

Abstract
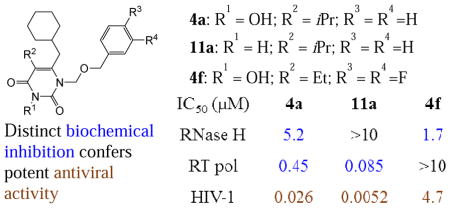
3-Hydroxypyrimidine-2,4-dione (HPD) represents a versatile chemical core in the design of inhibitors of human immunodeficiency virus (HIV) reverse transcriptase (RT)-associated RNase H and integrase strand transfer (INST). We report herein the design, synthesis and biological evaluation of an HPD subtype (4) featuring a cyclohexylmethyl group at the C-6 position. Antiviral testing showed that most analogues of 4 inhibited HIV-1 in the low nanomolar to submicromolar range, without cytotoxicity at concentrations up to 100 μM. Biochemically, these analogues dually inhibited both the polymerase (pol) and the RNase H functions of RT, but not INST. Co-crystal structure of 4a with RT revealed a nonnucleoside RT inhibitor (NNRTI) binding mode. Interestingly, chemotype 11, the synthetic precursor of 4 lacking the 3-OH group, did not inhibit RNase H while potently inhibiting pol. By virtue of the potent antiviral activity and biochemical RNase H inhibition, HPD subtype 4 could provide a viable platform for eventually achieving potent and selective RNase H inhibition through further medicinal chemistry.
Keywords: 3-Hydroxypyrimidine-2,4-dione (HPD); of human immunodeficiency virus (HIV); Inhibiting RNase H; Polymerase
Graphical Abstract
1. Introduction
HIV-1 infection is clinically managed [1] by highly active antiretroviral therapy (HAART) [1] which comprises four major classes of drugs: the nucleoside RT inhibitors (NRTIs) [2], NNRTIs [3], protease inhibitors (PIs) [4], and INST inhibitors (INSTIs) [5]. Long term HAART can be plagued by toxicity and the emergence of drug resistance. Structurally novel HIV inhibitors, particularly those with a distinct mechanism of action, could add significantly to the existing HAART repertoire. We have long been interested in exploring chemotypes amenable to structural elaboration toward inhibiting different targets of HIV-1 [6–12]. One such chemotype is the versatile HPD core (Figure 1) which was initially designed to dually inhibit RT pol and INST (subtype 1) [12]. Removal of the N-1 and C5 substituents and redesign of the C6 moiety led to subtype 2 which selectively inhibited the RNase H function of RT [8]. Introduction of a C-5 carboxamide moiety and replacement of the C-6 aryl with a small alkyl group resulted in subtype 3, which potently inhibited both RNase H and INST [9]. Among other viable modifications for the HPD core, the replacement of the phenyl ring at the C-6 position of subtype 1 with a cyclohexyl group generated a new subtype 4 (Figure 1b) which conferred highly potent antiviral activity with dual biochemical inhibition against HIV-1 RT pol and RNase H functions. We report herein the synthesis, antiviral, biochemical and biophysical characterizations of subtype 4. The key medicinal chemistry observation was that most analogues potently inhibited HIV in cell culture and that the biochemical inhibitory profile of RT hinges largely on the 3-OH group. The synthetic precursor of subtype 4, chemotype 11 which lacks the 3-OH group, did not inhibit the RNase H function of RT, though the higher potency against RT pol conferred better antiviral activity against HIV-1 when compared to 4.
Figure 1.

Variants of the HPD chemotype. (a) HPD subtypes 1—3 have been reported to inhibit RT and/or INST of HIV-1; (b) replacement of the C-6 phenyl ring of subtype 1 led to subtype 4 with substantially different biological activity profiles.
2. Results and Discussion
2.1 Chemistry
The synthesis of HPD subtype 4 is described in Scheme 1. The overall approach was adapted from the route reported for the synthesis of subtype 1 [12]. The synthesis entails a β-ketoester intermediate 6 which was previously prepared via a Blaise reaction between cyclohexylacetonitrile (7) and a bromoester (8) [13]. However, it turned out that the cost of starting material 7 is too high to allow the preparation of 6 at the scale necessary for analogue synthesis. In our synthesis, an alternative method starting from the much cheaper cyclohexylacetic acid (5) was used [14]. Acid 5 was first activated by treating with carbonyldiimidazole (CDI) and then reacted with the mono potassium salt of a substituted malonate. This procedure proved to be highly efficient in delivering β-ketoester intermediate 6 in large scale. Cyclization of 6 with thiourea yielded the 2-thioxopyrimidine-4-one intermediate 9, which upon S-alkylation and hydrolysis was converted to the key pyrimidine-2,4-dione intermediate 10. Subsequent selective N-1 alkylation of 10 via a bis-silylated pyrimidine intermediate with an in situ generated chloromethyl ether produced 11. Finally, the key N-3 OH group was introduced via a base-mediated oxidation with meta-chloroperoxybenzoic acid (mCPBA) to complete the synthesis of subtype 4.
Scheme 1a. Synthesis of HPD subtype 4.

aReagents and conditions: a) CDI, CH3CN, rt, 30 min, R1CH(CO2Et)(CO2K), Et3N–MgCl2, rt, overnight; b) Zn, THF; c) thiourea, EtONa, EtOH, refluxing for 8h; d) ClCH2COOH, AcOH, H2O, refluxing, overnight; e) CH3C(OTMS)=NTMS (BSA), substituted benzyl alcohol, (HCHO)n, TMSCl, TBAI ( cat.), CH2Cl2, rt, 70–90%; f) NaH, mCPBA, THF, rt.
2.2. Biology
All analogues of subtype 4 were tested in a MAGI antiviral assay[[15, 16]] in dose response fashion, as well as three biochemical assays against RT pol, RT RNase H and INST. The results are described in Table 1.
Table 1.
Biochemical and antiviral assay results of subtype 4
| Compd | Structure | RT IC50 a(μM) | INST IC50 a (μM) | MAGI Antiviral | ||
|---|---|---|---|---|---|---|
|
|
|
|||||
| RNase H | Pol | EC50b (μM) | CC50c (μM) | |||
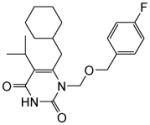
| 4ad |
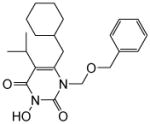
|
5.2 ± 0.8 | 0.45± 0.07 | >100 | 0.026 ± 0.002 | >100 |
| 4b |
|
2.7 ± 2 | 9.9 ± 0.04 | >100 | 2.4 ± 0.2 | >100 |
| 4c |
|
5.5 ± 0.2 | 2.3 ± 2 | >100 | 0.059 ± 0.004 | >100 |
| 4d |
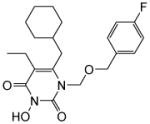
|
5.8 ± 0.2 | 1.5 ± 0.5 | >100 | 0.039 ± 0.008 | >100 |
| 4e |
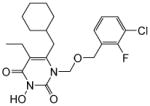
|
1.1 ± 0.3 | 6.5 ± 0.2 | >100 | 0.42 ± 0.1 | >100 |
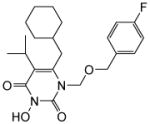
| 4f |
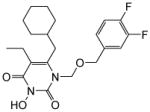
|
1.7 ± 0.5 | >10 | >100 | 4.1 ± 0.5 | >100 |
| 4g |
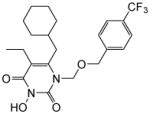
|
10 | 7.0 ± 2 | >100 | 2.4 ± 0.3 | >100 |
| 4h |
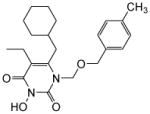
|
2.1 ± 1 | 6.5 ± 1 | >100 | 0.43 ± 0.06 | >100 |
| 4i |
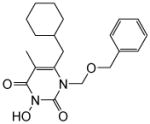
|
3.1 ± 1 | >10 | >100 | >15 | >100 |
| 4j |
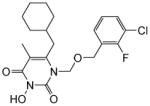
|
2.2 ± 0.9 | >10 | >100 | >15 | >100 |
| 4k |
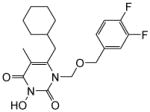
|
4.6 ± 0.4 | 10 | >100 | >15 | >100 |
| 4l |
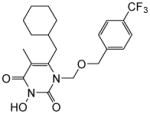
|
7.6 ± 0.6 | 10 | >100 | >15 | >100 |
| 4m |
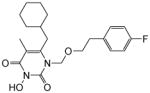
|
3.9 ± 0.2 | >10 | >100 | 10 ± 1.1 | >100 |
| 14 | -- | >10 | 0.42 ± 0.2 | -- | 0.011 ± 0.0001 | 58 ± 2 |
Concentration of compound inhibiting the target enzyme by 50%.
