

Abstract
Inhibition of specific protein-protein interactions is attractive for a range of therapeutic applications, but the large and irregularly shaped contact surfaces involved in many such interactions make it challenging to design synthetic antagonists. Here, we describe the development of backbone-modified peptides containing both α- and β-amino acid residues (“α/β-peptides”) that target the receptor-binding surface of vascular endothelial growth factor (VEGF). Our approach is based on the Z-domain, which adopts a three-helix bundle tertiary structure. We show how a two-helix “mini-Z-domain” can be modified to contain β and other non-proteinogenic residues while retaining the target-binding epitope using iterative non-natural residue incorporation. The resulting α/β-peptides are less susceptible to proteolysis than is their parent α-peptide, and some of these α/β-peptides match the full-length Z-domain in terms of affinity for receptor-recognition surfaces on the VEGF homodimer.
Keywords: α/β-peptides, foldamers, vascular endothelial growth factor, inhibitors
TOC image
α/β-Peptides with a helix-loop-helix tertiary structure that target the receptor-binding surface of VEGF were developed via an iterative replacement strategy from a starting peptide containing exclusively proteinogenic residues. This strategy might lead to α/β-peptides that adopt other tertiary structures.
Introduction
Protein-protein interactions represent attractive targets for modulation with designed agents, for both diagnostic applications and the treatment of disease.[1] Peptides of medium length can often be developed to bind target proteins with high affinity and selectivity, but if composition is limited to proteinogenic amino acid residues, then these peptides are highly susceptible to degradation by proteases.[2] Rapid proteolysis severely limits the applications of conventional peptides in biological systems. Modifying the peptide backbone, e.g., by replacing an α-amino acid residue with a β-amino acid residue, protects nearby amide groups from proteolysis;[2] however, α→β substitutions can also alter folding and recognition properties relative to the original all-α polypeptide.[3, 4] In recent years it has been discovered that α/β combinations containing 25–33% β residues distributed along the backbone can adopt helical conformations that closely mimic the α-helix found in natural proteins.[3, 5–8] Helical α/β-peptides of this type can mimic the recognition properties of a natural α-helix, thereby inhibiting protein-protein interactions in which one partner contributes a single α-helix to the interface, while avoiding rapid destruction by proteases.[5–10]
For many protein-protein interfaces of biomedical interest, each partner contributes a surface that is large and topologically irregular.[11] Such surfaces often cannot be effectively engaged by a single α-helix or α-helix-mimic, which would be too narrow to provide sufficient contact area for strong binding. We speculated that developing ligands for broad recognition surfaces on target proteins would require advancing the α/β-peptide strategy beyond mimicry of a lone α-helix. Vascular endothelial growth factor (VEGF) was selected as a testbed for this effort because of the extensive structural data available for VEGF bound to fragments of receptor proteins, to antibodies that target the receptor-recognition surface or to other polypeptide ligands.[12–15] The extent of this structural database reflects the biomedical significance of disrupting VEGF-receptor interactions.
VEGF occurs as a homodimer that binds primarily to two cell-surface receptor tyrosine kinases, VEGFR1 and VEGFR2, to mediate angiogenesis.[16] (In this report, “VEGF” refers to the VEGF-A isoform.) Binding of VEGF to its receptors leads to receptor dimerization and subsequent downstream signaling that promotes the survival, proliferation and migration of endothelial cells. VEGF signaling through VEGFR2 is thought to be the primary promotor of angiogenesis, while VEGFR1 is thought to regulate angiogenesis by sequestering VEGF and other growth factors, although the precise roles of VEGFR1 remain unclear. Antagonists of the VEGF-VEGFR interaction are currently used to treat various forms of cancer and wet macular degeneration.[17] The receptor-recognition site on VEGF is large and topologically irregular, with >800 Å2 of surface on VEGF buried upon binding to a single receptor (Figure 1A).[12] The large and relatively shallow surfaces that form the VEGF-VEGFR interface have made it difficult to develop small molecules to target this interaction.
Figure 1.
