

Abstract
Some forms of familial Alzheimer’s disease (FAD) are caused by mutations in presenilins (PSs), catalytic components of a γ-secretase complex that cleaves target proteins, including amyloid precursor protein (APP). Calcium (Ca2+) dysregulation in cells with these FAD-causing PS mutants has been attributed to attenuated store-operated Ca2+ entry [SOCE; also called capacitative Ca2+ entry (CCE)]. CCE occurs when STIM1 detects decreases in Ca2+ in the endoplasmic reticulum (ER) and activates ORAI channels to replenish Ca2+ stores in the ER. We showed that CCE was attenuated by PS1-associated γ-secretase activity. Endogenous PS1 and STIM1 interacted in human neuroblastoma SH-SY5Y cells, patient fibroblasts, and mouse primary cortical neurons. Forms of PS1 with FAD-associated mutations enhanced γ-secretase cleavage of the STIM1 transmembrane domain at a sequence that was similar to the γ-secretase cleavage sequence of APP. Cultured hippocampal neurons expressing mutant PS1 had attenuated CCE that was associated with destabilized dendritic spines, which were rescued by either γ-secretase inhibition or over-expression of STIM1. Our results indicate that γ-secretase activity may physiologically regulate CCE by targeting STIM1 and that restoring STIM1 may be a therapeutic approach in AD.
INTRODUCTION
Alzheimer’s disease (AD) is a common neurodegenerative disorder, and most cases are idiopathic and sporadic in nature (1). However, about 5% of AD is early onset caused by autosomal dominant inheritance of mutant amyloid precursor protein (APP) or mutant presenilins (PS1 or PS2) (2, 3); the latter are components of a γ-secretase protein complex that cleaves APP. Because sporadic AD (SAD) and familial AD (FAD) share similar neuropathological hallmarks, including deposition of amyloid plaques and neurofibrillary tangles, they may have common disease etiologies. The accumulation of amyloid plaques derived from γ-secretase cleavage of APP has led to the “amyloid cascade hypothesis” that has provided the rationale for therapeutic strategies in AD. Thus, current therapeutic approaches are designed either to reduce the formation of amyloid peptides (γ-secretase inhibitors) or to increase the clearance of accumulated plaques (vaccines against Aβ) (4, 5). However, several clinical trials have been aborted or showed no efficacy, suggesting that amyloid and tau accumulation may not be the proximal origin of the disease (4, 5).
An alternative hypothesis posits that disrupted neuronal calcium (Ca2+) homeostasis plays a central role in AD pathogenesis [for review, see (4, 6–8)]. The “Ca2+ disruption hypothesis” postulates that sustained disruption of intracellular Ca2+ homeostasis is a proximal cause of AD. Disrupted Ca2+ signaling may impose a slow feed-forward cascade that promotes amyloid and tau aggregation, oxidative stress, neuronal apoptosis, synaptic deficits, and memory loss (6, 9). Ca2+ signaling mechanisms that have been proposed to be disrupted in cells, including neurons, expressing FAD-mutant PS include enhanced Ca2+ release from the endoplasmic reticulum (ER) and attenuated capacitative Ca2+ entry [CCE; a process also referred to as store-operated Ca2+ entry (SOCE)] (6, 9–11). Several molecular mechanisms have been proposed to account for the enhanced ER Ca2+ release, including increased ER Ca2+ loading by increased SERCA (sarcoplasmic/ endoplasmic reticulum Ca2+-ATPase) activity (12), disruption of a putative Ca2+ leak mediated by PS proteins (13), and sensitization of inositol tri-sphosphate receptor channel activity (14, 15). Although the attenuation of CCE is well documented in several models of AD, including primary neurons from transgenic mice with FAD-linked PS mutations (10, 11), little is known about the mechanisms that underlie the CCE deficits in FAD. CCE is mediated by the oligomerization of stromal interaction molecule (STIM) upon ER Ca2+ store depletion and its subsequent activation of the plasma membrane ORAI Ca2+ channel (16, 17). PS1-associated γ-secretase activity may be important in this process because knockout of PS1 and PS2 or expression of catalytically inactive PS1 mutants (D257A or D385A) is associated with enhanced CCE (11, 18, 19).
Accordingly, here, we sought to elucidate the molecular mechanisms of Ca2+ entry deficits in cells expressing FAD-mutant PS1, including in skin fibroblasts from patients with FAD-associated mutations in PS1. We identified STIM1 as a target of PS1-containing γ-secretase activity that reduced STIM1 availability under normal conditions. Furthermore, we found that the FAD-associated mutation in PS1 enhanced γ-secretase cleavage of STIM1, reducing activation of ORAI1 and attenuating CCE. In hippocampal neurons, this attenuation resulted in dendritic spine deformity that was rescued by overexpression of STIM1 or inhibition of γ-secretase activity.
RESULTS
FAD-linked mutant PS1 impairs CCE
To elucidate the underlying mechanisms of CCE deficits in FAD, we generated SH-SY5Y neuroblastoma cell lines stably expressing either wild-type PS1 (PS1WT) or FAD-mutant PS1 (PS1-M146L) at comparable amounts, or enhanced green fluorescent protein (EGFP) as a control (fig. S1). Using single-cell Ca2+ imaging, application of the muscarinic acetylcholine receptor agonist carbachol in a Ca2+-free buffer elicited a transient increase in intracellular Ca2+ concentration ([Ca2+]i) that depleted the ER Ca2+ stores. We then triggered CCE by replenishing Ca2+ in the medium in the absence (Fig. 1A) or presence (Fig. 1B) of the γ-secretase inhibitor DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester). In the absence of DAPT, CCE was comparable in PS1WT- and EGFP-expressing cells (Fig. 1A) such that the peak [Ca2+]i (Fig. 1C) and initial rate of Ca2+ entry (Fig. 1D) were similar (table S1). However, CCE in PS1-M146L–expressing cells was attenuated (Fig. 1A), with both the peak [Ca2+]i (Fig. 1C) and initial rate of Ca2+ entry (Fig. 1D) reduced compared to the control and PS1WT-expressing cells (table S1). We observed similar attenuation of CCE in cells expressing either FAD PS1-A246E or two forms of PS1 with FAD-associated mutations specific to Chinese populations, PS1-V97L and PS1-A136G (fig. S2A and table S1). In human skin fibroblasts, we triggered CCE with the pro-inflammatory mediator bradykinin in Ca2+-free buffer followed by Ca2+ readdition. Consistently, CCE was attenuated in skin fibroblasts from human FAD patients harboring PS1-M146L or PS1-A246E mutants compared with two age-matched control fibroblasts without PS1 mutation (Fig. 1, E to G, and table S1), indicating that CCE deficits may be a conserved phenotype in cells expressing FAD-linked mutant PS1.
Fig. 1. γ-Secretase attenuates CCE.

(A) Cytoplasmic Ca2+ concentration ([Ca2+]i) in SH-SY5Y cells stably expressing empty vector (CTRL), PS1WT, or PS1-M146L. CCE was triggered by depleting ER Ca2+ stores using 50 μM carbachol in Ca2+-free HBSS (white bar), followed by readdition of 2 mM Ca2+ to the extracellular solution (dark gray bar). (B) Experiments like those described in (A) were performed with cells pre-treated with 1 μM DAPT for 48 hours. In (A) and (B), gray lines depict individual cellular [Ca2+]i; colored lines depict the mean [Ca2+]i. (C and D) Summary of peak [Ca2+]i and initial rate of [Ca2+]i change during CCE with or without DAPT treatment. (E and H) Analogous CCE experiments performed in human skin fibroblasts from FAD patients harboring PS1-M146L or PS1-A246E mutation or from two age-matched individuals without PS1 mutation. (F and G) Summary of peak [Ca2+]i and initial rate of Ca2+ entry during CCE with or without DAPT treatment. Experiments were repeated three times for each cell line, with 30 cells analyzed in each experiment. Bars indicate means ± SEM. See also table S1. *P < 0.01, unpaired Student’s t test within group. #P < 0.01, one-way analysis of variance (ANOVA) with Tukey’s tests compared to CTRL and PS1WT groups in SH-SY5Y or CTRL1 and CTRL2 groups in human fibroblasts.
Inhibition of γ-secretase activity by preincubation with DAPT completely restored CCE in mutant PS1 cells such that the peak [Ca2+]i and initial rate of Ca2+ entry were similar to control and PS1WT-expressing SH-SY5Y cells (Fig. 1, B to D) or age-matched control human fibroblasts (Fig. 1, F to H). DAPT potentiated CCE in control cells and cells expressing PS1WT (Fig. 1 and table S1). To verify that DAPT potentiated CCE as a result of inhibiting PS1-mediated γ-secretase activity, we evaluated CCE in clustered regularly interspaced short palindromic repeats (CRISPR)–mediated PS1 knockout (PS1KO) SH-SY5Y cells. Deleting PS1 potentiated CCE to an amount that was comparable to that observed in native SH-SY5Y cells treated with DAPT (fig. S2B and table S1). Together, these results suggested that FAD-linked mutant PS1 impairs CCE and that PS1-mediated γ-secretase activity inhibits CCE.