Concentration of compound inhibiting virus replication by 50%.
Concentration of compound resulting in 50% cell death. All assay results expressed as mean ± standard deviation from at least two independent experiments.
No pol inhibition was observed against RT mutants Y181C or K103N at concentrations up to 10 μM.
HPD 4 potently inhibited HIV-1
In the antiviral assays, all analogues with an isopropyl or ethyl group at the C5 position (4a–4h) showed strong inhibition against HIV-1 in the low nanomolar to low micromolar range, whereas those with a methyl substituent (4i–4m) did not inhibit HIV-1 at concentrations up to 15 μM (Table 1). Among the active analogues, 4a (EC50 = 26 nM), 4c (EC50 = 59 nM) and 4d (EC50 = 39 nM) exhibited antiviral potencies comparable to that of 14 (EC50 = 11 nM), a potent NNRTI. In addition, while 14 showed modest cytotoxicity (CC50 = 58 μM), toxicity was not observed with any of the subtype 4 analogues at concentrations up to 100 μM, suggesting that our new compounds possess a favorable therapeutic profile in cell culture. Importantly, the antiviral potency observed from the MAGI assay was confirmed in a distinct cytoprotection assay[16, 17] in which cytopathic effect of HIV-1 was measured. In this assay, 4c (EC50 = 0.56 μM), 4d (EC50 = 0.32 μM) and 4e (EC50 = 1.8 μM) all showed potent antiviral activity (data not listed in Table 1).
Subtype 4 dually inhibited RT pol and RNase H
Biochemical evaluations of our compounds resulted in a few major observations. First of all, none of the analogues inhibited INST at concentrations up to 100 μM (Table 1), whereas subtype 1 was previously reported to confer low micromolar inhibition against INST[12]. This is not surprising as inhibition of INST minimally requires a chelating triad and a hydrophobic aromatic group, typically a fluorobenzyl group[18]. In subtype 4 the benzyl group is replaced with a cyclohexylmethyl group, thus the lack of anti-INST activity. It is noteworthy that achieving selective RNase H inhibition over INST inhibition could be a challenge due to their similarity in active site fold and mechanism of catalysis[19]. By abrogating the inhibitory activity against INST, substituting the cyclohexyl group for the phenyl ring provides a distinct advantage toward achieving selective RNase H inhibition. Second, all analogues with an isopropyl or ethyl group at the C5 position (4a–4h) showed dual inhibition against RT pol and RNase H, except for compound 4f which did not inhibit RT pol. By contrast, analogues with a methyl substituent (4i–4m) only inhibited RT RNase H (Table 1). Importantly, with most compounds the RT pol inhibition appeared to correlate very well with the observed antiviral activity. As mentioned earlier, the methyl substitution at the C5 position conferred no antiviral activity at 15 μM and weak or no pol inhibition as observed with compounds 4i–4l, whereas the three most active analogues (4a, 4c and 4d) from the antiviral assay also exhibited the highest potency against RT pol. In addition, compound 4a inhibited RT pol with an IC50 (0.45 μM) similar to 14 (IC50 = 0.42 μM) and conferred a comparable antiviral activity (EC50 = 26 nM) to 14 (EC50 = 11 nM). Furthermore, significant inhibition was not observed with compound 4a at concentrations up to 10 μM against the Y181C or K103N mutants of RT (data not shown). All these findings strongly indicate that the observed antiviral activity is most likely via RT pol inhibition. The exceptions are compounds 4f and 4m with which the antiviral activity correlated with only the RNase H inhibition as no inhibition against RT pol was observed.
Subtype 4 binds to NNRTI binding pocket
To confirm the antiviral mechanism of action through RT pol inhibition, we solved a crystal structure of analogue 4a bound to RT at 2.7 Å. The structure revealed that analogue 4a binds at the NNRTI binding pocket of RT, reminiscent to that of the 1-[(2-Hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) type of NNRTIs [20, 21]. HEPT (12)[22, 23] was among the earliest NNRTIs and a few variants (Figure 3), such as MKC-442 (13)[20, 24] and TNK-651(14)[20] and TNK-6123 (15)[21], have been reported to inhibit HIV-1 with exceptional potencies.
Figure 3.
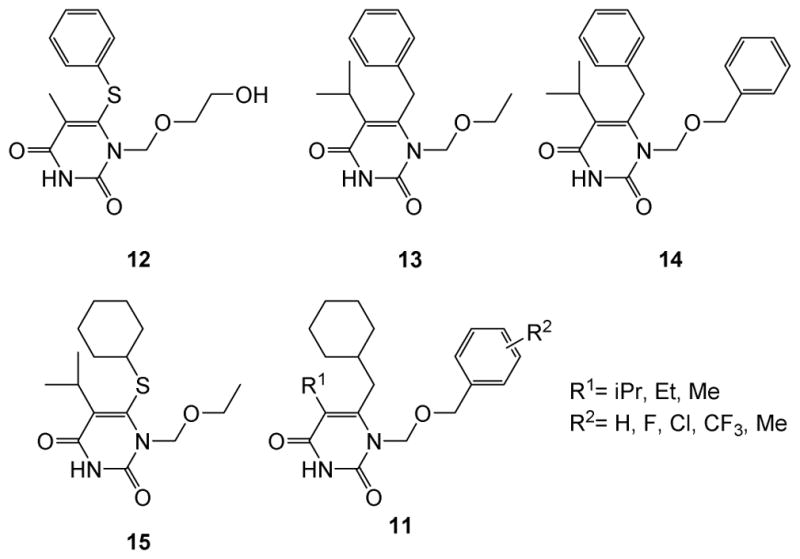
Structures of reported HEPT-based NNRTIs 12–15. Precursor 11 closely mimics 14 and could be a scaffold for novel NNRTIs.
The precursor 11 potently inhibited HIV-1 and RT pol
The immediate synthetic precursor of subtype 4, chemotype 11, closely mimics the HEPT variants, particularly 14. Therefore, we were prompted to test all analogues of 11. We first tested chemotype 11 in the MAGI antiviral assay. Remarkably, with the single exception of 11m, all analogues inhibited HIV-1 in low nanomolar to submicromolar range (Table 2). Particularly potent are 11a–d, 11f and 11h, which exhibited antiviral potencies (EC50 = 5.2–14 nM) comparable to or better than that observed with 14 (EC50 = 11 nM). Since 14 was among the most potent NNRTIs of the HEPT family, the exceptional antiviral potency may render 11 a suitable chemotype for antiviral development.
Table 2.
Biochemical and antiviral assay results of 3-deoxy precursor 11
| Compd | Structure | RT IC50 a (μM) | MAGI Antiviral | ||
|---|---|---|---|---|---|
|
| |||||
| RNH | Pol | EC50b (μM) | CC50c (μM) | ||
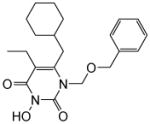
| 11a |
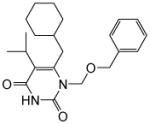
|
>10 | 0.085 ± 0.034 | 0.0052 ± 0.0024 | 23 ± 0.74 |
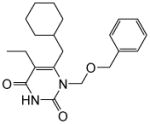
| 11b |
|
>10 | 0.30 ± 0.034 | 0.0072 ± 0.0029 | 31 ± 1.4 |
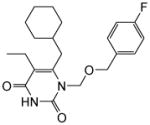
| 11c |
|
>10 | 0.22 ± 0.12 | 0.0062 ± 0.00020 | >50 |
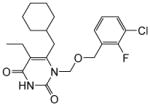
| 11d |
|
>10 | 0.48 ± 0.089 | 0.0075 ± 0.0010 | 42 ± 2.7 |
| 11e |
|
>10 | 1.5 ± 0.50 | 0.048 ± 0.029 | 32 ± 0.67 |
| 11f |
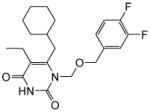
|
>10 | 0.82 ± 0.014 | 0.014 ± 0.0058 | 29 ± 0.78 |
| 11g |
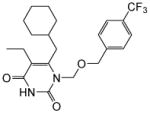
|
>10 | 0.81 ± 0.59 | 0.039 ± 0.012 | 28 ± 0.81 |
| 11h |
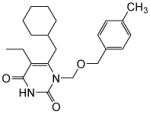
|
>10 | 0.36 ± 0.17 | 0.011 ± 0.0035 | 30 ± 1.30 |
| 11i |
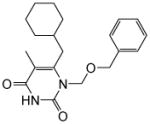
|
>10 | 6.8 ± 2.0 | 0.065 ± 0.017 | >100 |
| 11j |
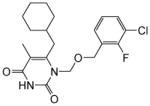
|
>10 | 10 | 0.83 ± 0.020 | 38 ± 3.2 |
| 11k |
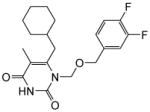
|
>10 | 10 | 0.21 ± 0.062 | 65 ± 3.5 |
| 11l |
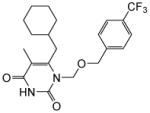
|
>10 | 10 | 0.80 ± 0.11 | >100 |
| 11m |
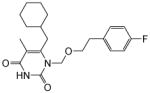
|
>10 | >10 | 5.3 ± 0.43 | 56 ± 0.83 |
| 14 | -- | >10 | 0.42 ± 0.24 | 0.011 ± 0.00010 | 58 ± 2.4 |
Concentration of compound inhibiting the target enzyme by 50%.