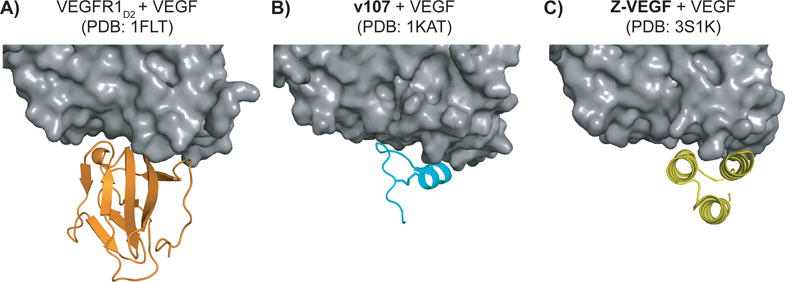
High-resolution structures of VEGF (grey) in complex with A) domain 2 of VEGFR1 (orange), B) phage-derived peptide v107 (teal), and C) Z-domain analogue Z-VEGF (yellow).
Our initial efforts to develop α/β-peptides that bind to the receptor-recognition sites on the VEGF homodimer were based on a family of disulfide-crosslinked 19-mer peptides discovered by Fairbrother et al. via phage-display.[18] An NMR-derived structure of the peptide designated v107 bound to the VEGF homodimer revealed that the 19-mer adopts a specific but largely non-helical conformation (Figure 1B).[15] We found that implementing α→β replacements in this scaffold could enhance resistance to proteolysis, but that these replacements caused an erosion in affinity for VEGF.[19] These observations led us to seek VEGF-binding peptides with higher α-helical content.
In 2011, Fedorova et al. reported a 59-residue peptide that adopts a three-helix-bundle tertiary structure and binds to the receptor-recognition sites on the VEGF homodimer (Z-VEGF, Figure 1C and Figure 2).[14] This engineered ligand was based on the “Z-domain”; the parent Z-domain peptide binds to a specific site on IgG via a broad surface formed by α-helices 1 and 2 of the three-helix bundle.[20] The Z-domain scaffold has become a popular basis for developing ligands for a variety of target proteins via screening of phage-display libraries in which residues on the composite helix 1+helix 2 surface are varied.[21, 22] In recent human clinical trials Z-domain-based imaging agents that target HER2 have been evaluated for diagnosis of breast cancer.[23]
Figure 2.
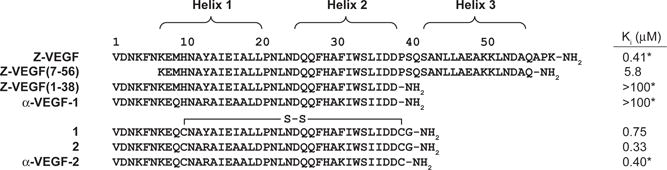
Primary sequences of two-helix α-peptides derived from Z-VEGF derivatives, along with associated Ki values, as determined by VEGF165 competition FP assay. Each cysteine is engaged in an intramolecular disulfide bond. Ki values marked with a * are values previously reported for these compounds.[26] See Figure S1 for derivation of Ki values and a discussion of experimental uncertainty in FP measurements.
We saw Z-VEGF as a potential starting point for developing an α/β-peptide that could block VEGF-mediated signaling and avoid rapid proteolysis. Specifically, we envisioned following the lead of Wells et al.[24, 25] to truncate the Z-domain to a disulfide-crosslinked helix-loop-helix tertiary structure that retained the binding surface that had been optimized via phage-display, and then incorporating unnatural residues into the distal sides of the helices (relative to the VEGF-recognition epitope). We have already described the final outcome of this effort, in which both β-amino acid residues and Aib residues were employed to generate a 39-mer α/β-peptide that retained high affinity for VEGF but displayed much less susceptibility to proteolysis than did Z-VEGF, the 59-residue parent Z-domain.[26] The resulting α/β scaffold proved amenable to the grafting of contact α-amino acid residues specific for other protein targets, to generate new α/β-peptides that retained the binding specificities of their parent Z-domain analogues.
Here we describe the design path that led to the optimized anti-VEGF α/β-peptides recently described.[26] We document an iterative strategy for identifying sites that tolerate α→β residue replacement without compromising affinity for VEGF. This approach is distinct from prior efforts to transform α-helical α-peptides into α/β-peptides with comparable conformations and binding properties, which have focused primarily on regular patterns of α→β-amino acid residue substitution implemented throughout the entire sequence. Those prior efforts were limited to mimicry of an isolated secondary structure (a single helix), while the approach that delivered high-affinity VEGF-binding α/β-peptides had to accommodate a specific tertiary structure. The challenges of implementing backbone modifications in the context of a discrete tertiary structure are highlighted by a recently described D-α-amino acid scan of a disulfide-crosslinked, two-helix Z-domain derivative;[27] single L→D residue replacements at most of the sites led to a dramatic decline in affinity for the target protein (IgG). We speculate that the principles illustrated by the iterative development process documented here may prove to be useful for generating target-specific α/β-peptide ligands based on other tertiary structures.