PS1 interacts with STIM1 in the ER
STIM-induced clustering of ORAI mediates CCE (16, 17). Some studies report that STIM1 and STIM2 abundance are reduced in cells expressing certain FAD-linked mutant forms of PS1 (19, 20). However, we observed that stable SH-SY5Y cell lines expressing either PS1WT or PS1-M146L had similar protein abundances of endogenous ORAI1 and STIM1, which were not affected by knocking out of PS1 (fig. S1). Because both PS1 and STIM1 are predominantly localized in the ER, we hypothesized that PS1 interacted with STIM1. We assessed the interaction between STIM1 and PS1 in SH-SY5Y cells cotransfected with STIM1 and empty vector, PS1WT, or PS1-M146L. Immunoprecipitation of STIM1 pulled down endogenous PS1 as well as expressed PS1WT and PS1M146L (Fig. 2A). As a control, immunoprecipitation of cystic fibrosis transmembrane conductance regulator (CFTR) from SH-SY5Y cells cotransfected with CFTR and empty vector, PS1WT, or PS1-M146L failed to pull down PS1 (fig. S3B). Notably, endogenous nicastrin, another component of γ-secretase complex, was present in STIM1 immunoprecipitates, with the amount pulled down significantly increased by overexpression of either WT or mutant PS1 (Fig. 2A). In contrast, STIM1 immunoprecipitation in PS1KO SH-SY5Y cells failed to coimmunoprecipitate nicastrin (fig. S3A), suggesting that the PS1-STIM1 interaction was responsible for the presence of nicastrin in the immunoprecipitated complex.
Fig. 2. PS1 interacts with STIM1.

(A) PS1 and STIM1 in SH-SY5Y cells coexpressing STIM1 with empty vector, PS1WT, or PS1-M146L. Input (1/20) shown in the first three lanes, IgG immunoprecipitates (IP: IgG) shown in the middle three lanes, and STIM1 immunoprecipitates (IP: STIM1) shown in the last three lanes. The graphs summarize band intensities normalized to input from three separate experiments. *P < 0.01, one-way ANOVA with Bonferroni’s test compared to empty vector control. IB, immunoblot. (B) Coimmunoprecipitation of endogenous PS1 with STIM1 antibody in mouse primary cortical neurons. Input (1/20) shown in the first lane, IgG immunoprecipitates shown in the second lane, and STIM1 immunoprecipitates shown in the last lane. Experiments were repeated three times, and band intensities of coimmunoprecipitated proteins were normalized with their respectiveinput intensities. (C) Representative micrographs showing the interaction between endogenous PS1 and STIM1 by in situ PLA in SH-SY5Y cells. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). In situ PLA performed in CRISPR-mediated PS1KO or STIM1KO SH-SY5Y cells as negative controls. Images were taken at ×20 magnification. Scale bar, 10 μm. Bars represent means ± SEM from three separate experiments, with 15 cells analyzed in each group. See also table S2. #P < 0.01, one-way ANOVA with Bonferroni’s test compared to PLA performed in normal SH-SY5Y cells using STIM1 and PS1 antibodies.
To establish that the interaction between PS1 and STIM1 occurred with endogenous proteins, we performed analogous coimmunoprecipitation experiment in primary neurons isolated from mouse brain cortex. Immunoprecipitation of endogenous STIM1 coprecipitated endogenous PS1 from the cortical neuron lysate (Fig. 2B). To further establish a physical interaction between STIM1 and PS1, we performed in situ proximity ligation assay (PLA) in nontransfected SH-SY5Y cells. PLA generates positive signals (red fluorescent dots) when proteins are in close proximity (<40 nm). We detected fluorescent dots by PLA using monoclonal antibodies against PS1 and STIM1 (Fig. 2C). Comparable numbers of fluorescent dots were detected when PLA was performed in SH-SY5Y cells using antibodies against PS1 and nicastrin (Fig. 2C and table S2). However, the number of fluorescent dots was significantly reduced in PS1KO or STIM1 knockout (STIM1KO) SH-SY5Y cells (Fig. 2C and table S2). Confocal imaging and three-dimensional (3D) reconstruction indicated that the interaction of PS1 and STIM1 was in the cytoplasm and surrounding the nucleus, which would be consistent with an ER distribution for both proteins (fig. S4). Together, these results demonstrated that PS1 interacts with STIM1 in overexpressed or endogenous systems, including mouse neurons.
FAD-linked mutant PS1 attenuates STIM1 oligomerization and ORAI1 recruitment
As the ER Ca2+ sensor, STIM1 undergoes oligomerization upon ER Ca2+ depletion and translocates to ER–plasma membrane junctions to activate ORAI1 (21). To determine whether PS1 affects STIM1 oligomerization after ER Ca2+ depletion, we cotransfected cyan fluorescent protein (CFP)–tagged and yellow fluorescent protein (YFP)–tagged STIM1 into SH-SY5Y cells also expressing PS1WT or PS1-M146L and monitored fluorescence resonance energy transfer (FRET) to detect interactions between the tagged STIM1 proteins (22). We used carbachol in the absence of extracellular Ca2+ to deplete ER Ca2+ stores and trigger STIM1 oligomerization, which enhances FRET between CFP and YFP. The FRET signal was normalized to the CFP donor and YFP acceptor fluorescence signals to account for variation in the expression of the tagged proteins in the cells. Carbachol-induced store depletion increased the normalized FRET signal in all of the SH-SY5Y lines (Fig. 3A). After 5 min of carbachol stimulation in Ca2+-free medium, CFP-STIM1 and YFP-STIM1 colocalized at the plasma membrane region of the control and PS1WT-expressing cells (Fig. 3B) as expected. In contrast, plasma membrane regions lacking CFP- and YFP-STIM1 co-localization were present in PS1-M146L–expressing cells (Fig. 3B). In addition, the magnitude of the increase in the normalized FRET signal was significantly lower in PS1-M146L–expressing cells than that observed in control cells or in the cells expressing PS1WT (Fig. 3C). Consistent with impaired oligomerization, puncta formation of YFP-STIM1 triggered by thapsigargin-induced store depletion was markedly reduced in PS1-M146L–expressing SH-SY5Y cells as compared with control or PS1WT-expressing cells (fig. S5).
Fig. 3. FAD-linked PS1 mutant impairs STIM1 oligomerization.
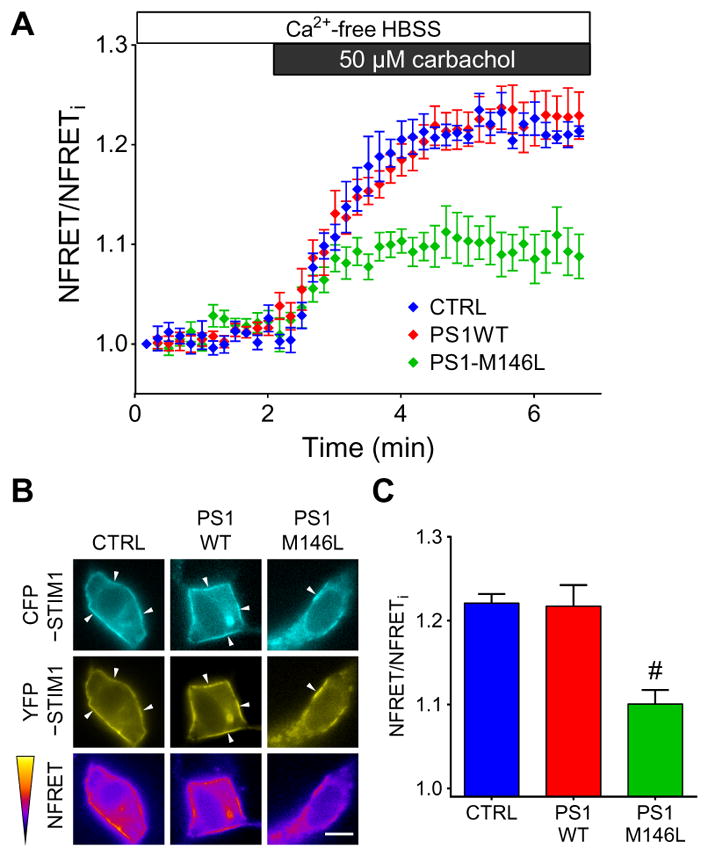
(A) FRET microscopy depicting oligomerization of STIM1 in SH-SY5Y cells stably expressing empty vector (CTRL), PS1WT, or PS1-M146L. FRET signals were triggered by carbachol-induced ER Ca2+ depletion in Ca2+-free buffer. FRET signals were normalized (NFRET) by CFP donor and YFP acceptor fluorescence intensities. (B) Representative images depicting localization and fluorescence intensities ofCFP, YFP, and FRET channels at time = 5 min of cells from the experiment shown in (A). Arrowheads indicate translocation of CFP- or YFP-tagged STIM1 after ER Ca2+ store depletion. Scale bar, 10 μm. (C) Summary of NFRET at 5 min. Experiments were repeated five times, and data are means ± SEM. *P < 0.01 as compared to either the CTRL or PS1WT group, using one-way ANOVA with Tukey’s multiple comparisons.