Concentration of compound inhibiting virus replication by 50%.
Concentration of compound resulting in 50% cell death. All assay results expressed as mean ± standard deviation from at least two independent experiments.
Biochemically, analogues of 11 did not inhibit RNase H at concentrations up to 10 μM, presumably due to the lack of chelating triad required for RNase H active site inhibitors [25]. This is in stark contrast to subtype 4 with which the 3-OH group forms a chelating triad with the two flanking carbonyls, resulting in the observed consistent inhibition of RNase H. However, the precursor scaffold 11 conferred substantially stronger inhibition against RT pol than chemotype 4. As shown in Table 2, all analogues with an isopropyl or ethyl group at the C5 position (11a–11h) showed nanomolar inhibition against RT pol, with the exception of compound 11e which inhibited RT pol with an IC50 of 1.5 μM. By contrast, analogues with a methyl substituent (11i–11m) did not inhibit RT pol. This structure-activity-relationship (SAR) trend is similar to that observed with chemotype 4 and consistent with the binding mode of HEPT NNRTIs which typically require a bulkier isopropyl or ethyl group at C5 position 1.[20] Furthermore, the crystal structure of HIV RT in complex with 4a provides a structural basis for the observed higher potency of 11a (5.2 nM) vs. 4a (26 nM). In the structure, the 3-hydroxy group of 4a is in close proximity to the protein backbone in the NNRTI binding pocket (main chain of K101, Figure 2), providing some steric hindrance for inhibitor binding. Compounds without the 3-hydroxy group, such as 11a, should be able to bind at the NNRTI binding pocket without this interference, similar to the previously reported crystal structures of HIV RT in complex with HEPT compounds 13[20] and 15[21]. It is also noteworthy that the correlation between pol inhibition and antiviral potency of 11 appeared to be as strong as it is with 14, suggesting that the antiviral potency of 11 is most likely due to the RT pol inhibition.
Figure 2.

Crystal structure of HIV-1 RT in complex with 4a (PDB ID: 5TUQ). Analogue 4a (cyan sticks) binds at the NNRTI binding pocket of RT (Fingers, blue; Palm, red; Thumb, green; Connection, yellow; RNase H, orange; p51, gray). Inset: cross-eyed stereo view of 4a at the NNRTI binding pocket with a 2.7 Å 2Fo-Fc electron density map at σ=1.0 in light blue.
3. Conclusion
By replacing the phenyl ring with a cyclohexyl group at the C-6 position of HPD subtype 1, we have generated an HPD subtype 4 which potently inhibited HIV-1. Biochemically, subtype 4 demonstrated dual inhibition against both the RNase H and the pol functions of RT without inhibiting INST. The antiviral SAR was found to better correlate with that of RT pol inhibition. Crystal structure of 4a bound RT and the lack of biochemical inhibition of 4a against Y181C and K103N RT mutants further corroborate an antiviral mechanism of action of 4 via RT pol inhibition. Interestingly, the synthetic precursor 11 inhibited HIV-1 and RT pol substantially better than subtype 4 without inhibiting INST or RNase H. The crystal structure of 4a bound to RT provided a structural basis for the observed increase in potency of chemotype 11 over 4. The best compounds of chemotypes 4 and 11 exhibited antiviral potencies comparable to that of 14, a well-known and highly potent NNRTI, and hence provide valuable additions to NNRTIs. Most importantly, the consistent biochemical inhibition against RNase H, the potent antiviral activity and the close correlation between antiviral potency and RNase H inhibition with analogues 4f and 4m render chemotype 4 a valuable platform for achieving potent antiviral phenotype through selective RNase H inhibition.
4. Experimental
4.1. General-Chemistry
All commercial chemicals were used as supplied unless otherwise indicated. Dry solvents were either purchased (CH3CN and EtOH) or dispensed under argon from an anhydrous solvent system with two packed columns of neutral alumina or molecular sieves (CH2Cl2, THF), Flash chromatography was performed on a Teledyne Combiflash RF-200 with RediSep columns (silica) and indicated mobile phase. All moisture sensitive reactions were performed under an inert atmosphere of ultra-pure argon with oven-dried glassware. 1H and 13C NMR spectra were recorded on a Varian 600 MHz spectrometer. Mass data were acquired on an Agilent TOF II TOS/MS spectrometer capable of ESI and APCI ion sources. Analysis of sample purity was performed on a Varian Prepstar SD-1 HPLC system with a Phenomenex Gemini, 5 micron C18 column (250mm × 4.6 mm). HPLC conditions: solvent A = H2O, solvent B = MeCN; flow rate = 1.0 mL/min; compounds were eluted with a gradient of 20% MeCN/H2O to 90% MeCN for 20 min. Purity was determined by total absorbance at 254 nm. All tested compounds have a purity ≥ 96%. In addition, all tested compounds were examined for potential pan assay interference and none was deemed prone to any of the potential mechanisms of interference: aggregation, redox activity, fluorescence, protein reactivity, singlet-oxygen quenching, member disruption, and decomposition in assay buffer to form reactive compound.
4.1.1. General Procedures for synthesis of 11a–11m
to a suspension of paraformaldehyde (17 mg, 0.57 mmol, 1.3 equiv) in TMSCl (1.0 mL) was added substituted benzyl alcohol (0.57 mmol, 1.3 equiv) at room temperature. The resulting suspension was stirred until a clear solution was achieved, removed solvent to give substituted aryl chloromethyl ether as oil, which can be used for next step directly. To the solution of 10a (110 mg, 0.44 mmol, 1.0 equiv) in anhydrous DCM (5mL), was added BSA ( 235 μM, 0.96 mmol, 2.2 equiv). The resulting reaction mixture was stirred at rt for 1 h. Then the fresh made substituted chloro-methyl-pheny ether and catalytic amount TBAI was added. This reaction was stirred at rt overnight, quenched by adding saturated NaHCO3 solution and extracted with CH2Cl2 (3 × 10 mL). Combined the organic phase, dried over Na2SO4, removed the solvent to give the crude product which purified by combiflash (Hex/EtOAc, 3:1) to give compound as white solid.
4.1.1.1.1-((Benzyloxy)methyl)-6-(cyclohexylmethyl)-5-isopropylpyrimidine-2,4(1H,3H)-dione (11a)
Yield: 90%; 1H NMR (600 MHz, CDCl3) δ 8.77 (s, 1H), 7.30 (m, 5H), 5.36 (s, 2H), 4.57 (s, 2H), 2.72 (m, 1H), 2.52 (d, J = 7.2 Hz, 2H), 1.66 (m, 5H), 1.38 (m, 1H), 1.22 (d, J = 6.0 Hz, 6H), 1.12 (m, 5H); 13C NMR (150 MHz, CDCl3) δ 162.3, 152.2, 150.0, 137.3, 128.4, 127.9, 127.8, 118.9, 72.2, 71.9, 40.8, 37.1, 35.2, 30.8, 28.5, 28.2, 22.4; HRMS (ESI+) calcd. for C22H31N2O3 [M+H]+ 371.2356, found 371.2354.
4.1.1.2. 6-(Cyclohexylmethyl)-1-(((4-fluorobenzyl)oxy)methyl)-5-isopropylpyrimidine-2,4(1H,3H)-dione (11b)
This compound was prepared as white solid following the procedure described for the preparation of 11a, Yield: 81%; 1H NMR (600 MHz, CDCl3) δ 8.84 (s, 1H), 7.22 (m, 2H), 6.96 (m, 2H), 5.40 (s, 2H), 4.53 (s, 2H), 2.72 (m, 1H), 2.52 (d, J = 6.0 Hz, 2H), 2.20 (m, 5H), 1.40 (m, 1H), 1.23 (d, J = 6.0 Hz, 6H), 1.12(m, 3H), 0.98 (m, 2H); HRMS (ESI+) calcd. for C22H30FN2O3 [M+H]+ 389.4723, found 389.4725.