Results
Design of two-helix analogues of Z-VEGF
Z-VEGF (Figure 2) is a 59-residue three-helix peptide that was developed by Fedorova et al. by randomizing 9 residues on helices 1 and 2 of the Z-domain scaffold via phage display, followed by selection for binding to the VEGF8-109 dimer.[14] A co-crystal structure of Z-VEGF with VEGF8-109 shows that the engineered Z-domain adopts the expected three-helix bundle tertiary structure and engages the receptor-recognition sites on the VEGF dimer through a surface formed by helices 1 and 2 of Z-VEGF (Figure 1C).[14] We hypothesized that α/β-peptide analogues of Z-VEGF might function as VEGF antagonists. Furthermore, because only helix 1 and helix 2 of Z-VEGF make contact with VEGF in the co-crystal structure, we sought to generate a two-helix α/β-peptide analogue that would mimic the helix 1-loop-helix 2 motif within the Z-VEGF tertiary structure.
Only residues 7–58 of the 59-mer ligand appear in the Z-VEGF+VEGF8-109 co-crystal structure.[14] For several other engineered Z-domains that bind to diverse protein partners, removal of N-terminal residues has a negligible effect on affinity for the target.[24, 25, 28] These precedents suggested to us that residues 1–6 of Z-VEGF might not be required for high-affinity binding to VEGF. We tested this hypothesis by evaluating the truncated α-peptide Z-VEGF(7-56) for binding to VEGF165. Affinity was assessed with a competition fluorescence polarization (FP) assay that we previously described, involving displacement of a fluorescently labelled peptide known to bind to the receptor-recognition surfaces on the VEGF165 dimer.[29] Full-length Z-VEGF displayed an equilibrium dissociation constant (Ki) of 0.41 μM in this assay, while Ki = 5.8 μM for Z-VEGF(7-56) (Figure 2, Figure S1). (See Figure S1 for derivation of Ki values and a discussion of experimental uncertainty in FP measurements.) The ~14-fold decline in affinity for VEGF165 resulting from these seemingly modest changes were surprising in light of reports that target affinity had been largely retained after more dramatic truncations of engineered Z-domains that engage other proteins.[24, 25, 28] The diminished affinity of Z-VEGF(7-56) suggests that the terminal residues of the 59-mer somehow encourage complexation with VEGF, despite being unresolved in the Z-VEGF+VEGF8-109 co-crystal structure. Efforts to design truncated and conformationally-constrained analogues of Z-VEGF(7-56) proved only modestly successful (Figure S2).
To ask whether residues 1–6 and 57–59 exert a large effect on the conformation of Z-VEGF, we compared the far-UV circular dichroism (CD) spectrum of this 59-mer with the data for Z-VEGF(7-56) (Figure 3). Both α-peptides displayed strong minima at ~208 nm and 222 nm, as expected for conformations that feature primarily α-helical secondary structure. The per-residue CD spectra of these two polypeptides overlay very closely, which suggests that residues 1–6 and 57–59 do not exert a strong influence on the extent of α-helicity in Z-VEGF.
Figure 3.