To evaluate the influence of PS1 on the formation of STIM1-ORAI1 complexes, we investigated puncta formation using total internal reflection fluorescence (TIRF) microscopy of coexpressed STIM1-GFP and ORAI1-mCherry in SH-SY5Y cells after ER Ca2+ depletion. The numbers of STIM1-ORAI1–colocalized puncta (yellow puncta) in empty vector control, PS1WT-, or PS1-M146L–expressing SH-SY5Y cells were normalized to their respective TIRF footprints (the area attached to the glass coverslip). Epifluorescence imaging revealed that STIM1-GFP and ORAI1-mCherry were localized in separate compartments when stores were filled. TIRF microscopy revealed only a few STIM1-GFP and ORAI1-mCherry puncta that did not generally colocalize in all of the SH-SY5Y lines (Fig. 4A). After store depletion by thapsigargin, the number of STIM1-GFP puncta formed was significantly increased, and they colocalized with ORAI1-mCherry puncta. Comparable numbers of colocalized STIM1-ORAI1 puncta (yellow puncta) were observed in control and PS1WT-expressing cells (Fig. 4B and table S3), whereas the number of STIM1-ORAI1 puncta per unit area was significantly reduced in PS1-M146L–expressing cells (Fig. 4, A and B). DAPT treatment increased the numbers of STIM1-ORAI1–colocalized puncta in control or PS1WT-expressing SH-SY5Y cells and increased those in PS1-M146L–expressing SH-SY5Y cells tovalues comparable to dimethyl sulfoxide (DMSO)–treated control or PS1WT-expressing cells (Fig. 4, A and B). Deletion of endogenous PS1 increased the number of STIM1-ORAI1–colocalized puncta compared with native SH-SY5Y cells, and this enhancement was not further increased by DAPT treatment (Fig. 4, C and D). STIM1-ORAI1 puncta were also reduced in number in skin fibroblasts from two FAD patients harboring mutant PS1-M146L or PS1-A246E compared with those from two age-matched controls. This deficit was rescued by treating cells with DAPT (fig. S6 and table S3). Together, these results suggested that impaired STIM1 oligomerization and reduced recruitment of ORAI1 underlie the CCE deficit observed in cells expressing FAD-linked PS1 mutants, and this CCE deficit is linked to PS1-mediated γ-secretase activity.
Fig. 4. FAD-linked PS1 mutant attenuates STIM1-ORAI1 interaction.

(A) Representative epifluorescence and TIRF images depicting the interaction of STIM1-GFP and ORAI1-mCherry in SH-SY5Y cells stably expressing empty vector (CTRL), PS1WT, or PS1-M146L before or after ER Ca2+ store depletion by 3 μM thapsigargin (TG) in the absence of extracellular Ca2+. Analogous experiments were performed with cells pretreated with 1 μM DAPT for 48 hours. (B) Quantification of STIM1-ORAI1–colocalized puncta in TIRF microscopy of SH-SY5Y cell lines shown in (A) normalized to their respective TIRF footprints before or after ER Ca2+ depletion. (C and D) ER Ca2+ depletion and TIRF microscopy experiments similar to (A) were performed in SH-SY5Y cells with endogenous PS1 knocked out (PS1KO) or native SH-SY5Y cells. Data are means ± SEM from three separate experiments, with 12 cells analyzed in each cell line and for each treatment. See also table S3. *P < 0.01 by Student’s t test compared to the respective DMSO treatment group. #P < 0.01 by one-way ANOVA with Tukey’s tests compared to either the CTRL and PS1WT group. #’P < 0.01 by Student’s t test compared to DMSO-treated native SH-SY5Y cells.
STIM1 is a substrate for γ-secretase
Our results showing reduced CCE in the PS1-mutant–expressing cells and FAD fibroblasts, together with observations of enhanced CCE in PS-deficient cells (11, 19), suggest that γ-secretase activity regulates CCE. Substrates for γ-secretase cleavage are type I transmembrane proteins, including APP (23). STIM1 is also a type I transmembrane protein that colocalizes and interacts with PS1 (Fig. 2). Accounting for their reversed topologies, the amino acid sequence of the transmembrane domain of STIM1 is similar to that of the transmembrane domain of APP, particularly near the APP cleavage sites that produce Aβ40 and Aβ42 (Fig. 5A). Therefore, we hypothesized that STIM1 may be a substrate for PS1-associated γ-secretase. We tested this by examining STIM1 Western blots for evidence of PS1-dependent cleavage. PS1-M146L–expressing SH-SY5Y cells had a weak STIM1-immunoreactive band with a molecular weight (~60 kDa) close to that expected for a predicted STIM1 C terminus if STIM1 was cleaved in its transmembrane domain (fig. S1 and Fig. 5B). We found two STIM1-immunoreactive bands with lower molecular weight in films developed with long exposure time (Fig. 5B). These bands were not detected in STIM1KO SH-SY5Y cells (Fig. 5B). To verify that the lower molecular weight band was a STIM1 cleavage product, we treated cells with the γ-secretase inhibitor DAPT or the proteasome inhibitor MG132 (24). Exposure of SH-SY5Y cells to the proteasome inhibitor MG132 increased the intensity of the lower molecular weight STIM1-immunoreactive bands (both upper and lower), whereas treating cells with the γ-secretase inhibitor DAPT with or without MG132 decreased the intensity of the lower band (Fig. 5B). These results suggest that STIM1 is a substrate of PS1-associated γ-secretase.
Fig. 5. FAD-linked PS1 mutant enhances γ-secretase cleavage of STIM1.

(A) Schematic depiction of the cellular localization and orientation of STIM1 and APP, and sequence alignment of the transmembrane domains of STIM1 and APP. Aβ40 and Aβ42 cleavage sites are indicated. Orientation of transmembrane domains reflects native topologies. (B) Representative immunoblots of STIM1 in STIM1KO SH-SY5Y or cells transduced with empty vector (CTRL), PS1WT, or PS1-M146L treated with the proteasome inhibitor MG132, the γ-secretase inhibitor DAPT, or both, respectively. Arrow indicates STIM1-immunoreactive band detected with longer exposure. Actin served as the loading control. Experiments were repeated five times. Bars indicate means ± SEM. *P < 0.01 and #P < 0.01 compared with respective DMSO or MG132 treatment group within the same cell line using Student’s t test. (C) In vitro fluorogenic assay of γ-secretase activity of solubilized SH-SY5Y cells for APP, STIM1, or STIM1 in reversed orientation [STIM1 (Rev)] peptides in the presence of increasing concentrations of DAPT. Itgβ1 and NPR-A transmembrane peptides served as controls. Experiments were repeated five times. (D) Summary of proteolysis of synthetic transmembrane peptides of (C) by γ-secretase activity in SH-SY5Y cells stably expressing empty vector (CTRL), PS1WT, or PS1-M146L. Experiments were repeated five times. Bars indicate means ± SEM. #P < 0.01 compared to CTRL and PS1WT groups using one-way ANOVA with Tukey’s multiple comparisons.
In another approach, we assessed STIM1 cleavage using an in vitro fluorogenic γ-secretase cleavage assay (25), using SH-SY5Y cell homogenates and fluorogenic synthetic peptides of the STIM1 transmembrane region. An APP transmembrane region peptide served as the positive control, whereas those of the proteins integrin β1 (Itgβ1) and natriuretic peptide receptor-A (NPR-A) served as the negative controls (Fig. 5C and fig. S7) (26). DAPT inhibited γ-secretase cleavage of APP, STIM1, and reversed STIM [STIM1 (Rev)] peptides in a concentration-dependent manner (Fig. 5C), whereas the Itgβ1 and NPR-A peptides were cleaved to a much lower extent and in a relatively DAPT-insensitive manner (Fig. 5C). Proteolytic cleavage of both APP and STIM1 peptides, but not of the Itgβ1 and NPR-A peptides, was enhanced in the presence of homogenates from PS1-M146L–expressing cells (Fig. 5D). These results suggested that, like APP, STIM1 is a substrate of γ-secretase and that FAD-associated mutant PS1 exhibits enhanced activity toward STIM1.
