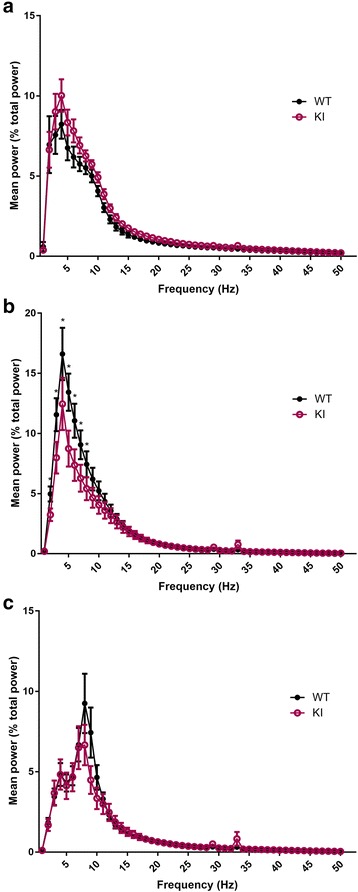
Power spectral profiles of WT and Nlgn3R451C mutant mice. The power of the individual frequency band (1 Hz bins) was normalized by expressing it as % of the average of total power (1-56 Hz for all epochs). Repeated measure two-way ANOVA using “genotype” and “frequency” as factors revealed significant genotype-frequency interaction for NREM sleep (F = 2.856, p <0.001). The NREM (B) spectral profile of Nlgn3R451C (KI) mice differed significantly between 2-8 Hz from WT mice (WT, n = 7; KI, n = 7; all p <0.05). More specifically, Nlgn3R451C mice had reduced powers between 2-8 Hz during NREM sleep (b) in comparison to WT mice. No significant genotype effect or genotype-frequency interactions were found for wake (a) and REM (c) states
Table 1.
Summary statistics for wake, NREM sleep, and REM sleep
| Df | Delta | Theta | Alpha | Sigma | Beta | |
|---|---|---|---|---|---|---|
| Wake | ||||||
| Gen | 1 | F = 2.317 | F = 2.916 | F = 1.224 | F = 7.363 | F = 9.024 |
| p = 0.131 | p = 0.091 | p = 0.271 | p = 0.008** | p = 0.003** | ||
| Time | 7 | F =1.601 | F =0.327 | F =2.717 | F = 1.597 | F = 2.226 |
| p =0.144 | p =0.940 | p =0.013* | p = 0.146 | p = 0.039* | ||
| Inter | 7 | F = 0.470 | F = 0.092 | F = 0.061 | F = 0.136 | F = 0.046 |
| p = 0.854 | p =0.999 | p = 1.000 | p = 0.995 | p = 1.000 | ||
| NREM | ||||||
| Gen | 1 | F = 20.981 | F = 30.622 | F = 6.162 | F = 0.140 | F = 0.457 |
| p <0.001*** | p <0.001*** | p = 0.015* | p = 0.709 | p = 0.501 | ||
| Time | 7 | F = 2.612 | F = 0.623 | F = 0.127 | F = 0.221 | F = 0.430 |
| p = 0.016* | p = 0.735 | p = 0.996 | p = 0.980 | p = 0.881 | ||
| Inter | 7 | F = 0.184 | F = 0.120 | F = 0.021 | F = 0.023 | F = 0.027 |
| p = 0.988 | p = 0.997 | p = 1.000 | p = 1.000 | p = 1.000 | ||
| REM | ||||||
| Gen | 1 | F = 0.885 | F = 1.586 | F = 12.069 | F = 2.343 | F = 4.829 |
| p = 0.349 | p = 0.211 | p < 0.001*** | p = 0.129 | p = 0.030* | ||
| Time | 7 | F = 0.350 | F = 0.083 | F = 0.120 | F = 0.245 | F = 0.115 |
| p = 0.928 | p = 0.999 | p = 0.997 | p = 0.973 | p = 0.997 | ||
| Inter | 7 | F = 0.118 | F = 0.134 | F = 0.348 | F = 0.401 | F = 0.209 |
| p = 0.997 | p = 0.995 | p = 0.930 | p = 0.899 | p = 0.983 | ||
“Genotype” and “time” were used as factors to examine the effects of genotype (Gen) and time (3 h intervals) on spectral power for each behavioural state (WT n = 7, KI n = 7). The table displays the degree of freedom (Df), F values, and significance levels (p) for the factors and the genotype-time interactions (Inter) in the analysis. * p <0.05. ** p <0.01. *** p <0.001
Fig. 6.