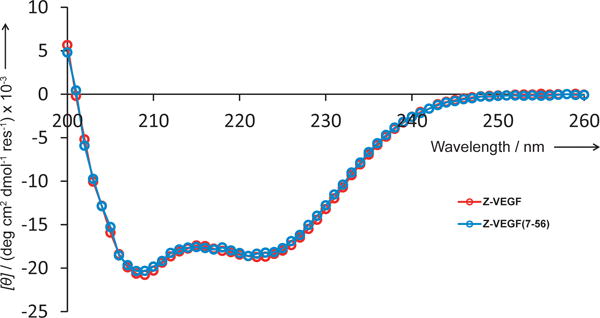
Far-UV CD spectrum for Z-VEGF (7-56) (blue) at 75 μM in PBS, pH 7.5. The previously reported spectrum for Z-VEGF (red) is shown for comparison.[26]
In light of the results obtained with peptide Z-VEGF(7-56), we designed two-helix analogues of Z-VEGF that retained residues 1–6 (Figure 2). As previously reported,[26] Z-VEGF(1-38) showed no detectable binding to VEGF. Since removal of helix 3 was expected to reduce conformational stability, as seen in prior truncation efforts based on Z-domain analogues,[24, 25, 28] we next incorporated five α-amino acid residue substitutions that had previously been identified by phage display[24, 28] as stabilizing for a helix-loop-helix tertiary fold. For our Z-VEGF-based design, structural alignment of Z-VEGF with a two-helix IgG-targeting Z-domain analogue[25] dictated these replacements to be Tyr13→Arg, Ile17→Ala, Leu20→Asp, Phe31→Lys and Leu35→Ile. These substitutions, along with a Met9→Gln substitution to avoid undesired oxidation, yielded α-VEGF-1 (Figure 2). The substitution at position 9 was based on a two-helix, disulfide-stabilized IgG-binding Z-domain analogue that contains Gln at the equivalent position.[24, 25] However, in contrast to prior Z-domain analogue truncation efforts,[24, 25, 28] we observed that the incorporation of these helix-stabilizing substitutions did not cause recovery of protein affinity in the absence of helix 3, as α-VEGF-1 did not show any affinity for VEGF in the competition FP assay (Figure 2).
In an attempt to further stabilize the intended helix 1-loop-helix 2 tertiary structure, we next incorporated a disulfide bond into the design of our two-helix Z-VEGF analogues (peptides 1 and 2, Figure 2), a strategy that has been previously successful for other two-helix Z-domain analogues.[25, 28] The residues for cysteine replacement were chosen based on structural alignment of the Z-VEGF crystal structure[14] with the solution NMR structure of a two-helix, disulfide-constrained Z-domain analogue targeting IgG.[25] This alignment showed that His10 and Pro39 lie at the positions in the helix1-helix 2 segment of Z-VEGF that correspond to the oxidized cysteine residues in the two-helix IgG-targeting Z-domain analogue. Our design included a glycine residue at the C-terminus as a spacer between the linker to the solid support and the last cysteine residue during solid-phase peptide synthesis. A C-terminal Gly is found also in other peptides described below. This feature was included in an effort to maximize synthesis yields.
We found that α-peptides 1 and 2, which are the disulfide-constrained analogues of Z-VEGF(1-38) and α-VEGF-1, respectively, bind to VEGF165 with affinity comparable to that of the Z-VEGF 59-mer (Figure 2). α-Peptide α-VEGF-2, which differs from 2 only in lacking the C-terminal Gly residue, bound to VEGF with affinity comparable to that of 2, according to the competition FP assay. This result indicates that the C-terminal Gly of 2 has a negligible effect in terms of binding to VEGF.
Iterative incorporation of β-amino acid residues and Aib residues based on scaffold peptide 2
α-Peptide 2 was used as the starting point for a systematic evaluation of α→β residue substitutions with the goal of generating α/β-peptides that bind tightly to VEGF but are minimally susceptible to proteolysis. We have previously shown that the substitution of α residues with the homologous β3 residues (bearing the same side chain, Figure 4B) in an ααβαααβ pattern can generate α/β-peptides that structurally and functionally mimic isolated α-helices that are recognized by specific partner proteins.[6, 9, 30] Based on these precedents, we asked whether this pattern of substitutions in a single helix of the helix-loop-helix tertiary structure would be tolerated in the present system. α/β-Peptide 3 contains three α→β3 substitutions in positions that are predicted not to make contact intermolecularly with VEGF or intramolecularly with helix 1 (Figure 4); these substitution sites should occur on the solvent-exposed face of the peptide when bound to VEGF. This α/β-peptide, however, showed very little affinity for VEGF according to the FP assay (Figure 4, Figure S1).
Figure 4.