4.1.1.3. 1-((Benzyloxy)methyl)-6-(cyclohexylmethyl)-5-ethylpyrimidine-2,4(1H,3H)-dione (11c)
This compound was prepared as a white solid following the procedure described for the preparation of 11a. Yield: 73%; 1HNMR (600 MHz, CDCl3) δ 7.34 (m, 5H), 5.43 (s, 2H), 4.64 (s, 2H), 2.58 (d, J = 6.0 Hz, 2H), 2.83 (t, J = 7.2 Hz, 2H), 2.43 (q, J = 7.2 Hz, 2H), 1.72 (m, 6H), 1.49 (m, 1H), 1.27 (m, 4H), 1.06 (t, J = 7.8 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 163.4, 152.3, 150.4, 137.3, 128.4, 127.9, 127.8, 116.2, 72.9, 71.8, 38.5, 35.1, 33.2, 26.2, 26.0, 19.4, 13.6; HRMS (ESI+) calcd. for C21H29N2O3 [M+H]+ 357.2229, found 357.2228.
4.1.1.4. 6-(Cyclohexylmethyl)-5-ethyl-1-(((4-fluorobenzyl)oxy)methyl)pyrimidine-2,4(1H,3H)-dione (11d)
This compound was prepared as a white solid following the procedure described for the preparation of 11a. Yield: 63%; 1H NMR (600 MHz, CDCl3) δ 8.76 (s, 1H), 7.15 (t, J = 8.4 Hz, 2H), 6.88 (t, J = 8.4 Hz, 2H), 5.25 (s, 2H), 4.45 (s, 2H), 2.42 (d, J = 7.2 Hz, 2H), 2.27 (q, J = 6.6 Hz, 2H), 1.59 (m, 6 H), 1.38 (m, 1H), 1.03 (m, 4H), 0.91 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 163.1, 152.1, 150.4, 133.0, 129.6, 116.2, 115.4, 115.2, 72.8, 71.0, 38.6, 35.1, 33.2, 26.3, 26.0, 19.4, 13.6; HRMS (ESI+) calcd. for C21H28FN2O3 [M+H]+ 375.2110, found 375.2109.
4.1.1.5. 6-(Cyclohexylmethyl)-1-((4-fluorophenethoxy)methyl)-3-hydroxy-5-methylpyrimidine-2,4(1H,3H)-dione (11e)
This compound was prepared as a white solid following the procedure described for the preparation of 11a. Yield: 53%; 1HNMR (600 MHz, CDCl3) δ 8.60 (s, 1H), 7.29 (t, J = 6.6 Hz, 1H), 7.21 (m, 1H), 7.01 (t, J = 7.8 Hz, 1H), 5.37 (s, 2H), 4.63 (s, 2H), 2.51 (d, J = 6.0 Hz, 2H), 2.36 (q, J = 7.2 Hz, 2H), 1.67 (m, 5H), 1.41 (m, 1H), 1.12 (m, 3H), 0.99 (m, 5H); 13C NMR (150 MHz, CDCl3) δ 162.1, 154.5, 151.1, 149.4, 129.5, 127.5, 125.1, 123.5, 120.2, 115.3, 72.0, 64.5, 37.6, 34.1, 32.2, 25.3, 25.0, 18.5, 12.6; HRMS (ESI+) calcd. for C21H27FClN2O3 [M+H]+ 409.1727, found 409.1725.
4.1.1.6. 6-(Cyclohexylmethyl)-1-(((3,4-difluorobenzyl)oxy)methyl)-5-ethylpyrimidine-2,4(1H,3H)-dione (11f)
This compound was prepared as a white solid following the procedure described for the preparation of 11a. Yield: 70%; 1H NMR (600 MHz, CDCl3) δ 8.43 (s, 1H), δ 7.09 (m, 2H), 6.96 (m, 1H), 5.34 (s, 2H), 4.52 (s, 2H), 2.52 (d, J =7.2 Hz, 2H), 2.37 (q, J =7.2 Hz, 2H), 1.68 (m, 5H), 1.43 (m, 1H), 1.13 (m, 5H), 1.09 (t, J =7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 163.3, 152.4, 150.2, 134.5, 123.6, 117.2, 117.1, 116.7, 116.5, 116.4, 72.7, 70.4, 38.5, 35.1, 33.2, 26.3, 26.0, 19.4, 13.5; HRMS (ESI+) calcd. for C21H27F2N2O3 [M+H]+ 393.2019, found 393.2022.
4.1.1.7. 6-(Cyclohexylmethyl)-5-ethyl-1-(((4-(trifluoromethyl)benzyl)oxy)methyl)-pyrimidine-2,4(1H,3H)-dione (11g)
This compound was prepared as a white solid following the procedure described for the preparation of 11a. Yield: 47%; 1H NMR (600 MHz, CDCl3 δ 8.22 (s, 1H), 7.53(d, J =8.4 Hz,, 2H), 7.37 (d, J =8.4 Hz, 2H), 5.36 (s, 2H), 4.63 (s, 2H), 2.52 (d, J =7.2 Hz 2H), 2.53 (q, J = 7.2 Hz, 2H), 1.67 (m, 5H), 1.44 (m, 1H), 1.12 (m, 5H), 0.98 (t, J =7.8 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 162.3, 151.4, 149.2, 140.4, 126.8, 124.3, 115.4, 71.9, 70.0, 37.6, 34.1, 25.3, 25.0, 18.4, 12.6; HRMS (ESI+) calcd. for C22H28F3N2O3 [M+H]+ 425.2117, found 425.2115.
4.1.1.8. 6-(Cyclohexylmethyl)-5-ethyl-1-(((4-methylbenzyl)oxy)methyl)pyrimidine-2,4(1H,3H)-dione (11h)
This compound was prepared as a white solid following the procedure described for the preparation of 11a. Yield: 65%; 1HNMR (600 MHz, CDCl3) δ 8.01 (s, 1H), 7.13 (d, J = 7.8 Hz, 2H), 7.07 (d, J = 7.8 Hz, 2H), 5.31 (s, 2H), 4.52 (s, 2H), 2.49 (d, J = 7.2 Hz, 2H), 2.32 (q, J = 6.6 Hz, 2H), 2.26 (s, 3H), 1.65 (m, 6H), 1.41 (m, 1H), 1.09 (m, 4H), 0.99 (t, J =7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 163.4, 152.3, 150.4, 137.7, 134.2, 129.1, 128.0, 116.2, 72.7, 71.6, 38.6, 35.0, 33.1, 26.3, 26.0, 21.1, 19.4, 13.6; HRMS (ESI+) calcd. for C22H31N2O3 [M+H]+ 371.2390, found 371.2387.
4.1.1.9. 1-((Benzyloxy)methyl)-6-(cyclohexylmethyl)-5-methylpyrimidine-2,4(1H,3H)-dione (11i)
This compound was prepared as a white solid following the procedure described for the preparation of 11a. Yield: 33%; 1HNMR (600 MHz, CDCl3) 8.58 (s, 1H), 7.27 (m, 5H), 5.34 (s, 2H), 4.56 (s, 2H), 2.53 (d, J = 7.2 Hz, 2H), 1.85 (s, 3H), 1.65 (m, 7H), 1.43 (m, 1H), 1.11 (m, 3H), 0.98 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 160.8, 149.2, 148.4, 134.6, 125.8, 125.4, 125.2, 107.6, 70.1, 69.0, 35.9, 33.1, 30.7, 23.6, 23.4, 9.2; HRMS (ESI+) calcd. for C20H27N2O3 [M+H]+ 343.2059, found 343.2066.
4.1.1.10. 1-(((3-Chloro-2-fluorobenzyl)oxy)methyl)-6-(cyclohexylmethyl)-5-methylpyrimidine-2,4(1H,3H)-dione (11j)
This compound was prepared as a white solid following the procedure described for the preparation of 11a.1Yield: 62%; HNMR (600 MHz, CDCl3) δ 8.10 (s, 1H), 7.29 (m, 1H), 7.21 (m, 1H), 7.01 (t, J = 7.2 Hz, 1H), 5.37 (s, 2H), 4.62 (s, 2H), 2.53 (d, J = 7.2 Hz, 2H), 1.87 (s, 3H), 1.67 (m, 5H), 1.44 (m, 1H), 1.12 (m, 3H), 1.00 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 162.7, 151.2, 149.8, 129.4, 127.5, 125.0, 123.5, 120.0 109.4, 71.8, 64.4, 37.4, 34.6, 32.2, 25.2, 25.0, 10.8; HRMS (ESI+) calcd. for C20H25FClN2O3 [M+H]+ 395.1599, found 395.1589.
4.1.1.11. 6-(Cyclohexylmethyl)-1-(((3,4-difluorobenzyl)oxy)methyl)-5-methylpyrimidine-2,4(1H,3H)-dione (11k)
This compound was prepared as a white solid following the procedure described for the preparation of 11a. 1Yield: 52%; HNMR (600 MHz, CDCl3) δ8.82 (s, 1H), 7.08 (m, 2H), 6.97 (s, 1H), 5.35 (s, 2H), 4.41 (s, 2H), 2.54 (d, J = 7.2 Hz, 2H), 1.89 (s, 3H), 1.67 (m, 6H), 1.48 (m, 1H), 1.13 (m, 4H); HRMS (ESI+) calcd. for C20H25F2N2O3 [M+H]+ 379.1888, found 379.1880.