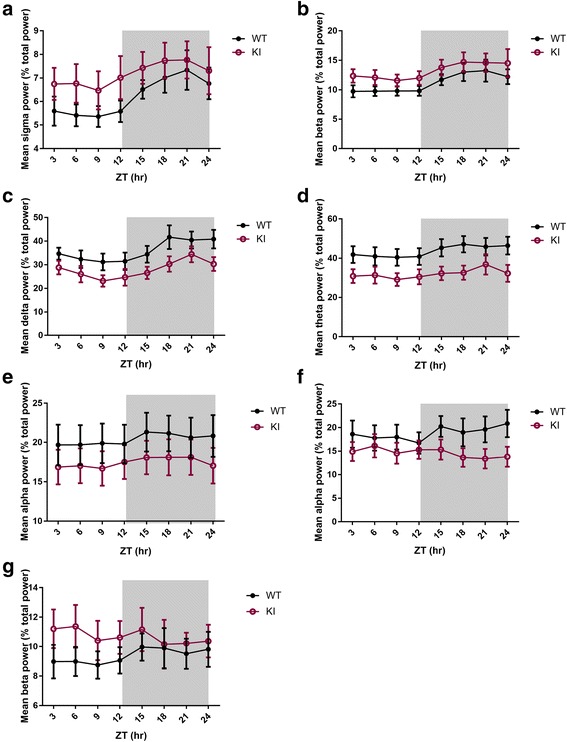
Altered power spectral profiles in Nlgn3R451C mutant mice. The power of the individual frequency band was normalized by expressing it as % of the mean of total power (1-56 Hz for all epochs). The mean normalized power of each 3-h interval (of both days) is displayed over the 24-h period. “Genotype” and “time” were used as factors to examine their effects on delta, theta, alpha, sigma, and beta powers in each behavioural vigilance state (only data with significant genotype effect are displayed here). During wakefulness, Nlgn3R451C (KI) mice exhibited significantly higher sigma (a) and beta (b) powers than WT mice (p <0.05). During NREM sleep, Nlgn3R451C mice showed suppressed delta (c), theta (d), and alpha (e) powers than WT mice (p <0.05). During REM sleep, Nlgn3R451C mice exhibited significantly lower alpha (f) and higher beta (g) powers than WT (p <0.05). No significant interactions between genotype and time were found (all p >0.05)
Discussion
In this study, we recorded EEG-EMG activity in the Nlgn3R451C KI mice and their WT littermates to investigate whether the Nlgn3 R451C mutation is involved in sleep regulation. We found that both WT and Nlgn3R451C mutant mice spent more time sleeping during the light phase compared to the dark, consistent with the typical time distribution of vigilance states in nocturnal rodents [40]. Nlgn3R451C mutant mice were also not different from their WT controls in the total amount of time as well as the total number and durations of episodes for each sleep/wake state, suggesting that Nlgn3R451C mutation alone may not be sufficient to cause reduced sleep time or frequent waking observed in patients with ASD [3, 4, 11]. However, Nlgn3R451C mutant mice exhibited significantly altered EEG power spectra profiles, suggesting that this mutation may contribute to alterations in the quality of sleep/wake states.