γ-Secretase or STIM1 overexpression rescues impaired spine morphology in FAD-linked mutant PS1–expressing neurons
Memory loss, a hallmark feature of AD, is likely due to the elimination of mature dendritic spines, and abnormal Ca2+ homeostasis destabilizes mature (mushroom-shaped) spines (27). We quantified the number and density of mature spines defined as those with mushroom appearance and head-to-neck ratio of >1.4 [according to (27, 28)] in primary rat hippocampal neurons or neurons transfected with PS1WT, PS1-M146L, or vector control (Fig. 6A). We also assessed the effect of DAPT (Fig. 6B) and STIM1 overexpression (Fig. 6C) on mature spine density and number. The densities of mature spines and the percent of neurites (identified by cotransfected EGFP) with mature spines were similar in neurons transfected with empty vector (CTRL) or PS1WT (Fig. 6D and table S4). In contrast, both features were reduced in the neurons expressing PS1-M146L (Fig. 6, A and D, and table S4), in agreement with a previous report (20). To determine whether attenuated CCE due to enhanced γ-secretase cleavage of STIM1 contributed to the decrease in dendritic spines, we examined spine morphology in neurons treated with DAPT or expressing STIM1, both of which were expected to rescue defective CCE. Both interventions restored normal mature spine density and the percentage of neurites with mature spines in PS1-M146L–expressing hip-pocampal neurons (Fig. 6, B to D, and table S4). These results suggested that CCE contributes to dendritic spine maintenance and that proteolysis of STIM1 by FAD-associated mutant PS1 may provide a molecular mechanism for dendritic spine loss and thus memory loss in AD.
Fig. 6. Mutant PS1 reduces the number of mature dendritic spines in cultured hippocampal neurons.

(A) Confocal images of spine morphology of cultured rat hippocampal neurons transfected with CTRL (empty vector), PS1WT, or PS1-M146L together with EGFP as a transfection marker. Enlarged image in lower panel depicts a representative dendrite (white box). Scale bars, 10 μm (white) or 2 μm (red). (B) Effect of DAPT treatment (48 hours) on spine morphology. (C) Effect of STIM1 overexpression on spine morphology. (D) Summary of spine density and population in indicated cultured rat hippocampal neurons. Experiments were repeated three times for each group, with a total of 36 dendrites analyzed for each group. See also table S4. Bars indicate means ± SEM. #P < 0.01 compared with CTRL and PS1WT groups using one-way ANOVA with Tukey’s multiple comparison. *P < 0.01 in neurons treated with 1 μM DAPT or with STIM1 overexpression compared to control group using one-way ANOVA with Bonferroni’s multiple comparisons.
DISCUSSION
Mounting evidence suggests that disrupted cellular Ca2+ homeostasis plays a proximal role in FAD pathogenesis (9, 29). Defective CCE that leads to altered Ca2+ homeostasis is associated with the presence of FAD-associated mutant PS1 (29), yet the molecular mechanisms have not been fully elucidated. Excessive filling of ER Ca2+ stores has been proposed as an underlying mechanism for CCE attenuation (10). However, many studies report that the ER Ca2+ stores in cells individually expressing various FAD-associated mutant PS1 remain unchanged or even decreased (14, 30–32), suggesting that CCE deficits may not be tightly correlated with the resting ER Ca2+ store content.
Another hypothesis is that CCE may be related to γ-secretase activity (11, 33). PS is the catalytic subunit of γ-secretase, which is essential for the proteolytic cleavage of certain type I transmembrane proteins, including APP and Notch (34). CCE is attenuated in cells expressing PS mutants that have enhanced γ-secretase activity (29). Here, we observed attenuated CCE in cells, including skin fibroblasts from FAD patients, expressing the FAD-associated mutants PS1-M146L, PS1-V97L, PS1-A136G, and PS1-A246E (Fig. 1 and fig. S2), all of which have enhanced γ-secretase activity (35, 36). Conversely, CCE was potentiated in the CRISPR-mediated PS1KO cells (fig. S2), as well as in PS-deficient neurons and in cells expressing catalytically dead mutant PS1-D257A and PS1-D385A (11). Furthermore, we and others have observed that CCE can be potentiated by γ-secretase inhibitors (33). Together, these data suggested that CCE is inhibited by γ-secretase activity, although the mechanisms have been unknown.
CCE is activated by store depletion–induced oligomerization of ER-localized STIM1 and its interaction with plasma membrane–localized ORAI1 (16, 17). STIM proteins are altered in abundance in PS-deficient cells and in cells harboring FAD-linked mutant PS (19, 20). STIM1 abundance is increased in PS-deficient mouse embryonic fibroblasts (MEFs) (19), whereas STIM2 abundance is decreased in these MEFs and in FAD mutant PS1–expressing human B lymphocytes (19) and in mouse neurons with PS1-M146V knocked in (20). Although changes in STIM abundance have been suggested to be due to changes in transcription or protein stability (19, 20), there has been no proposed mechanism linking PS to these processes. Here, we demonstrated that PS1 interacted with STIM1 in both overexpression and endogenous systems (Fig. 2 and fig. S4). We defined STIM1 as a substrate of the γ-secretase (Fig. 5). STIM1 is a type I trans-membrane protein, the substrate of PS-associated γ-secretase (23, 37). Although γ-secretase cleavage occurs at the plasma membrane, APP can also be processed intracellularly by γ-secretase, which generates a substantial pool of aggregation-prone intracellular Aβ42 (38–40). Although the transmembrane domain sequence of STIM1 is not identical to that of APP (Fig. 5A), γ-secretase has a promiscuous substrate dependence (41), and our in vitro γ-secretase cleavage assay demonstrated that peptides encompassing a sequence in the STIM1 transmembrane domain resembling the cleavage sequence of APP were cleaved by γ-secretase in either normal or reversed orientations (Fig. 5). In addition, we observed that expression of FAD-linked mutant PS1 with enhanced γ-secretase activity increased STIM1 cleavage. Although we have not examined the cleavage of STIM2, the sequence similarities of the transmembrane domains of STIM1 and STIM2 suggest that STIM2 could also be targeted by γ-secretase. Such an effect may account for the previous reported reduction in STIM protein abundance (19, 20). FAD-mutant PS1–mediated enhanced STIM1 cleavage was associated with diminished CCE, whereas a γ-secretase inhibitor enhanced CCE in normal cells. In addition, the γ-secretase inhibitor or knocking out PS1, as well as STIM1 overexpression, rescued impaired CCE in FAD-PS1–expressing cells. Enhanced STIM1 cleavage in cells expressing FAD mutant PS1 was correlated with reduced STIM1 oligomerization (Fig. 3 and fig. S5) and localization at the plasma membrane and interaction with ORAI1 (Fig. 4 and fig. S6) after store depletion. Nevertheless, and somewhat surprisingly, steady-state STIM1 abundance was not grossly diminished by normal or modified γ-secretase activity. Thus, γ-secretase–mediated cleavage appeared to largely affect a pool of STIM1 that is normally recruited for efficient CCE. STIM1 is localized to the plasma membrane (42) and acidic Ca2+ stores (43). Plasma membrane STIM1 is involved in store-independent arachidonate-regulated Ca2+ entry (42), whereas those in acidic organelles regulate acidic Ca2+ store refilling (44). Whether these other pools of STIM1 are subject to γ-secretase–mediated cleavage will need to be investigated further. Nonetheless, our findings suggest a physiological role of PS1/γ-secretase in regulating CCE by limiting the available pool of STIM1. Depending on cell type, different γ-secretase activity and pools of STIM1 may have more or less pronounced effects on total STIM1 abundance than observed here.
Reduced STIM2 abundance destabilizes mature dendritic spines in mice bearing FAD mutant PS (20). Our analyses agree with this previous study and provide mechanistic insight into this effect. CCE is vital to replete ER Ca2+ stores in resting neurons (45), and it plays a role in many neuronal functions (46), such as quick recharging of ER Ca2+ stores during rapid neurotransmission (47) and regulating neurotransmitter release and synaptic plasticity (48). Our results suggest that FAD-linked mutant PS1 impairs STIM1 function by enhancing its cleavage that leads to den-dritic spine deformity in hippocampal neurons. Mature spine destabilization in FAD-linked mutant PS1–expressing neurons suggests a pathogenic linkage between γ-secretase activity, impaired CCE, and cognitive impairment, a pathological hallmark of AD (49, 50).
Predicated on the amyloid cascade hypothesis, many γ-secretase modifiers (GSMs) have been developed to treat AD, but, to date, none have shown evidence of therapeutic efficacy (51). Although γ-secretase inhibitors reduce Aβ amounts in the cerebrospinal fluid and improve cognitive functions in AD mouse models (52), a γ-secretase inhibitor has not promoted cognitive improvement in a human clinical trial (53). The disappointing clinical outcomes of GSMs in humans may be a consequence on intervening too late in the process of disease pathogenesis, or it could be due to lack of understanding of the complicated and multi-faceted functions of γ-secretase (51). γ-Secretase activity is important in various cellular pathways because its substrates are not limited to APP and also include Notch (23, 54), which is essential to numerous cellular processes. Our results suggest that γ-secretase activity is important in regulating the CCE-relevant STIM1. Furthermore, they demonstrated that STIM1 cleavage and impaired CCE resulted from enhanced γ-secretase activity associated with FAD-linked mutant forms of PS1. Inhibition of γ-secretase activity restored CCE, but potentiation of CCE in normal cells suggests that an appropriate dose of GSMs is required to titrate normal CCE function. For example, because no STIM1-specific GSMs have been developed, and because all current GSMs have been developed to target APP or Notch cleavage, use of these nonspecific GSMs may overinhibit STIM cleavage, leading to potentiation of CCE and exacerbation of Ca2+ dysregulation in FAD. Neuronal Ca2+ homeostasis is important for normal Aβ processing, reactive oxygen species formation, neuronal apoptosis, and other cellular processes (55–57). Abnormal CCE, attenuated or accentuated, may impair these processes (58). A Notch-sparing GSM has already been developed (59). The identification of STIM1 as a γ-secretase substrate provides possibilities for knowledge-based design of STIM-specific GSM to correct CCE deficits, with therapeutic implications in AD.