4.1.1.12. 6-(Cyclohexylmethyl)-5-methyl-1-(((4-(trifluoromethyl)benzyl)oxy)methyl) pyrimidine-2,4(1H,3H)-dione (11l)
Yield: 43%; This compound was prepared as a white solid following the procedure described for the preparation of 11a; 1HNMR (600 MHz, CDCl3) δ 8.42 (s, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 7.2 Hz, 2H), 5.36 (s, 2H), 4.62 (s, 2H), 2.54 (d, J = 7.2 Hz, 2H), 1.87 (s, 3H), 1.67 (m, 5H), 1.42 (m, 1H), 1.13 (m, 5H); HRMS (ESI+) calcd. for for C21H26F3N2O3 [M+H]+ 411.1955, found 411.1954.
4.1.1.3. 6-(Cyclohexylmethyl)-1-((4-fluorophenethoxy)methyl)-5-methylpyrimidine-2,4(1H,3H)-dione (11m)
Yield: 50%; This compound was prepared as a white solid following the procedure described for the preparation of 11a; 1HNMR (600 MHz, CDCl3) δ 8.20 (s, 1H), 7.18 (t, J = 7.2 Hz, 2H), 6.95 (J = 8.4 Hz, 2H), 5.24 (s, 2H), 3.70 (t, J = 6.0 Hz, 2H), 2.75 (t, J = 6.0 Hz, 2H), 2.40 (d, J = 6.6 Hz, 2H), 1.84 (s, 3H), 1.67 (m, 2H), 1.61 (m, 4H), 1.41 (m, 1H), 1.15 (m, 2H), 0.99 (m, 2H); HRMS (ESI+) calcd. for for C21H28FN2O3 [M+H]+ 375.2140, found 375.2141.
4.1.2. General Procedures for synthesis of 4a–4m
To a solution of 11a (60 mg, 0.162 mmol ) in 3 mL of THF was added NaH (60% dispension in merinal oil) (65 mg, 1.62 mmol) at 0 °C. This reaction mixture was allowed to warm to room temperature over 1h., and then cooled to 0 °C followed by the addition of m-CPBA (223 mg, 1.3 mmol). After stirring for 0.5 h, this reaction mixture was stirred at rt overnight. The reaction was quenched by adding 10 mL of H2O, the aqueous phase was then extracted with ethyl acetate (10 mL × 3). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The resultant residue was subjected to combiflash (Hex/EtOAc, 1:1) to give compound 4a (42 mg, 70 %).
4.1.2.1. 1-((Benzyloxy)methyl)-6-(cyclohexylmethyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (4a)
Yield: 70 %; 1H NMR (600 MHz, CDCl3) δ 7.22 (m, 5H), 5.41 (s, 2H), 4.57 (s, 2H), 2.72 (m, 1H), 2.53 (d, J = 6.0 Hz, 2H), 1.63 (m, 5H), 1.37 (m, 1H), 1.24 (d, J = 6.0 Hz, 6H), 1.39 (m, 3H), 0.94 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 162.2, 152.2. 150.0. 137.3, 128.4, 127.9, 127.8, 118.9, 72.9, 71.7, 38.6, 34.9, 33.0, 28.6, 26.3, 26.0, 20.2; HRMS (ESI+) calcd. for C22H31N2O4 [M+H]+ 387.2269, found 387.2272.
4.1.2.2. 6-(Cyclohexylmethyl)-1-(((4-fluorobenzyl)oxy)methyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (4b)
This compound was prepared following the procedure described for the preparation of 4a. Yield: 70%; 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J = 6.6 Hz, 2H), 7.03 (t, J = 8.4 Hz, 2H), 5.50 (s, 2H), 4.62 (s, 2H), 2.88 (m, 1H), 2.63 (d, J = 7.2 Hz, 2H), 1.75 (m, 2H), 1.68 (m, 3H), 1.46 (m, 1H), 1.31 (d, J = 6.6 Hz, 6H), 1.20 (m, 3H), 1.06 (m, 2H); HRMS (ESI+) calcd. for C22H30FN2O4 [M+H]+ 405.2227, found 405.2237.
4.1.2.3. 1-((Benzyloxy)methyl)-6-(cyclohexylmethyl)-5-ethyl-3-hydroxypyrimidine-2,4(1H,3H)-dione (4c)
This compound was prepared following the procedure described for the preparation of 4a. Yield: 74%; 1HNMR (600 MHz, CDCl3) δ 7.22 (m, 5H), 5.39 (s, 2H), 4.57 (s, 2H), 2.51 (d, J = 6.6 Hz, 2H), 2.34 (q, J = 7.2 Hz, 2H), 1.64 (m, 6H), 1.42 (m, 1H), 1.09 (m, 3H), 0.98 (m, 4H); HRMS (ESI+) calcd. for C21H29N2O4 [M+H]+ 373.4580, found 373.4586.
4.1.2.4. 6-(Cyclohexylmethyl)-5-ethyl-1-(((4-fluorobenzyl)oxy)methyl)-3-hydroxypyrimidine-2,4(1H,3H)-dione (4d)
This compound was prepared following the procedure described for the preparation of 4a. Yield: 66%; 1H NMR (600 MHz, CDCl3) δ 8.84 (s, 1H), 7.22 (t, J = 6.6 Hz, 2H), 6.94 (m, 2H), 5.39 (s, 2H), 4.53 (s, 2H), 2.50 (d, J = 6.0 Hz, 2H), 2.36 (m, 2H), 1.65 (m, 5H), 1.40 (m, 1H), 1.12 (m, 5H), 0.98 (t, J = 7.2 Hz, 3H); HRMS (ESI+) calcd. for C21H28FN2O4 [M+H]+ 391.4485, found 391.4469.
4.1.2.5. 1-(((3-Chloro-2-fluorobenzyl)oxy)methyl)-6-(cyclohexylmethyl)-5-ethyl-3-hydroxypyrimidine-2,4(1H,3H)-dione (4e)
This compound was following the procedure described for the preparation of 4a. Yield: 68%; 1HNMR (600 MHz, CDCl3) δ 7.26 (m, 1H), 7.19 (m, 1H), 6.98 (m, 1H), 5.42 (s, 2H), 4.63 (s, 2H), 2.51 (d, J = 7.2 Hz, 2H), 2.36 (m, 2H), 1.64 (m, 5H), 1.41 (m, 1H), 1.11 (m, 3H), 0.99 (m, 5H); 13C NMR (150 MHz, CDCl3) δ 162.0, 156.1, 151.0, 149.4, 129.5, 127.5, 125.1, 123.5, 120.1, 115.3, 72.0, 64.5, 37.6, 34.0, 32.2, 25.3, 25.0, 18.5, 12.6, HRMS (ESI+) calcd. for C21H27FClN2O4 [M+H]+ 425.1680, found 425.1681.
4.1.2.6. 6-(Cyclohexylmethyl)-1-(((3,4-difluorobenzyl)oxy)methyl)-5-ethyl-3-hydroxypyrimidine-2,4(1H,3H)-dione (4f)
This compound was prepared as a white solid following the procedure described for the preparation of 4a. 1Yield: 63%; HNMR (600 MHz, CDCl3) δ 7.03 (m, 2H), 6.96 (m,1H), 5.41 (s, 2H), 4.54 (s, 2H), 2.52 (m, 2H), 2.39 (m, 2H), 1.65 (m, 2H), 1.60 (m, 3H), 1.42 (m, 1H), 1.09 (m, 3H), 1.00 (m, 5H); HRMS (ESI+) calcd. for C21H27F2N2O4 [M+H]+ 409.4389, found 409.4394.
4.1.2.7. 6-(Cyclohexylmethyl)-5-ethyl-3-hydroxy-1-(((4-(trifluoromethyl)benzyl)oxy) methyl)pyrimidine-2,4(1H,3H)-dione (4g)
This compound was prepared following the procedure described for the preparation of 4a. Yield: 60%; 1H NMR (600 MHz, CDCl3) δ 7.52 (d, J =7.8 Hz,, 2H), 7.37 (d, J =8.4 Hz, 2H), 5.45 (s, 2H), 4.66 (s, 2H), 2.54 (d, J =7.2 Hz 2H), 2.41 (q, J = 7.8 Hz, 2H), 1.66 (m, 5H), 1.41 (m, 1H), 1.09 (m, 5H), 0.98 (t, J =7.8 Hz, 3H); HRMS (ESI-) calcd. for C22H26F3N2O4 [M-H]− 439.1744, found 439.1745.