During wakefulness, the Nlgn3R451C mice exhibited increased sigma and beta spectral powers compared to WT. The significance of these changes is unknown. Delta power during NREM sleep was significantly reduced in Nlgn3R451C mutant mice, which may reflect heightened arousal and ‘lighter’ sleep in the mutants [35, 45]. The mechanism for the reduced delta power is unknown, but it may be related to altered GABA inhibitory transmission. Delta oscillations are generated by the thalamo-corticol network and are regulated by GABAergic transmission [38, 46–49], and interestingly, a reduction in E/I ratio in the somatosensory cortex was reported in the Nlgn3R451C mutant mice due to increased inhibitory synaptic transmission without changes in excitatory transmission in this region [24, 30]. Consistent with this finding, suppression of delta oscillations was also observed following the administration of diazepam, a GABA agonist [38, 50]. Theta oscillations, which are predominantly generated by the hippocampal network [40, 51, 52], were also reduced in Nlgn3R451C mutant mice during NREM sleep. Previously, Nlgn3R451C mutants mice were demonstrated to have increased excitatory glutamatergic synaptic transmission without alterations in inhibitory GABAergic transmission in the hippocampus, resulting in an augmented E/I ratio in this region [30]. Surprisingly, a reduction in E/I ratio via diazepam administration also reduced theta oscillations during NREM sleep [38]. As diazepam increases GABAergic transmission, which is not affected in the Nlgn3R451C mutant hippocampus [30], these data suggest that Nlgn3 may regulate NREM sleep theta oscillations via mechanisms independent of GABAergic signalling. Alpha oscillations, which are believed to be generated by cortico-cortical and thalamo-cortical networks [40], were reduced during both NREM and REM sleep in Nlgn3R451C mutant mice. Alpha oscillations are thought to reflect input from dorsal anterior cingulate cortex, anterior insula, and thalamus that relay sensory information to the cortex, signalling the brain of external stimuli [43, 53]. Increased alpha power during REM sleep therefore may reflect micro-arousal during REM sleep and possibly contributing to REM sleep instability [39, 43, 54]. Hence, the reduced alpha power in Nlgn3R451C KI mice might indicate more stable REM sleep in these mutants. Although the significance of alpha power reduction for sleep/wake regulation remains unclear, it is consistent with the increased cortical GABAergic transmission as diazepam also suppresses alpha oscillations [50]. In human, beta oscillations (15-30 Hz) are reflective of cortical arousal within sleep [37, 45]. Although, the changes in beta power in patients with insomnia are variable during REM sleep [45], several reports have demonstrated increased beta power in REM sleep in patients with primary insomnia [37, 41, 55]. It is plausible the increased beta oscillations during REM sleep may also suggest poor sleep quality in Nlgn3R451C mice. The simultaneously reduced alpha power and increased beta power may indicate the simultaneous activation of both wake-promoting and sleep-promoting mechanisms in Nlgn3R451C mice.
It is interesting to note that the alterations in spectral powers in Nlgn3R451C mice are quite different from those found in Nlgn1KO mice [31]. For example, Nlgn1KO mice showed decreased high delta, theta, alpha powers during wakefulness while showing a trend of increased delta power during NREM, whereas Nlgn3R451C mutant mice exhibited increased sigma and beta bands during wakefulness and reduced delta, theta, and alpha powers during NREM sleep. These results suggest that the role of neuroligins in sleep/wake regulation is dependent on specific members of the neuroligin gene family. Consistent with this possibility, members of the neuroligin family show different patterns of expression and modes of regulation [21, 22]. However, it is possible that in Nlgn3R451C mutant mice, there may be developmental compensations that alter the expression of other proteins, including Nlgn 1, which could complicate the interpretation of the results. It is also possible that members of the neuroligin family cross-talk so that the deletion of one member affects the functionality of the others. Further studies are needed to elucidate whether and how these interactions and specificity are achieved and their impact on sleep/wake states by using acute manipulations and a combination of knockout mice lacking one or more neuroligins. Lastly, we found a trend of reduced NREM sleep in Nlgn3R451C mice (Fig. 3). Future studies using higher number of mice may help better elucidate the effect of Nlgn3R451C on NREM sleep.
Acknowledgements
We thank all members of Jia lab for their technical assistance and comments on the manuscript. In particular, we would like to thank Ruhan Wei for genotyping and EEG visual analysis, Mengyuan Zhu, Su Jin Lee for genotyping, Neil Yang for his help with surgery, and Shouping Zhang for his help with mice colony maintenance. We would like to thank Dr. Wojtek Kostelecki for his expertise with matlab, and Lia Mesbah-Oskui and Alex Shephard for training the author in stereotaxic surgery.
Funding
This work was supported by grants from the Canadian Institutes of Health Research (CIHR, MOP119421, ZPJ) and Canadian Natural Science and Engineering Research Council (NSERC, RGPIN341498, ZPJ).
Availability of data and materials
Not applicable.