In summary, our results from the current study suggest a molecular mechanism of CCE deficits in which FAD-mutant PS1 enhances γ-secretase cleavage of STIM1, reducing recruitment of ORAI1 that results in impaired CCE (Fig. 7). Sustained attenuation of CCE leads to spine deformity in FAD-mutant PS1–expressing hippocampal neurons that can be rescued by a γ-secretase inhibitor or overexpression of STIM proteins [Fig. 6 and (20)]. Our findings indicate a physiological role of PS1/γ-secretase in modulating the availability of STIM1 for CCE and suggest that identification of STIM1 as a substrate of γ-secretase provides a therapeutic target for the treatment of AD.
Fig. 7. Hypothetic model of spine destabilization induced by mutant PS1–associated CCE attenuation.

STIM1 is a type I transmembrane protein that senses Ca2+ concentrations in the ER. It oligomerizes and translocates to near–plasma membrane regions and interacts with and activates ORAI1 to sustain extracellular Ca2+ entry upon ER Ca2+ store depletion. The transmembrane (TM) domain of STIM1 is highly similar to that of APP and is cleaved by γ-secretase. In normal neurons, PS1WT modulates CCE by inactivating STIM1 through γ-secretase cleavage (left panel). In FAD-PS1 neurons, mutant PS1 enhances γ-secretase cleavage of STIM1, resulting in the reduction of functional STIM1 oligomers with consequent diminished ORAI1 channel activation, leading to attenuated CCE. Sustained CCE reduction may lead to spine deformity and result in memory loss in AD.
MATERIALS AND METHODS
Cell culture and stable cell line generation
The human neuroblastoma cell line SH-SY5Y was cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and 50 U of penicillin-streptomycin mixture (Invitrogen) at 37°C, gassed with 5% CO2. Human embryonic kidney (HEK) 293T cell was cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and penicillin-streptomycin mixture (Invitrogen) at 37°C, gassed with 5% CO2. Stable SH-SY5Y stable cell lines were generated by retrovirus transduction as previously described (60). In brief, complete coding sequence of human PS1WT was subcloned into retroviral vector pΔMX-IRES2-EGFP (15) or pBABE-puro (Addgene; #1764). PS1-M146L and other FAD-linked point mutants (PS1-V97L, PS1-A136G, and PS1-A246E) were then generated using QuikChange Site-Directed Mutagenesis Kit (Stratagene). The resulting plasmids were confirmed by DNA sequencing [Beijing Genomics Institute (BGI)]. Retroviruses were produced in HEK293T by transfecting cells with retroviral vector containing the gene of interest, together with the VSV-G and packaging plasmid pUMVC (Addgene; #8454 and #8449). SH-SY5Y cells were infected by the respective retrovirus, and transduced cells were selected by fluorescence-activated cell sorting or puromycin selection.
Human skin fibroblasts from FAD patients harboring PS1-M146L (AG07872) or PS1-A246E (AG06840) or age-matched controls without PS1 mutation (CTRL1, AG06869; CTRL2, AG09181) were obtained from the Coriell Institute for Medical Research (Camdem, NJ). The fibroblasts were cultured in Eagle’s minimum essential medium supplemented with Earle’s salts, nonessential amino acids, and 15% fetal bovine serum and penicillin-streptomycin mixture (Invitrogen) at 37°C, gassed with 5% CO2.
Generation of CRISPR-mediated knockout SH-SY5Y cells
CRISPR/Cas9-based genome editing was performed on SH-SY5Y cells to generate PS1 or STIM1KO lines. In brief, specific CRISPR guide RNAs (gRNAs) were generated with the assistance of in silico tool provided by the manufacturer (Invitrogen). The efficiency of each gRNA was assayed and screened using GeneArt Genomic Cleavage Detection Kit (Invitrogen; #A24372). After the confirmation of optimal target genomic cleavage, the following gRNAs were selected for knocking out the endogenous PS1 or STIM1 in SH-SY5Y cells: PS1, ATTATCTAATGGACGACCCC; STIM1, CAAACTGATGGACGATGATG.
The GeneArt CRISPR Nuclease Vector (Invitrogen; #A21175) was used to deliver CRISPR nuclease and gRNA to the SH-SY5Y cells using Lipofectamine 3000 Reagent (Invitrogen; #L3000015) according to the manufacturer’s protocol. Successful transformants, which carried CD4 surface antigen, were enriched by Dynabeads CD4 Positive Isolation Kit (Invitrogen; #11331D). The enriched CD4-positive cells were subjected to limited dilution for clonal selection. Isolated cell colonies arising from a single cell were picked by cloning cylinder (Sigma-Aldrich; #CLS31666) for the confirmation of gene deletion by DNA sequencing (BGI). Knockout of endogenous PS1 or STIM1 was then verified by Western blots.
Western blotting
Cells were washed twice with ice-cold phosphate-buffered saline (PBS) [137 mM NaCl, 10 mM Na2HPO4, 2 mM KH2PO4, 2.7 mM KCl (pH 7.4)] and then lysed on ice with lysis buffer [50 mM tris-HCl (pH 8.0), 150 mM NaCl, 1% Triton X-100, Roche cOmplete protease inhibitor cocktail] for 15 min. Cell lysate was homogenized, and supernatant was collected after centrifugation at 18,000g for 10 min at 4°C. Protein concentrations were determined by Bio-Rad Bradford assays using bovine serum albumin (BSA) as standard. Equal volumes of Laemmli 2× buffer [4% SDS, 10% β-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue, 0.125 M tris-HCl (pH 6.8)] and protein lysatewere mixed and denatured in boiling water for 10 min. Protein samples (25 μg) were subjected to SDS–polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes. Membranes were immunoblotted with primary antibodies and washed three times with tris-buffered saline 0.2% Tween 20 [150 mM NaCl, 50 mM tris-HCl (pH 7.6)] for 10 min followed by incubation with the corresponding horseradish peroxidase (HRP)–conjugated secondary antibody (Bio-Rad). Chemiluminescence emitted upon addition of HRP substrate (Millipore) was captured by x-ray film (Fujifilm) or Gel Doc XR+ System (Bio-Rad). The following primary antibodies were used: CFTR (Cystic Fibrosis Foundation Therapeutics; #596), nicastrin (R&D Systems Inc.; #MAB53781), STIM1 (ProSci Inc.; #4119), ORAI1 (ProSci Inc.; #4041), PS1 (EMD Millipore; #MAB5232), and actin (EMD Millipore; #MAB1501R).
Coimmunoprecipitation
SH-SY5Y cells were cotransfected with CFTR or STIM1 together with pIRES2-EGFP empty vector (Clontech Laboratories Inc.), pIRES2-EGFP-PS1WT, or pIRES2–EGFP–PS1-M146L. Two micrograms of mouse STIM1 antibody (Santa Cruz Biotechnology; #sc-66173), 2 μg of mouse CFTR antibody (Cystic Fibrosis Foundation Therapeutics; #596), or equivalent amount of mouse immunoglobulin G (IgG) isotype control (Thermo Fisher Scientific; #10400C) was conjugated with 1.5 mg of protein G–conjugated Dynabeads according to the manufacturer’s instructions (Invitrogen; #10003D). Antibody-conjugated Dynabeads were incubated with protein lysates (500 μg of protein) for 2 hours with gentle agitation. The beads were retained after washing with PBS three times. Laemmli buffer was added to dissociate immunoprecipitates, and Western blottings against PS1, nicastrin, actin, STIM1, or CFTR were performed accordingly.
Single-cell Ca2+ imaging and CCE measurement
Single-cell Ca2+ imaging was used to measure cytoplasmic Ca2+ concentration [Ca2+]i as described (14, 60). SH-SY5Y cells or human skin fibroblasts were plated and cultured on poly-D-lysine–coated coverslips to 80% confluency. Cells were loaded with 2 μM fura-2 AM in Hepes-buffered Hanks’ balanced salt solution (Hepes-HBSS) with 1% BSA for 30 min at room temperature. Fura-2 AM–loaded SH-SY5Y cells were mounted in an open bath imaging chamber (RC-21B; Warner Instruments) and visualized with a Nikon Eclipse Ti microscope using a 20× objective (Nikon CFI S Fluor Objective). Cells were alternatively excited at 340 and 380 nm, and emitted fluorescence at 510 nm was captured by a SPOT RT3 CCD (charge-coupled device) digital microscope camera (SPOT Imaging Solutions) every 5 s controlled by MetaFluor software (Molecular Devices). Ratios of emitted fluorescence were converted to [Ca2+]i according to the Grynkiewicz equation, as previously described (15, 61). CCE was triggered by depleting ER Ca2+ stores using 50 μM carbachol in SH-SY5Y cells or 100 nM bradykinin in human fibroblasts in Ca2+-free Hepes-HBSS followed by readdition of 2 mM Ca2+ to Hepes-HBSS.