4.1.2.8. 6-(Cyclohexylmethyl)-5-ethyl-3-hydroxy-1-(((4-methylbenzyl)oxy)methyl) pyrimidine-2,4(1H,3H)-dione (4h)
This compound was prepared following the procedure described for the preparation of 4a. Yield: 71%; 1HNMR (600 MHz, CDCl3) δ 7.11 (d, J = 7.2 Hz, 2H), 7.03 (d, J = 7.2 Hz, 2H), 5.36 (s, 2H), 4.52 (s, 2H), 2.45 (d, J = 7.2 Hz, 2H), 2.28 (q, J = 6.6 Hz, 2H), 2.23 (s, 3H), 1.65 (m, 6H), 1.35 (m, 1H), 1.06 (m, 3H), 0.91 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 160.2, 148.3, 137.5, 134.3, 133.6, 129.0, 128.0, 114.5, 73.7, 71.6, 38.4, 35.0, 33.1, 26.2, 26.0, 21.1, 19.9, 13.2; HRMS (ESI+) calcd. for C22H31N2O4 [M+H]+ 387.4846, found 387.4852.
4.1.2.9. 1-((Benzyloxy)methyl)-6-(cyclohexylmethyl)-3-hydroxy-5-methylpyrimidine-2,4(1H,3H)-dione (4i)
This compound was prepared following the procedure described for the preparation of 4a. Yield: 77%; 1HNMR (600 MHz, CDCl3) 7.23 (m, 5H), 5.43 (s, 2H), 4.57 (s, 2H), 2.53 (d, J = 6.0 Hz, 2H), 1.90 (s, 3H), 1.64 (m, 5H), 1.41 (m, 1H), 1.10 (m, 3H), 0.96 (m, 2H); HRMS (ESI+) calcd. for C20H27N2O4 [M+H] − 358.2022, found 358.2025.
4.1.2.10. 1-(((3-Chloro-2-fluorobenzyl)oxy)methyl)-6-(cyclohexylmethyl)-3-hydroxy-5-methylpyrimidine-2,4(1H,3H)-dione (4j)
This compound was prepared following the procedure described for the preparation of 4a. Yield: 62%; 1HNMR (600 MHz, CDCl3) δ 7.29 (t, J = 7.2 Hz, 1H), 7.21 (m, 1H), 6.99 (t, J = 7.2 Hz, 1H), 5.44 (s, 2H), 4.64 (s, 2H), 2.54 (d, J = 6.6 Hz, 2H), 1.93 (s, 3H), 1.65 (m, 5H), 1.42 (m, 1H), 1.11 (m, 5H); 13C NMR (150 MHz, CDCl3) δ 159.2, 157.2, 155.5, 148.7, 148.3, 130.6, 128.5, 125.9, 124.5, 108.3, 73.7, 65.9, 38.5, 35.5, 33.2, 26.2, 25.9, 12.1; HRMS (ESI+) calcd. for C20H25ClFN2O4 [M+H]+ 411.1542, found 411.1550.
4.1.2.11. 6-(Cyclohexylmethyl)-1-(((3,4-difluorobenzyl)oxy)methyl)-3-hydroxy-5-methyl pyrimidine-2,4(1H,3H)-dione (4k)
This compound was prepared following the procedure described for the preparation of 4a. Yield: 65%; 1HNMR (600 MHz, CDCl3) δ 7.10 (m, 2H), 6.97 (m, 2H), 5.41 (s, 2H), 4.53 (s, 2H), 2.54 (d, J = 6.0 Hz, 2H), 1.94 (s, 3H), 1.67 (m, 6H), 1.44 (m, 1H), 1.18 (m, 4H); HRMS (ESI+) calcd. for C20H25F2N2O4 [M+H]+ 395.1832, found 395.1838.
4.1.2.12. 6-(Cyclohexylmethyl)-3-hydroxy-5-methyl-1-(((4-(trifluoromethyl)benzyl)oxy) methyl)pyrimidine-2,4(1H,3H)-dione (4l)
This compound was prepared following the procedure described for the preparation of 4a. Yield: 58%; 1HNMR (600 MHz, CDCl3) δ 8.64 (s, 1H), 7.52 (d, J = 7.2 Hz, 2H), 7.37 (d, J = 7.8 Hz, 2H), 5.45 (s, 2H), 4.64 (s, 2H), 2.56 (d, J = 6.6 Hz, 2H), 1.94 (s, 3H), 1.65 (m, 2H), 1.61 (m, 2H), 1.46 (m, 1H), 1.11 (m, 4H), 0.99 (m, 2H); HRMS (ESI+) calcd. for C21H26FN2O4 [M+H]+ 427.1733, found 427.1735.
4.1.2.13. 6-(Cyclohexylmethyl)-1-((4-fluorophenethoxy)methyl)-3-hydroxy-5-methyl pyrimidine-2,4(1H,3H)-dione (4m)
This compound was prepared following the procedure described for the preparation of 4a. Yield: 68%; 1HNMR (600 MHz, CDCl3) δ 7.03 (m, 2H), 6.87 (t, J = 7.2 Hz, 2H), 5.32 (s, 2H), 3.72 (t, J = 6.0 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.41 (d, J = 6.0 Hz, 2H), 1.91 (s, 3H), 1.65 (m, 5H), 1.40 (m, 1H), 1.06 (m, 3H), 0.96 (m, 2H); HRMS (ESI+) calcd. for C21H28FN2O4 [M+H]+ 391.2089, found 391.2093.
4.2. Biology
4.2.1. RNase H assay
RNase H activity was measured essentially as previously described [26]. Full-length HIV RT was incubated with the RNA/DNA duplex substrate HTS-1 (RNA 5′-gaucugagccugggagcu-3′-fluorescein annealed to DNA 3′-CTAGACTCGGACCCTCGA-5′-Dabcyl), a high sensitivity duplex that assesses non-specific internal cleavage of the RNA strand. Results were analyzed using GraphPad Prism (GraphPad Software, San Diego, CA) for nonlinear regression to fit dose-response data to logistic curve models.
4.2.2. RT pol assay
RT pol assays were carried out in 96-well plates by measuring the extension of an 18 nucleotide DNA primer (5′-GTCACTGTTCGAGCACA-3′) annealed to a 100 nucleotide DNA template (5′-ATGTGTGTGCCCGTCTGTTGTGTGACTCTGGTAACTAGAGATCCCTCAGACCC TTTTAGTCAGTGTGGAATATCTCATAGCTTGGTGCTCGAACAGTGAC-3′). Reactions containing 20 nM RT, 40 nM template/primer, and 10 μM deoxynucleotide triphosphates (dNTPs) in a buffer containing 50 mM Tris (pH 7.8) and 50 mM NaCl were initiated by the addition of 6 mM MgCl2. Reactions contained 1% DMSO and increasing concentrations of compounds. DNA synthesis was carried out for 30 min at 37 °C, and reactions were arrested by the addition of 100 mM EDTA. The QuantiFluor dsDNA System (Promega) was used to quantify the amount of formed double-stranded DNA. Reactions were read at ex/em 504/531 nm in a PerkinElmer EnSpire Multilabel plate reader. Results were analyzed using “Prism” software (GraphPad Software, San Diego, CA) for nonlinear regression to fit dose-response data to logistic curve models.
4.2.3. INST assay
HIV integrase was expressed and purified as previously reported [10]. Inhibition assays were performed using a modified protocol of our reported method 1[10]. Briefly, 2.1 μL of compound suspended in DMSO was placed in duplicate into a Black 96 well non-binding plate (Corning). Compounds were plated in duplicate to a final concentration of 0.13 – 100 μM. To each well of the plate 186.9 μL of reaction mixture without DNA substrate was added (10 mM HEPES pH 7.5, 10% glycerol w/v, 10 mM MnCl2, 1 mM DTT, 1 μM integrase). The enzyme was incubated with inhibitor for 10 min at 25 °C after which the reaction was initiated by the addition of 21 μL of 500 nM oligo (5′ biotin ATGTGGAAAATCTCTAGCA annealed with ACTGCTAGAGATTTTCCACAT 3′ Cy5). Reactions were incubated at 37 °C for 30 min and then quenched by the addition of 5.2 μL 500 mM EDTA. Each reaction was moved (200 μL) to a MultiScreen HTS PCR plate (Millipore) containing 20 μL streptavidin agarose beads (Life Technologies) and incubated with shaking for 30 min. A vacuum manifold was used to remove the reaction mixture and the beads were similarly washed 3 times with wash buffer (.05% SDS, 1 mM EDTA in PBS). The plates were further washed 3 times with 200 μL 50 mM NaOH to denature DNA not covalently linked to the biotin modification. For each denaturation step the plate was incubated with shaking at 25 °C for 5 min and the NaOH was removed by centrifugation at 1000 g for 1 min. The reaction products were eluted from the beads by the addition of 150 μL formamide. The plate was incubated at 25 °C for 10 min and read directly at 635/675 in a SpectraMax i3 plate reader (Molecular Devices).