Author’s contributions
JL designed and performed the experiments and wrote the paper. KG and RH helped with the design of the experiments, performed data analysis, and edited the paper. MC guided the conceptualization/technical procedures of the experiments and edited the paper. YS performed visual analysis of sleep-wake states and contributed to data analysis. ZJ designed the experiment, interpreted the data, and wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All protocols and experimental procedures used for this study were approved by the Animal Lab Services (LAS) ethics committee of the Hospital for Sick Children, Toronto, Canada.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ASD
Autism spectrum disorders
- E/I
Excitation to inhibition
- EEG
Electroencephalography
- EMG
Electromyography
- GABA
γ-aminobutyric acid
- i.p.
Intraperitoneal
- KI
Knock-in
- Nlgn
Neuroligin
- NREM
Non-rapid-eye-movement
- REM
Rapid-eye-movement
- s.c.
subcutaneous
- WT
Wild-type
Contributor Information
Jackie J. Liu, Email: jiu.liu@mail.utoronto.ca
Kevin P. Grace, Email: kevin.grace@mail.utoronto.ca
Richard L. Horner, Email: richard.horner@utoronto.ca
Miguel A. Cortez, Email: miguel.cortez@sickkids.ca
Yiwen Shao, Email: yiwen.shao@mail.utoronto.ca.
Zhengping Jia, Email: zhengping.jia@sickkids.ca.
References
- 1.Bailey A, Phillips W, Rutter M. Autism: towards an integration of clinical, genetic, neuropsychological, and neurobiological perspectives. J Child Psychol Psychiatry. 1996;37(1):89–126. doi: 10.1111/j.1469-7610.1996.tb01381.x. [DOI] [PubMed] [Google Scholar]
- 2.Polšek D, et al. Recent developments in neuropathology of autism spectrum disorders. Transl Neurosci. 2011;2(3):256–264. doi: 10.2478/s13380-011-0024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen S, et al. The relationship between sleep and behavior in autism spectrum disorder (ASD): a review. J Neurodev Disord. 2014;6(1):44. doi: 10.1186/1866-1955-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cortesi F, et al. Sleep in children with autistic spectrum disorder. Sleep Med. 2010;11(7):659–664. doi: 10.1016/j.sleep.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 5.Couturier JL, et al. Parental perception of sleep problems in children of normal intelligence with pervasive developmental disorders: prevalence, severity, and pattern. J Am Acad Child Adolesc Psychiatry. 2005;44(8):815–822. doi: 10.1097/01.chi.0000166377.22651.87. [DOI] [PubMed] [Google Scholar]
- 6.Goldman SE, et al. Parental sleep concerns in autism spectrum disorders: variations from childhood to adolescence. J Autism Dev Disord. 2012;42(4):531–538. doi: 10.1007/s10803-011-1270-5. [DOI] [PubMed] [Google Scholar]
- 7.Krakowiak P, et al. Sleep problems in children with autism spectrum disorders, developmental delays, and typical development: a population-based study. J Sleep Res. 2008;17(2):197–206. doi: 10.1111/j.1365-2869.2008.00650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Owens JA, Spirito A, McGuinn M. The Children’s sleep habits questionnaire (CSHQ): psychometric properties of a survey instrument for school-aged children. Sleep. 2000;23(8):1043–1051. [PubMed] [Google Scholar]
- 9.Richdale AL. Sleep problems in autism: prevalence, cause, and intervention. Dev Med Child Neurol. 1999;41(1):60–66. doi: 10.1017/S0012162299000122. [DOI] [PubMed] [Google Scholar]
- 10.Souders MC, et al. Sleep behaviors and sleep quality in children with autism spectrum disorders. Sleep. 2009;32(12):1566–1578. doi: 10.1093/sleep/32.12.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giannotti F, et al. An investigation of sleep characteristics, EEG abnormalities and epilepsy in developmentally regressed and non-regressed children with autism. J Autism Dev Disord. 2008;38(10):1888–1897. doi: 10.1007/s10803-008-0584-4. [DOI] [PubMed] [Google Scholar]