FRET microscopy
FRET microscopy was performed as described (62). In brief, stable SH-SY5Y cell lines were cotransfected with SP-CFP-STIM1 and SP-YFP-STIM1 (Addgene; #18857 and #18858) and observed under 40× objective (Nikon CFI S Fluor Objective). Images of CFP, YFP, and FRET channels were captured by MetaFluor (Molecular Devices)–controlled filter wheels with standard FRET filter sets (#89002; Chroma Technology Corp.). Ca2+-free Hepes-HBSS, carbachol, and Ca2+ were perfused locally to the zone of interest with a gravity flow perfusion pencil (AutoMate Scientific). CFP, YFP, and FRET images were obtained every 10 s. Normalized FRET was calculated and normalized as described (63), and images were saved for analysis by ImageJ with pixel-to-pixel calculation (64).
In situ PLA
In situ PLA was used to determine the interaction of endogenous PS1 and STIM1 in native nontransfected SH-SY5Y cells according to the manufacturer’s instructions (Duolink PLA kit; Olink Bioscience). In brief, cells at 70% confluence were fixed with 4% paraformaldehyde (PFA) for 15 min, permeabilized by 0.2% Triton buffer for 10 min, and blocked in blocking buffer (supplied in the kit) at 37°C for 30 min. Endogenous STIM1 and PS were probed with rabbit PS monoclonal antibody (#S182; Sigma-Aldrich) and mouse STIM1 monoclonal antibody (#610954; Becton Dickinson Co.). SH-SY5Y cells with STIM1KO or PS1KO were used as negative controls. Analogous in situ PLA assay was performed using antibodies against PS1 (#S182; Sigma-Aldrich) and nicastrin (#MAB53781; R&D Systems) to demonstrate positive interaction. After incubation with primary antibodies, corresponding PLA secondary probes were added and incubated at 37°C for 1 hour. Hybridization of the complementary DNA strands was facilitated by incubating with ligation buffer with DNA ligase at 37°C for 30 min. The ligated DNA strand was then amplified and labeled with specific fluorescent DNA probes in amplification buffer at 37°C for 100 min. The coverslip was then mounted on a glass slide using mounting medium with DAPI. The fluorescent-labeled PLA complex was visualized by X-Y 2D laser scan in a Zeiss LSM 710 Upright Confocal Microscope [Li Ka Shing (LKS) Faculty of Medicine Imaging Core, Carl Zeiss] with 63× oil immersion objective.
Confocal imaging of STIM1 puncta
SH-SY5Y cells were plated and cultured on poly-D-lysine–coated coverslips and transfected with human STIM1-YFP (Addgene; #19754). In some experiments, cells were incubated with 3 μM thapsigargin in Ca2+-free Hepes-HBSS for 15 min. In other experiments, the cells were placed in Hepes-HBSS with 2 mM Ca2+ and incubated for 15 min. Cells were then visualized with an Olympus IX71 microscope (Olympus) using a 60× oil immersion objective. Confocal images were collected by X-Y 2D laser scanning by an Olympus Fluoview confocal system FV300 (Olympus). Quantification of STIM1 puncta was performed as described (65–67). In brief, confocal images of single, isolated cells were selected. The images were thresholded, and mean intensity of puncta greater than 3 SDs was analyzed. Number of puncta after store depletion in cell was determined by ImageJ using particle analysis function with the following parameters: size (0.1 to 2.5 μm2) and circularity (0 to 1). Data are means ± SEM.
TIRF microscopy
Interaction of STIM1 and ORAI1 after ER Ca2+ depletion was studied by TIRF microscopy. SH-SY5Y cells or human fibroblast were plated and cultured on glass-bottom microwell dishes (#P35G-1.5-20-C; MatTek). The cells were allowed to grow on the dish until 70% confluence was reached. Cells were then cotransfected with STIM1-GFP and ORAI11-mCherry plasmids (68). Forty-eight hours after transfection, cells were incubated in Hepes-HBSS with 2 mM Ca2+ (resting state) or 3 μM thapsigargin to deplete ER Ca2+. Cells were then fixed with 4% PFA for 15 min before they were subjected to imaging. Simultaneous wide-field and TIRF imaging were performed with a Zeiss Axio Observer Z1 microscope with Roper iLas2 360° ring TIRF illuminator and 100× oil immersion objective. Sapphire 488-nm and 561-nm (100 mW) lasers (Coherent Inc.) were used. STIM1 and ORAI1 puncta were captured by an electron-multiplying CCD camera (Evolve 512) with fixed evanescent light penetration (80-nm depth) using the calibrated incident angles of each laser beam.
Investigation on STIM1 cleavage with proteasome inhibitors and in vitro fluorogenic γ-secretase cleavage assay
Cleavage of endogenous STIM1 in SH-SY5Y cells was assessed by Western blots. Stable SH-SY5Y cell lines were treated with 5 μM DAPT for 12 hours. DMSO of equal volume was used as control. In some experiments, a proteasome inhibitor (2 μM MG132; Sigma-Aldrich) was added alone or together with DAPT to prevent protein degradation 12 hours before cell lysis. The cells were then lysed with lysis buffer and subjected to Western blot analysis. STIM1 was probed with an antibody from Cell Signaling (#4916).
To demonstrateγ-secretasecleavage of STIM1,aninvitro fluorogenicassay was performed with CHAPSO (3-[(3-cholamidopropyl) dimethylammonio]-2-hydroxy-1-propanesulfonate)–solubilized γ-secretase complex (25). Short transmembrane peptides of several type I transmembrane proteins were synthesized (GL Biochem), and the fluorogenic MCA group and the coupled quencher DNP were covalently linked at each terminus as shown: APP: {MCA} – [GGVVIATV] – K – {DNP} – RRR; STIM1: {MCA} – [VVSIVIGVGG] –K – {DNP} – RRR; STIM1 (Rev): {MCA} – [GGVGIVISVV] – K – {DNP} –RRR; Itgβ1: {MCA} – [AGVVAGIV] – K – {DNP} – RRR; NPR-A: {MCA} – [GSLSLLGI] – K – {DNP} – RRR.
With the presence of the quencher DNP, no fluorescence would be generated and vice versa. Peptide cleavage events were reflected by the change of MCA fluorescence. In brief, SH-SY5Y cells expressing PS1WTor mutant PS1-M146L were washed with ice-cold PBS and centrifuged at 500g for 5 min. Cell pellets were homogenized with a Dounce homogenizer in lysis buffer containing 5 mM tris-HCl (pH 7.4), 5 mM EDTA, 5 mM EGTA, and protease inhibitor cocktail (Roche cOmplete). The homogenates were centrifuged at 800g for 10 min to remove nuclei and large debris. Supernatants were solubilized with CHAPSO buffer (0.25%) containing 20 mM Hepes (pH 7.0), 2 mM EGTA, 150 mM KCl, and protease inhibitor cocktail (Roche) for 2 hours. CHAPSO-solubilized membrane (250 μg) was incubated with each of the 4 μM synthetic fluorogenic peptides. γ-Secretase cleavage of peptides was allowed to react overnight, and fluorescence with excitation at 344 nm and emission 443 nm was read with Tecan M200 plate reader.
Primary rat hippocampal neuron culture and spine morphology analysis
All animal protocols and procedures described were performed in compliance with the Principles of Laboratory Animal Care (National Institutes of Health publication no. 86-23, revised 1985) and were approved by the University of Hong Kong Committee on the Use of Live Animals in Teaching and Research. Primary hippocampal neurons were prepared and cultured as described (69). In brief, embryonic hippocampal neurons were obtained from embryonic day 18 rat embryos and were plated on poly-D-lysine–coated coverslips. The cells were cultured in neurobasal medium supplemented with 2% B27 and 0.5 mM glutamine (Invitrogen). Transfection of PS1 was performed at 10 days in vitro (DIV) using calcium phosphate precipitation method, and emerald green fluorescent protein plasmid was cotransfected as a positive marker (69). To determine dendritic spine morphology, immunocytochemistry of PS1-transfected rat hippocampal neurons was performed as described (69). In brief, transfected cells were fixed at 16 DIV with 4% PFA in 4% sucrose and permeabilized by gelatin dilution buffer [0.2% gelatin, 0.5% Triton X-100 in 0.8 M NaCl, 30 mM phosphate buffer (pH 7.4)] (70). Anti-GFP (#A-11120; Invitrogen) antibody and the corresponding Alexa Fluor 488 IgG2a secondary antibody (Invitrogen) were used to label transfected neurons. Neurons were then visualized with an Olympus Fluoview confocal system (FV300) (Olympus) using a 60× oil immersed objective and 488-nm argon laser. Emission filters were set accordingly to collect the green channel. Confocal images were collected from X-Y-Z 2D serial laser scanning with 0.235 μm per optical section. Images were analyzed by ImageJ, and maximum Z-projection was performed to stack images from different focal planes. Spine morphological analysis was performed as described (69). Four representative neurons from each coverslip were selected for analysis, and three basal or secondary apical dendritic spines were subjected to analysis in each selected neuron. ImageJ was used to measure spine head diameter and the length of the dendrites. Spines were categorized into mature and immature spine using head-to-neck ratio as described. Head-to-neck ratio of >1.4 was defined as mature, mushroom spine (27, 28).