Expression and purification of the recombinant IN in Escherichia coli were performed as previously reported[27, 28] with addition of 10% glycerol to all buffers. Preparation of oligonucleotide substrates has been described.[29] Integrase reactions were performed in 10 μL with 400 nM of recombinant IN, 20 nM of 5′-end [32P]-labeled oligonucleotide substrate and inhibitors at various concentrations. Solutions of 10% DMSO without inhibitors were used as controls. Reactions were incubated at 37 °C (60 minutes) in buffer containing 50 mM MOPS pH 7.2, 7.5 mM MgCl2, and 14.3 mM 2-mercaptoethanol. Reactions were stopped by addition of 10 μL of loading dye (10 mM EDTA, 98% deionized formamide, 0.025% xylene cyanol and 0.025% bromophenol blue). Reactions were then subjected to electrophoresis in 20% polyacrylamide–7 M urea gels. Gels were dried and reaction products were visualized and quantitated with a Typhoon 8600 (GE Healthcare). Densitometric analyses were performed using ImageQuant from Molecular Dynamics Inc. The concentrations at which enzyme activity was reduced by 50% (IC50) were determined using “Prism” software (GraphPad Software) for nonlinear regression to fit dose-response data to logistic curve models.
4.2.4. Antiviral MAGI assays
MAGI assays were carried out using P4R5 indicator cells essentially as previously described [16]. P4R5 cells were cultured in 96-well microplates with 4×103 cells per well and maintained in DMEM/10% FBS supplemented with puromycin (1 μg/ml). Cells were incubated with either 1% DMSO or varying concentrations of the drugs for 24h and then exposed to HIV (MOI of 1.25) followed by an additional incubation period of 48h. The extent of infection was assessed using a fluorescence-based β-galactosidase detection assay, as previously described with minor modifications [30]. After the 48h incubation period, cells were lysed and substrate 4-methylumbelliferyl-galactoside (MUG) was added. The β-galactosidase produced during infection acts on the substrate MUG and yields a fluorescent product 4-methylumbelliferone (4-MU) that could be detected fluorimetrically with excitation wavelength 365 nm and emission wavelength 446 nm.
4.2.5. HIV-1 cytoprotection assay[17]
The HIV cytoprotection assay used CEM-SS cells and the IIIB strain of HIV-1. Briefly, virus and cells were mixed in the presence of test compound and incubated for 6 days. The virus was pre-titered such that control wells exhibit 70 to 95% loss of cell viability due to virus replication. Therefore, antiviral effect or cytoprotection was observed when compounds prevent virus replication. Each assay plate contained cell control wells (cells only), virus control wells (cells plus virus), compound toxicity control wells (cells plus compound only), compound colorimetric control wells (compound only) as well as experimental wells (compound plus cells plus virus). Cytoprotection and compound cytotoxicity were assessed by MTS (CellTiter® 96 Reagent, Promega) and the EC50 (concentration inhibiting virus replication by 50%), CC50 (concentration resulting in 50% cell death) and a calculated TI (therapeutic index CC50/EC50) were provided. Each assay included AZT as a positive control.
4.2.6. HIV-1 RT/4a crystallization and data collection
HIV-1 RT containing C280S mutations in the p66 and p51 subunits was expressed and purified as previously described [31, 32]. Co-crystallization of HIV-1 RT with 4a was achieved by mixing a solution of RT at a final concentration of 11 mg/ml, 50 mM MgCl2, 5 mM tris(2-carboxyethyl)phosphine (TCEP) HCl, 0.5% β-octylglucoside, and 1 mM 4a in a 1:1 ratio with a solution of 15% PEG 3500, 0.1 M sodium potassium phosphate, 5% ethylene glycol, and 0.1 M MES pH 6.1. Large, blocky crystals grew by hanging drop vapor diffusion at 18 °C in 2–3 days.
HIV-1 RT/4a co-crystals were additionally soaked with 1 mM 4a, 5 mM TCEP HCl, 0.5% β-octylglucoside, 8% ethylene glycol, and 50 mM MgCl2 for 20 minutes before brief cryoprotection in a solution containing 23% ethylene glycol and 4% trimethylamine N-oxide, followed by fast cryo-cooling in liquid nitrogen. Data collected at Beamline 23-ID-B of the Advanced Photon Source at Argonne National Laboratory was processed and scaled to 2.7 Å resolution using XDS [33]. The HIV-1 RT/4a crystals were of space group C2, and had unit cell dimensions of a = 161.9 Å, b = 72.4 Å, c = 107.3 Å and β = 99.9°. One RT molecule was present in the asymmetric unit, and the Matthews coefficient was 2.7 Å3/Da, corresponding to a solvent content of 54.8% [34]. Crystal data and statistics are listed in SI Table 1.
4.3. Phase determination and structure refinement
The structure was determined by molecular replacement using Phaser [35], using a high resolution crystal structure of an HIV-1 RT/NNRTI complex (Protein Data Bank code 4G1Q) as the initial search model[36]. Rigid-body, simulated annealing (to remove bias), atomic displacement parameter (ADP), real-space and restrained refinement were performed using Phenix [37]. Several cycles of model building and refinement were carried out using Coot [38] and Phenix or Refmac [39], respectively. The final RT/4a structure was validated using MolProbity [40]. Final refinement statistics are listed in SI Table 1.
Supplementary Material
Highlights.
HPD subtype featuring a C-6 cyclohexylmethyl group potently inhibited HIV-1.
Dual biochemical inhibition against RNase H and pol.
Antiviral activity correlates better with RT pol inhibition for most analogues and with RNase H with two analogues 4f and 4m.
Analogues lacking the 3-OH inhibited HIV-1 and RT pol, but not RNase H.
Acknowledgments
This research was supported by the National Institutes of Health (AI100890) and partially by the Center for Drug Design, University of Minnesota. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.
Abbreviations
- HPD
3-Hydroxypyrimidine-2,4-dione
- HIV
human immunodeficiency virus
- RT
reverse transcriptase
- INST
integrase strand transfer
- Pol
polymerase
- NNRTI
nonnucleoside RT inhibitor
- HAART
highly active antiretroviral therapy
- NRTI
nucleoside reverse transcriptase inhibitor
- PIs
protease inhibitors
- CDI
carbonyldiimidazole
- mCPBA
meta-chloroperoxybenzoic acid
- HEPT
1-[(2-Hydroxyethoxy)methyl]-6-(phenylthio)thymine
- SAR
structure-activity-relationship
- dNTP
deoxynucleotide triphosphate
- MUG
4-methylumbelliferyl-galactoside
- 4-MU
4-methylumbelliferone
- TCEP
tris(2-carboxyethyl)phosphine
- ADP
atomic displacement parameter
Appendix A. Supplementary data
Synthesis and characterization data, including 1H NMR and MS data, of intermediates 6–10. This material is available free of charge via the Internet at t http://dx.doi.org/10.1016/j.ejmech.2016.12.030
PDB ID
The atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank (PDB ID: 5TUQ). Authors will release the atomic coordinates and experimental data upon article publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Yeni P. Update on HAART in HIV. J Hepatol. 2006;44(1 Suppl):S100–103. doi: 10.1016/j.jhep.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 2.Cihlar T, Ray AS. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antiviral Res. 2010;85(1):39–58. doi: 10.1016/j.antiviral.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 3.de Bethune MP. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989–2009) Antiviral Res. 2010;85(1):75–90. doi: 10.1016/j.antiviral.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh AK, Osswald HL, Prato G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J Med Chem. 2016;59(11):5172–5208. doi: 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park TE, Mohamed A, Kalabalik J, Sharma R. Review of integrase strand transfer inhibitors for the treatment of human immunodeficiency virus infection. Expert Rev Anti Infect Ther. 2015;13(10):1195–1212. doi: 10.1586/14787210.2015.1075393. [DOI] [PubMed] [Google Scholar]