- 12.Richdale AL, Schreck KA. Sleep problems in autism spectrum disorders: prevalence, nature, & possible biopsychosocial aetiologies. Sleep Med Rev. 2009;13(6):403–411. doi: 10.1016/j.smrv.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Schreck KA, Mulick JA, Smith AF. Sleep problems as possible predictors of intensified symptoms of autism. Res Dev Disabil. 2004;25(1):57–66. doi: 10.1016/j.ridd.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Johnson KP, Malow BA. Sleep in children with autism spectrum disorders. Curr Neurol Neurosci Rep. 2008;8(2):155–161. doi: 10.1007/s11910-008-0025-y. [DOI] [PubMed] [Google Scholar]
- 15.Gail Williams P, Sears LL, Allard A. Sleep problems in children with autism. J Sleep Res. 2004;13(3):265–268. doi: 10.1111/j.1365-2869.2004.00405.x. [DOI] [PubMed] [Google Scholar]
- 16.Malow BA, et al. Characterizing sleep in children with autism spectrum disorders: a multidimensional approach. Sleep. 2006;29(12):1563–1571. doi: 10.1093/sleep/29.12.1563. [DOI] [PubMed] [Google Scholar]
- 17.Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol. 2009;19(2):231–234. doi: 10.1016/j.conb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Maćkowiak M, Mordalska P, Wędzony K. Neuroligins, synapse balance and neuropsychiatric disorders. Pharmacol Rep. 2014;66(5):830–835. doi: 10.1016/j.pharep.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Pizzarelli R, Cherubini E. Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast. 2011;2011:297153. doi: 10.1155/2011/297153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dean C, Dresbach T. Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006;29(1):21–9. doi: 10.1016/j.tins.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Lisé MF, El-Husseini A. The neuroligin and neurexin families: from structure to function at the synapse. Cell Mol Life Sci. 2006;63(16):1833–1849. doi: 10.1007/s00018-006-6061-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455(7215):903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varoqueaux F, et al. Neuroligins determine synapse maturation and function. Neuron. 2006;51(6):741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Tabuchi K, et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318(5847):71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baudouin SJ, et al. Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science. 2012;338(6103):128–132. doi: 10.1126/science.1224159. [DOI] [PubMed] [Google Scholar]
- 26.Comoletti D, et al. The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J Neurosci. 2004;24(20):4889–4893. doi: 10.1523/JNEUROSCI.0468-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan J, et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol Psychiatry. 2005;10(4):329–332. doi: 10.1038/sj.mp.4001629. [DOI] [PubMed] [Google Scholar]
- 28.Radyushkin K, et al. Neuroligin-3-deficient mice: model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 2009;8(4):416–425. doi: 10.1111/j.1601-183X.2009.00487.x. [DOI] [PubMed] [Google Scholar]
- 29.Jaramillo TC, et al. Autism-related neuroligin-3 mutation alters social behavior and spatial learning. Autism Res. 2014;7(2):264–272. doi: 10.1002/aur.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Etherton M, et al. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc Natl Acad Sci U S A. 2011;108(33):13764–13769. doi: 10.1073/pnas.1111093108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El Helou J, et al. Neuroligin-1 links neuronal activity to sleep-wake regulation. Proc Natl Acad Sci U S A. 2013;110(24):9974–9979. doi: 10.1073/pnas.1221381110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, et al. Drosophila neuroligin 4 regulates sleep through modulating GABA transmission. J Neurosci. 2013;33(39):15545–15554. doi: 10.1523/JNEUROSCI.0819-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaushal N, et al. Socially isolated mice exhibit a blunted homeostatic sleep response to acute sleep deprivation compared to socially paired mice. Brain Res. 2012;1454:65–79. doi: 10.1016/j.brainres.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kotagal S, Broomall E. Sleep in children with autism spectrum disorder. Pediatr Neurol. 2012;47(4):242–251. doi: 10.1016/j.pediatrneurol.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 35.Franken P, Malafosse A, Tafti M. Genetic variation in EEG activity during sleep in inbred mice. Am J Physiol. 1998;275(4 Pt 2):R1127–R1137. doi: 10.1152/ajpregu.1998.275.4.R1127. [DOI] [PubMed] [Google Scholar]