Statistical analyses
All data were analyzed by GraphPad Prism (GraphPad Software). Unless specified, data are means ± SEM. Unpaired Student’s t test was used when comparing drug treatment with DMSO treatment within the same cell type. Multiple comparisons between groups were performed by one-way ANOVA. Bonferroni’s test was used for post hoc comparison with control group. Tukey’s tests were used for post hoc comparisons with control and PS1WT groups. P < 0.05 was considered statistically significant.
Supplementary Material
Fig. S1. Mutant PS1 or PS1 knockout does not grossly affect STIM1 or ORAI1 abundance in SH-SY5Y cells.
Fig. S2. Impaired CCE in mutant FAD-PS1–expressing cells or PS1 knockout cells.
Fig. S3. PS1 interacts with STIM1 but not with CFTR.
Fig. S4. In situ PLA demonstrates that endogenous PS1 and STIM1 interact.
Fig. S5. FAD-linked mutant PS1 impairs STIM1 puncta formation.
Fig. S6. Mutant PS1 attenuates the STIM1-ORAI1 interaction in fibroblasts from FAD patients.
Fig. S7. In vitro fluorogenic γ-secretase cleavage assay.
Table S1. The effects of PS1 on CCE.
Table S2. The amount of PLA red fluorescent dots in SH-SY5Y cells.
Table S3. The effects of PS1 on the STIM1-ORAI1 interaction after ER Ca2+ depletion by thapsigargin.
Table S4. The effects of PS1 on spine stability in isolated rat hippocampal neurons.
Acknowledgments
We thank the Imaging Core Facility of the LKS Faculty of Medicine for imaging support and H. Zhi (Biostatistics and Clinical Methodology Research Unit, University of Hong Kong) for statistical analysis advice.
Funding: This work was supported by General Research Fund of Hong Kong HKU763811M, National Natural Science Foundation of China grant 31200883, and Seed Funding for Basic Sciences of the University of Hong Kong 201101159010 (to K.-H.C.) and by NIH grant MH059937 (to J.K.F.). W.-H.C. was supported by an award from the Undergraduate Research Fellowship Programme of the University of Hong Kong.
Footnotes
Author contributions: K.-H.C. and J.K.F. conceived the research. B.C.-K.T. and K.-H.C. designed the research and analyzed data. B.C.-K.T., C.S.-K.L., and W.-H.C. performed experiments. K.-O.L. assisted in primary neuron culture and spine morphogenesis studies. B.C.-K.T., J.K.F., and K.-H.C. wrote the article.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: PS1KO or STIM1KO SH-SY5Y cells are available from K.-H.C. with a material transfer agreement with the Technology Transfer Office of the University of Hong Kong.
REFERENCES AND NOTES
- 1.Hardy J. A hundred years of Alzheimer’s disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 2.Hutton M, Hardy J. The presenilins and Alzheimer’s disease. Hum Mol Genet. 1997;6:1639–1646. doi: 10.1093/hmg/6.10.1639. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. The genetics and molecular pathology of Alzheimer’s disease: Roles of amyloid and the presenilins. Neurol Clin. 2000;18:903–922. doi: 10.1016/s0733-8619(05)70232-2. [DOI] [PubMed] [Google Scholar]
- 4.Chakroborty S, Stutzmann GE. Calcium channelopathies and Alzheimer’s disease: Insight into therapeutic success and failures. Eur J Pharmacol. 2014;739:83–95. doi: 10.1016/j.ejphar.2013.11.012. [DOI] [PubMed] [Google Scholar]
- 5.Mullane K, Williams M. Alzheimer’s therapeutics: Continued clinical failures question the validity of the amyloid hypothesis-but what lies beyond? Biochem Pharmacol. 2013;85:289–305. doi: 10.1016/j.bcp.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 6.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 8.Smith IF, Green KN, LaFerla FM. Calcium dysregulation in Alzheimer’s disease: Recent advances gained from genetically modified animals. Cell Calcium. 2005;38:427–437. doi: 10.1016/j.ceca.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 9.Berridge MJ. Calcium hypothesis of Alzheimer’s disease. Pflugers Arch. 2010;459:441–449. doi: 10.1007/s00424-009-0736-1. [DOI] [PubMed] [Google Scholar]
- 10.Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol. 2000;149:793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 12.Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J Cell Biol. 2008;181:1107–1116. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee S-F, Hao Y-H, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheung KH, Mei L, Mak DOD, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease–linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung KH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Veliçelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 18.Akbari Y, Hitt BD, Murphy MP, Dagher NN, Tseng BP, Green KN, Golde TE, LaFerla FM. Presenilin regulates capacitative calcium entry dependently and independently of γ-secretase activity. Biochem Biophys Res Commun. 2004;322:1145–1152. doi: 10.1016/j.bbrc.2004.07.136. [DOI] [PubMed] [Google Scholar]
- 19.Bojarski L, Pomorski P, Szybinska A, Drab M, Skibinska-Kijek A, Gruszczynska-Biegala J, Kuznicki J. Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer’s disease. Biochim Biophys Acta. 2009;1793:1050–1057. doi: 10.1016/j.bbamcr.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Sun S, Zhang H, Liu J, Popugaeva E, Xu N-J, Feske S, White CL, III, Bezprozvanny I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron. 2014;82:79–93. doi: 10.1016/j.neuron.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 2007;104:9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navarro-Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ, Prakriya M. STIM1–Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 2008;586:5383–5401. doi: 10.1113/jphysiol.2008.162503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lleó M. Activity of γ-secretase on substrates other than APP. Curr Top Med Chem. 2008;8:9–16. doi: 10.2174/156802608783334060. [DOI] [PubMed] [Google Scholar]
- 24.Lee DH, Goldberg AL. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 25.Wang L-F, Zhang R, Xie X. Development of a high-throughput assay for screening of γ-secretase inhibitor with endogenous human, mouse or Drosophila γ-secretase. Molecules. 2009;14:3589–3599. doi: 10.3390/molecules14093589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Proteomic profiling of γ-secretase substrates and mapping of substrate requirements. PLOS Biol. 2008;6:e257. doi: 10.1371/journal.pbio.0060257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris KM. Calcium from internal stores modifies dendritic spine shape. Proc Natl Acad Sci USA. 1999;96:12213–12215. doi: 10.1073/pnas.96.22.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McNair K, Spike R, Guilding C, Prendergast GC, Stone TW, Cobb SR, Morris BJ. A role for RhoB in synaptic plasticity and the regulation of neuronal morphology. J Neurosci. 2010;30:3508–3517. doi: 10.1523/JNEUROSCI.5386-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bojarski L, Herms J, Kuznicki J. Calcium dysregulation in Alzheimer’s disease. Neurochem Int. 2008;52:621–633. doi: 10.1016/j.neuint.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Brunello L, Zampese E, Florean C, Pozzan T, Pizzo P, Fasolato C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J Cell Mol Med. 2009;13:3358–3369. doi: 10.1111/j.1582-4934.2009.00755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCombs JE, Gibson EA, Palmer AE. Using a genetically targeted sensor to investigate the role of presenilin-1 in ER Ca2+ levels and dynamics. Mol Biosyst. 2010;6:1640–1649. doi: 10.1039/c001975e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zatti G, Burgo A, Giacomello M, Barbiero L, Ghidoni R, Sinigaglia G, Florean C, Bagnoli S, Binetti G, Sorbi S, Pizzo P, Fasolato C. Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium. 2006;39:539–550. doi: 10.1016/j.ceca.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Shideman CR, Reinardy JL, Thayer SA. γ-Secretase activity modulates store-operated Ca2+ entry into rat sensory neurons. Neurosci Lett. 2009;451:124–128. doi: 10.1016/j.neulet.2008.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc Natl Acad Sci USA. 2003;100:6382–6387. doi: 10.1073/pnas.1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bentahir M, Nyabi O, Verhamme J, Tolia A, Horré K, Wiltfang J, Esselmann H, De Strooper B. Presenilin clinical mutations can affect γ-secretase activity by different mechanisms. J Neurochem. 2006;96:732–742. doi: 10.1111/j.1471-4159.2005.03578.x. [DOI] [PubMed] [Google Scholar]