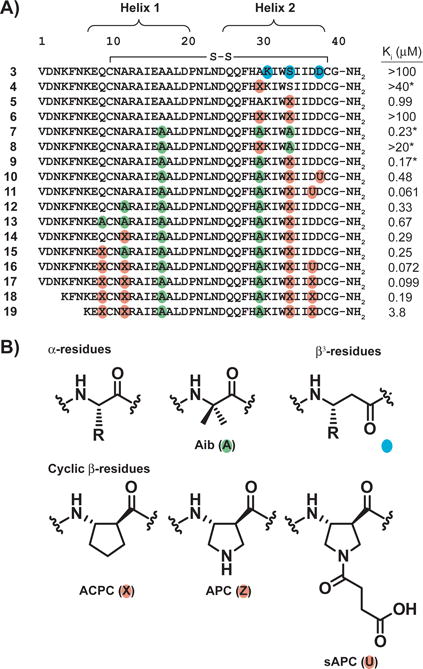
A) Primary sequences of α/β-peptide derivatives of 2 and α-VEGF-2, along with associated Ki values, as determined by VEGF165 competition fluorescence polarization (FP) assay. Non-natural residues are indicated by colored circles. Each cysteine is engaged in an intramolecular disulfide bond. Ki values marked with a * are values previously reported for these compounds.[26] B) Structures of a generic α residue, the Aib residue (green), a generic β3 residue (teal), and the cyclic β residues ACPC, APC, and sAPC (orange). See Figure S1 for derivation of Ki values and a discussion of experimental uncertainty in FP measurements.
Incorporation of β3-amino acid residues into helical structures can sometimes lead to a decreased affinity for target proteins, likely due to the increased backbone flexibility of a β3 residue relative to an α residue (a β3 residue contains one more bond with a low torsional barrier relative to an α residue).[5, 31] In such cases, the incorporation of cyclically-constrained β-amino acid residues such as ACPC, APC or sAPC (Figure 4B) sometimes increases affinity,[10, 32] presumably by restricting the Cα-Cβ torsion. We therefore implemented single cyclic β residue substitutions at solvent-exposed positions of helix 2, replacing uncharged Ala30 or Ser34 with ACPC, to give α/β-peptides 4 and 5, respectively. α/β-Peptide 4 showed very low affinity for VEGF by FP (Ki > 40 μM), while 5 showed significant affinity (Ki = 0.99 μM). α/β-Peptide 6, which contains both the Ala30→ACPC and Ser34→ACPC substitutions, showed very weak binding to VEGF (Ki > 100 μM). These data suggest that some solvent-exposed positions in helix 2, such as position 34, can tolerate α→cyclic β replacement but others, such as position 30, cannot.
Our next designs explored the use of Aib residues rather than β residues as non-proteinogenic substitutions. Aib has a strong helical propensity,[33] and this residue can hinder the action of proteases at nearby peptide bonds,[34] although less effectively than α→β substitutions.[31] Peptide 7, which contains three Aib residues, bound to VEGF with high affinity (Ki = 0.23 μM), possibly because Aib residues at these positions increase the helicity of the two-helix peptide. α/β-Peptide 8, which contains the Ala30→ACPC substitution in addition to two Aib substitutions, showed very low affinity for VEGF, mirroring the results from 4 and 6, which also place ACPC at position 30. However, α/β-peptide 9, which contains the Ser34→ACPC substitution in addition to two Aib residue replacements, showed high affinity for VEGF (Ki = 0.17 μM).
We used 9 as a starting point for additional replacements into the C-terminal end of helix 2 and into helix 1. Peptides 10 and 11 contain an Asp→sAPC substitution, which preserves the acidic side chain, in addition to those substitutions in 9. Both showed substantial affinity for VEGF, but 11 bound more tightly (Ki = 0.061 μM for 11 versus 0.48 μM for 10), indicating that the Asp37→sAPC substitution is more favorable than Asp38→sAPC. In a parallel set of studies based on 9 we examined non-natural substitutions into helix 1. Peptide 12 has an Aib substitution at position 12, and 13 has Aib substitutions at positions 9 and 12; both bound VEGF with affinity comparable to that of 9. α/β-Peptides 14 and 15 were prepared to evaluate α→cyclic β residue substitution at positions 9 or 12. Both of these sites proved amenable to these modifications, and α/β-peptide 16, containing ACPC at both sites and the Asp37→sAPC substitution identified above, bound to VEGF with high affinity (Ki = 0.072 μM in the competition FP assay). The building block for the sAPC residue requires multistep organic synthesis, but Fmoc-(S,S)-ACPC is commercially available; therefore, we examined 17, which contains ACPC at position 37. α/β-Peptide 17 showed affinity for VEGF that was comparable to affinity displayed by 16.
We next sought to evaluate which residues at the N-terminus of α/β-peptide 17 were necessary for tight binding to VEGF. An analogue of 17 lacking residues 1–3 bound tightly to VEGF (α/β-peptide 18, Ki = 0.19 μM), but a significant drop in affinity was observed when residues 1–6 were removed (α/β-peptide 19, Ki = 3.8 μM).