- 36.Kitaoka K, et al. Vitamin A deficiency induces a decrease in EEG delta power during sleep in mice. Brain Res. 2007;1150:121–130. doi: 10.1016/j.brainres.2007.02.077. [DOI] [PubMed] [Google Scholar]
- 37.Spiegelhalder K, et al. Increased EEG sigma and beta power during NREM sleep in primary insomnia. Biol Psychol. 2012;91(3):329–333. doi: 10.1016/j.biopsycho.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Kopp C, et al. Modulation of rhythmic brain activity by diazepam: GABA(A) receptor subtype and state specificity. Proc Natl Acad Sci U S A. 2004;101(10):3674–3679. doi: 10.1073/pnas.0306975101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parrino L, et al. Cyclic alternating pattern (CAP): the marker of sleep instability. Sleep Med Rev. 2012;16(1):27–45. doi: 10.1016/j.smrv.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 40.Fenzl T, et al. Sleep disturbances in highly stress reactive mice: modeling endophenotypes of major depression. BMC Neurosci. 2011;12:29. doi: 10.1186/1471-2202-12-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merica H, Blois R, Gaillard JM. Spectral characteristics of sleep EEG in chronic insomnia. Eur J Neurosci. 1998;10(5):1826–1834. doi: 10.1046/j.1460-9568.1998.00189.x. [DOI] [PubMed] [Google Scholar]
- 42.Colas D, Cespuglio R, Sarda N. Sleep wake profile and EEG spectral power in young or old senescence accelerated mice. Neurobiol Aging. 2005;26(2):265–273. doi: 10.1016/j.neurobiolaging.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 43.Simor P, et al. Fluctuations between sleep and wakefulness: wake-like features indicated by increased EEG alpha power during different sleep stages in nightmare disorder. Biol Psychol. 2013;94(3):592–600. doi: 10.1016/j.biopsycho.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 44.Campbell, IG, EEG recording and analysis for sleep research. Curr Protoc Neurosci. 2009. Chapter 10: p. Unit10.2. doi:10.1002/0471142301.ns1002s49. [DOI] [PMC free article] [PubMed]
- 45.Krystal AD, et al. NREM sleep EEG frequency spectral correlates of sleep complaints in primary insomnia subtypes. Sleep. 2002;25(6):630–640. [PubMed] [Google Scholar]
- 46.McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- 47.Steriade M. The corticothalamic system in sleep. Front Biosci. 2003;8:d878–d899. doi: 10.2741/1043. [DOI] [PubMed] [Google Scholar]
- 48.Sejnowski TJ, Destexhe A. Why do we sleep? Brain Res. 2000;886(1-2):208–223. doi: 10.1016/S0006-8993(00)03007-9. [DOI] [PubMed] [Google Scholar]
- 49.Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726–731. doi: 10.1016/S0166-2236(00)02002-6. [DOI] [PubMed] [Google Scholar]
- 50.Tobler I, et al. Diazepam-induced changes in sleep: role of the alpha 1 GABA(A) receptor subtype. Proc Natl Acad Sci U S A. 2001;98(11):6464–6469. doi: 10.1073/pnas.111055398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Louie K, Wilson MA. Temporally structured replay of awake hippocampal ensemble activity during rapid eye movement sleep. Neuron. 2001;29(1):145–156. doi: 10.1016/S0896-6273(01)00186-6. [DOI] [PubMed] [Google Scholar]
- 52.Buzsáki G, et al. Hippocampal network patterns of activity in the mouse. Neuroscience. 2003;116(1):201–211. doi: 10.1016/S0306-4522(02)00669-3. [DOI] [PubMed] [Google Scholar]
- 53.Sadaghiani S, et al. Intrinsic connectivity networks, alpha oscillations, and tonic alertness: a simultaneous electroencephalography/functional magnetic resonance imaging study. J Neurosci. 2010;30(30):10243–10250. doi: 10.1523/JNEUROSCI.1004-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cantero JL, Atienza M, Salas RM. Spectral features of EEG alpha activity in human REM sleep: two variants with different functional roles? Sleep. 2000;23(6):746–750. [PubMed] [Google Scholar]
- 55.Perlis ML, et al. Temporal and stagewise distribution of high frequency EEG activity in patients with primary and secondary insomnia and in good sleeper controls. J Sleep Res. 2001;10(2):93–104. doi: 10.1046/j.1365-2869.2001.00247.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.