- 6.Kankanala J, Kirby KA, Liu F, Miller L, Nagy E, Wilson DJ, Parniak MA, Sarafianos SG, Wang Z. Design, Synthesis, and Biological Evaluations of Hydroxypyridonecarboxylic Acids as Inhibitors of HIV Reverse Transcriptase Associated RNase H. J Med Chem. 2016;59(10):5051–5062. doi: 10.1021/acs.jmedchem.6b00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vernekar SKV, Liu Z, Nagy E, Miller L, Kirby KA, Wilson DJ, Kankanala J, Sarafianos SG, Parniak MA, Wang Z. Design, Synthesis, Biochemical, and Antiviral Evaluations of C6 Benzyl and C6 Biarylmethyl Substituted 2-Hydroxylisoquinoline-1,3-diones: Dual Inhibition against HIV Reverse Transcriptase-Associated RNase H and Polymerase with Antiviral Activities. J Med Chem. 2015;58(2):651–664. doi: 10.1021/jm501132s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang J, Liu F, Nagy E, Miller L, Kirby KA, Wilson DJ, Wu BL, Sarafianos SG, Parniak MA, Wang Z. 3-Hydroxypyrimidine-2,4-diones as Selective Active Site Inhibitors of HIV Reverse Transcriptase-Associated RNase H: Design, Synthesis, and Biochemical Evaluations. J Med Chem. 2016;59(6):2648–2659. doi: 10.1021/acs.jmedchem.5b01879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu B, Tang J, Wilson DJ, Huber AD, Casey MC, Ji J, Kankanala J, Xie J, Sarafianos SG, Wang Z. 3-Hydroxypyrimidine-2,4-dione-5-N-benzylcarboxamides Potently Inhibit HIV-1 Integrase and RNase H. J Med Chem. 2016 doi: 10.1021/acs.jmedchem.6b00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Bennett EM, Wilson DJ, Salomon C, Vince R. Rationally designed dual inhibitors of HIV reverse transcriptase and integrase. J Med Chem. 2007;50(15):3416–3419. doi: 10.1021/jm070512p. [DOI] [PubMed] [Google Scholar]
- 11.Tang J, Maddali K, Metifiot M, Sham YY, Vince R, Pommier Y, Wang Z. 3-Hydroxypyrimidine-2,4-diones as an Inhibitor Scaffold of HIV Integrase. J Med Chem. 2011;54(7):2282–2292. doi: 10.1021/jm1014378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang J, Maddali K, Dreis CD, Sham YY, Vince R, Pommier Y, Wang Z. N-3 Hydroxylation of Pyrimidine-2,4-diones Yields Dual Inhibitors of HIV Reverse Transcriptase and Integrase. ACS Med Chem Lett. 2011;2(1):63–67. doi: 10.1021/ml1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He YP, Long J, Zhang SS, Li C, Lai CC, Zhang CS, Li DX, Zhang DH, Wang H, Cai QQ, Zheng YT. Synthesis and biological evaluation of novel dihydroaryl/alkylsulfanyl-cyclohexylmethyl-oxopyrimidines (S-DACOs) as high active anti-HIV agents. Bioorg Med Chem Lett. 2011;21(2):694–697. doi: 10.1016/j.bmcl.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Clay RJ, Collom TA, Karrick GL, Wemple J, Safe A. Economical Method for the Preparation of Beta-Oxo Esters. Synthesis. 1993;(3):290–292. [Google Scholar]
- 15.Kimpton J, Emerman M. Detection of Replication-Competent and Pseudotyped Human-Immunodeficiency-Virus with a Sensitive Cell-Line on the Basis of Activation of an Integrated Beta-Galactosidase Gene. J Virol. 1992;66(4):2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sirivolu VR, Vernekar SKV, Ilina T, Myshakina NS, Parniak MA, Wang ZQ. Clicking 3 ′-Azidothymidine into Novel Potent Inhibitors of Human Immunodeficiency Virus. J Med Chem. 2013;56(21):8765–8780. doi: 10.1021/jm401232v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 18.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464(7286):232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nowotny M. Retroviral integrase superfamily: the structural perspective. EMBO rep. 2009;10(2):144–151. doi: 10.1038/embor.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hopkins AL, Ren JS, Esnouf RM, Willcox BE, Jones EY, Ross C, Miyasaka T, Walker RT, Tanaka H, Stammers DK, Stuart DI. Complexes of HIV-1 reverse transcriptase with inhibitors of the HEPT series reveal conformational changes relevant to the design of potent non-nucleoside inhibitors. J Med Chem. 1996;39(8):1589–1600. doi: 10.1021/jm960056x. [DOI] [PubMed] [Google Scholar]
- 21.Hopkins AL, Ren JS, Tanaka H, Baba M, Okamato M, Stuart DI, Stammers DK. Design of MKC-442 (emivirine) analogues with improved activity against drug-resistant HIV mutants. J Med Chem. 1999;42(22):4500–4505. doi: 10.1021/jm990192c. [DOI] [PubMed] [Google Scholar]
- 22.Miyasaka T, Tanaka H, Baba M, Hayakawa H, Walker RT, Balzarini J, De Clercq E. A novel lead for specific anti-HIV-1 agents: 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine. J Med Chem. 1989;32(12):2507–2509. doi: 10.1021/jm00132a002. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka H, Baba M, Hayakawa H, Sakamaki T, Miyasaka T, Ubasawa M, Takashima H, Sekiya K, Nitta I, Shigeta S, et al. A new class of HIV-1-specific 6-substituted acyclouridine derivatives: synthesis and anti-HIV-1 activity of 5- or 6-substituted analogues of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) J Med Chem. 1991;34(1):349–357. doi: 10.1021/jm00105a055. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka H, Takashima H, Ubasawa M, Sekiya K, Inouye N, Baba M, Shigeta S, Walker RT, De Clercq E, Miyasaka T. Synthesis and antiviral activity of 6-benzyl analogs of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) as potent and selective anti-HIV-1 agents. J Med Chem. 1995;38(15):2860–5. doi: 10.1021/jm00015a008. [DOI] [PubMed] [Google Scholar]
- 25.Cao L, Song W, De Clercq E, Zhan P, Liu X. Recent progress in the research of small molecule HIV-1 RNase H inhibitors. Curr Med Chem. 2014;21(17):1956–1967. doi: 10.2174/0929867321666140120121158. [DOI] [PubMed] [Google Scholar]
- 26.Parniak MA, Min KL, Budihas SR, Le Grice SF, Beutler JA. A fluorescence-based high-throughput screening assay for inhibitors of human immunodeficiency virus-1 reverse transcriptase-associated ribonuclease H activity. Anal Biochem. 2003;322(1):33–39. doi: 10.1016/j.ab.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 27.Leh H, Brodin P, Bischerour J, Deprez E, Tauc P, Brochon JC, LeCam E, Coulaud D, Auclair C, Mouscadet JF. Determinants of Mg2+-dependent activities of recombinant human immunodeficiency virus type 1 integrase. Biochemistry-Us. 2000;39(31):9285–9294. doi: 10.1021/bi000398b. [DOI] [PubMed] [Google Scholar]
- 28.Metifiot M, Maddali K, Naumova A, Zhang X, Marchand C, Pommier Y. Biochemical and pharmacological analyses of HIV-1 integrase flexible loop mutants resistant to raltegravir. Biochemistry-Us. 2010;49(17):3715–3722. doi: 10.1021/bi100130f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Semenova EA, Johnson AA, Marchand C, Davis DA, Yarchoan R, Pommier Y. Preferential inhibition of the magnesium-dependent strand transfer reaction of HIV-1 integrase by alpha-hydroxytropolones. Mol Pharmacol. 2006;69(4):1454–1460. doi: 10.1124/mol.105.020321. [DOI] [PubMed] [Google Scholar]
- 30.Abram ME, Parniak MA. Virion instability of human immunodeficiency virus type 1 reverse transcriptase (RT) mutated in the protease cleavage site between RT p51 and the RT RNase H domain. J Virol. 2005;79(18):11952–11961. doi: 10.1128/JVI.79.18.11952-11961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirby KA, Marchand B, Ong YT, Ndongwe TP, Hachiya A, Michailidis E, Leslie MD, Sietsema DV, Fetterly TL, Dorst CA, Singh K, Wang Z, Parniak MA, Sarafianos SG. Structural and inhibition studies of the RNase H function of xenotropic murine leukemia virus-related virus reverse transcriptase. Antimicrob Agents Chemother. 2012;56(4):2048–2061. doi: 10.1128/AAC.06000-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bauman JD, Das K, Ho WC, Baweja M, Himmel DM, Clark AD, Jr, Oren DA, Boyer PL, Hughes SH, Shatkin AJ, Arnold E. Crystal engineering of HIV-1 reverse transcriptase for structure-based drug design. Nucleic Acids Res. 2008;36(15):5083–5092. doi: 10.1093/nar/gkn464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kabsch W, XDS Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matthews BW. Determination of protein molecular weight, hydration, and packing from crystal density. Methods Enzymol. 1985;114:176–187. doi: 10.1016/0076-6879(85)14018-8. [DOI] [PubMed] [Google Scholar]
- 35.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuroda DG, Bauman JD, Challa JR, Patel D, Troxler T, Das K, Arnold E, Hochstrasser RM. Snapshot of the equilibrium dynamics of a drug bound to HIV-1 reverse transcriptase. Nat Chem. 2013;5(3):174–181. doi: 10.1038/nchem.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 40.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.