- 36.Fang B, Jia L, Jia J. Chinese Presenilin-1 V97L mutation enhanced Aβ42 levels in SH-SY5Y neuroblastoma cells. Neurosci Lett. 2006;406:33–37. doi: 10.1016/j.neulet.2006.06.072. [DOI] [PubMed] [Google Scholar]
- 37.Zheng L, Stathopulos PB, Li GY, Ikura M. Biophysical characterization of the EF-hand and SAM domain containing Ca2+ sensory region of STIM1 and STIM2. Biochem Biophys Res Commun. 2008;369:240–246. doi: 10.1016/j.bbrc.2007.12.129. [DOI] [PubMed] [Google Scholar]
- 38.De Strooper B, Simons M, Multhaup G, Van Leuven F, Beyreuther K, Dotti CG. Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J. 1995;14:4932–4938. doi: 10.1002/j.1460-2075.1995.tb00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skovronsky DM, Pijak DS, Doms RW, Lee VM. A distinct ER/IC γ-secretase competes with the proteasome for cleavage of APP. Biochemistry. 2000;39:810–817. doi: 10.1021/bi991728z. [DOI] [PubMed] [Google Scholar]
- 40.Hartmann T, Bieger SC, Brühl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Distinct sites of intracellular production for Alzheimer’s disease Aβ40/42 amyloid peptides. Nat Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- 41.Mayeux R, Honig L, Tang M-X, Manly J, Stern Y, Schupf N, Mehta P. Plasma Aβ40 and Aβ42 and Alzheimer’s disease: Relation to age, mortality, and risk. Neurology. 2003;61:1185–1190. doi: 10.1212/01.wnl.0000091890.32140.8f. [DOI] [PubMed] [Google Scholar]
- 42.Shuttleworth TJ, Thompson JL, Mignen O. STIM1 and the noncapacitative ARC channels. Cell Calcium. 2007;42:183–191. doi: 10.1016/j.ceca.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zbidi H, Jardin I, Woodard GE, Lopez JJ, Berna-Erro A, Salido GM, Rosado JA. STIM1 and STIM2 are located in the acidic Ca2+ stores and associates with Orai1 upon depletion of the acidic stores in human platelets. J Biol Chem. 2011;286:12257–12270. doi: 10.1074/jbc.M110.190694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.López JJ, Jardin I, Bobe R, Pariente JA, Enouf J, Salido GM, Rosado JA. STIM1 regulates acidic Ca2+ store refilling by interaction with SERCA3 in human platelets. Biochem Pharmacol. 2008;75:2157–2164. doi: 10.1016/j.bcp.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 45.Hooper R, Rothberg BS, Soboloff J. Neuronal STIMulation at rest. Sci Signal. 2014;7:pe18. doi: 10.1126/scisignal.2005556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Usachev YM, Thayer SA. Ca2+ influx in resting rat sensory neurones that regulates and is regulated by ryanodine-sensitive Ca2+ stores. J Physiol. 1999;519(Pt 1):115–130. doi: 10.1111/j.1469-7793.1999.0115o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Putney JW., Jr Capacitative calcium entry in the nervous system. Cell Calcium. 2003;34:339–344. doi: 10.1016/s0143-4160(03)00143-x. [DOI] [PubMed] [Google Scholar]
- 48.Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 49.Bhatt DH, Zhang S, Gan WB. Dendritic spine dynamics. Annu Rev Physiol. 2009;71:261–282. doi: 10.1146/annurev.physiol.010908.163140. [DOI] [PubMed] [Google Scholar]
- 50.Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Strooper B. Lessons from a failed γ-secretase Alzheimer trial. Cell. 2014;159:721–726. doi: 10.1016/j.cell.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 52.Comery TA, Martone RL, Aschmies S, Atchison KP, Diamantidis G, Gong X, Zhou H, Kreft AF, Pangalos MN, Sonnenberg-Reines J, Marquis KL. Acute γ-secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2005;25:8898–8902. doi: 10.1523/JNEUROSCI.2693-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Imbimbo BP, Giardina GAM. γ-Secretase inhibitors and modulators for the treatment of Alzheimer’s disease: Disappointments and hopes. Curr Top Med Chem. 2011;11:1555–1570. doi: 10.2174/156802611795860942. [DOI] [PubMed] [Google Scholar]
- 54.Yang T, Arslanova D, Gu Y, Augelli-Szafran C, Xia W. Quantification of gamma-secretase modulation differentiates inhibitor compound selectivity between two substrates Notch and amyloid precursor protein. Mol Brain. 2008;1:15. doi: 10.1186/1756-6606-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dreses-Werringloer U, Lambert J-C, Vingtdeux V, Zhao H, Vais H, Siebert A, Jain A, Koppel J, Rovelet-Lecrux A, Hannequin D, Pasquier F, Galimberti D, Scarpini E, Mann D, Lendon C, Campion D, Amouyel P, Davies P, Foskett JK, Campagne F, Marambaud P. A polymorphism in CALHM1 influences Ca2+ homeostasis, Aβ levels, and Alzheimer’s disease risk. Cell. 2008;133:1149–1161. doi: 10.1016/j.cell.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsukamoto A, Kaneko Y. Thapsigargin, a Ca2+-ATPase inhibitor, depletes the intracellular Ca2+ pool and induces apoptosis in human hepatoma cells. Cell Biol Int. 1993;17:969–970. doi: 10.1006/cbir.1993.1021. [DOI] [PubMed] [Google Scholar]
- 57.Keller JN, Guo Q, Holtsberg F, Bruce-Keller A, Mattson MP. Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J Neurosci. 1998;18:4439–4450. doi: 10.1523/JNEUROSCI.18-12-04439.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 59.Jia Q, Deng Y, Qing H. Potential therapeutic strategies for alzheimer’s disease targeting or beyond β-amyloid: Insights from clinical trials. BioMed Res Int. 2014;2014:837157. doi: 10.1155/2014/837157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Müller M, Cardenas C, Mei L, Cheung KH, Foskett JK. Constitutive cAMP response element binding protein (CREB) activation by Alzheimer’s disease presenilin- driven inositol trisphosphate receptor (InsP3R) Ca2+ signaling. Proc Natl Acad Sci USA. 2011;108:13293–13298. doi: 10.1073/pnas.1109297108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 62.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feige JN, Sage D, Wahli W, Desvergne B, Gelman L. PixFRET, an ImageJ plug-in for FRET calculation that can accommodate variations in spectral bleed-throughs. Microsc Res Tech. 2005;68:51–58. doi: 10.1002/jemt.20215. [DOI] [PubMed] [Google Scholar]
- 65.Gwozdz T, Dutko-Gwozdz J, Zarayskiy V, Peter K, Bolotina VM. How strict is the correlation between STIM1 and Orai1 expression, puncta formation, and ICRAC activation? Am J Physiol. 2008;295:C1133–C1140. doi: 10.1152/ajpcell.00306.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiao B, Coste B, Mathur J, Patapoutian A. Temperature-dependent STIM1 activation induces Ca2+ influx and modulates gene expression. Nat Chem Biol. 2011;7:351–358. doi: 10.1038/nchembio.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gruszczynska-Biegala J, Pomorski P, Wisniewska MB, Kuznicki J. Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLOS One. 2011;6:e19285. doi: 10.1371/journal.pone.0019285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Srikanth S, Jung H-J, Kim K-D, Souda P, Whitelegge J, Gwack Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat Cell Biol. 2010;12:436–446. doi: 10.1038/ncb2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lai KO, Wong ASL, Cheung MC, Xu P, Liang Z, Lok KC, Xie H, Palko ME, Yung WH, Tessarollo L, Cheung ZH, Ip NY. TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory. Nat Neurosci. 2012;15:1506–1515. doi: 10.1038/nn.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31:115–130. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Mutant PS1 or PS1 knockout does not grossly affect STIM1 or ORAI1 abundance in SH-SY5Y cells.
Fig. S2. Impaired CCE in mutant FAD-PS1–expressing cells or PS1 knockout cells.
Fig. S3. PS1 interacts with STIM1 but not with CFTR.
Fig. S4. In situ PLA demonstrates that endogenous PS1 and STIM1 interact.
Fig. S5. FAD-linked mutant PS1 impairs STIM1 puncta formation.
Fig. S6. Mutant PS1 attenuates the STIM1-ORAI1 interaction in fibroblasts from FAD patients.
Fig. S7. In vitro fluorogenic γ-secretase cleavage assay.
Table S1. The effects of PS1 on CCE.
Table S2. The amount of PLA red fluorescent dots in SH-SY5Y cells.
Table S3. The effects of PS1 on the STIM1-ORAI1 interaction after ER Ca2+ depletion by thapsigargin.
Table S4. The effects of PS1 on spine stability in isolated rat hippocampal neurons.