Protease stability assays
α/β-Peptide 16 incorporates four α→cyclic β and two Aib substitutions, each of which may reduce this oligomer’s susceptibility to protease degradation.[5, 10, 34] To evaluate this susceptibility, we incubated 40 μM of 16 with 10 μg/mL proteinase K, a promiscuous and aggressive protease,[35] and quantified the amount of intact oligomer remaining by high performance liquid chromatography (HPLC) (Figure 5, Figure S3). A parallel evaluation was conducted with α-peptide α-VEGF-2 to provide a basis for comparison.
Figure 5.
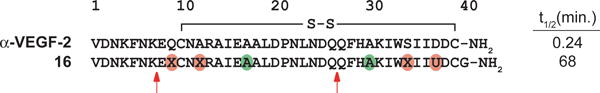
Susceptibility of 40 μM α-peptide α-VEGF-2 and α/β-peptide 16 to 10 μg/mL proteinase K in TBS, pH 7.5 with 5% DMSO at room temperature. Red arrows indicate early cleavage sites for α/β-peptide 16, as assessed by MALDI-TOF MS of the reaction mixture after 64 minutes.
α-VEGF-2 was rapidly degraded by proteinase K and displayed a half-life of 0.24 minutes in this assay. In contrast, α/β-peptide 16 displayed a half-life of 68 minutes under the same conditions (>280-fold increase in half-life relative to α-VEGF-2), which is consistent with our prediction that multiple α→cyclic β and Aib substitutions scattered across the sequence should discourage protease action. We examined several quenched aliquots from the reaction of 16 with proteinase K by matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS) to identify cleavage sites based on observation of masses that correspond to hypothetical products of peptide bond hydrolysis (Figure S4). Although we were not able to assign every fragment in the MALDI-TOF spectra, we identified masses that correspond to products resulting from hydrolysis of the Gln26-Gln27 amide bond and the Lys7-Glu8 amide bond for the proteinase K digestion of 16 (Figure 6, red arrows). These results suggest that 16 may be most susceptible to proteolysis near the central loop in helix 2 (Gln26-Gln27) and near the N-terminus (Lys7-Glu8).
Figure 6.

Primary sequences of α/β-peptide derivatives of 16 and 17, along with associated Ki values, as determined by VEGF165 competition fluorescence polarization (FP) assay. Non-natural residues are indicated by colored circles, as depicted in Figure 4B. Each cysteine is engaged in an intramolecular disulfide bond. Ki values marked with a * are values previously reported for these compounds.[26] See Figure S1 for derivation of Ki values and a discussion of experimental uncertainty in FP measurements.
Final α/β-peptide designs
Closely related α/β-peptides 16 and 17 showed high affinity for VEGF, and 16 was significantly less susceptible to protease degradation than was the parent α-peptide, α-VEGF-2. However, the high number of hydrophobic ACPC and Aib residues incorporated into 17–19 may encourage non-specific interactions with other proteins when these peptides are introduced into biological systems. In an effort to reduce hydrophobicity, we prepared 20, the analogue of 17 that contains basic APC residues in place of hydrophobic ACPC residues at positions 12 and 34 (Figure 6). In addition, 20 differs from 17 in lacking the C-terminal Gly residue. The affinity of α/β-peptide 20 for VEGF proved to be indistinguishable from the affinity of 17 (Ki = 0.10 μM for 20).
Our final design goal was to incorporate additional non-proteinogenic residues into the two regions of 20 containing long stretches of α residues, the N-terminus and the central loop, to discourage protease action in these regions. Because there is no high-resolution structural information regarding possible interactions between the N-terminal segment and VEGF, we focused on α→β3 replacements, which preserve the original side chains. The molecule previously designated α/β-VEGF-1 contains two such replacements, at positions 2 and 5, and binds to VEGF with affinity indistinguishable from that of 16, 17 or 20 (Ki for α/β-VEGF-1 = 0.11 μM, Figure 6).[26] An analogue of α/β-VEGF-1 lacking residues 1–3, α/β-peptide 21, displayed comparable affinity for VEGF (Ki = 0.15 μM); this observation is consistent with the binding similarities for 17 and 18.
The region from Ala18 to His29 in α/β-VEGF-1 contains only proteinogenic α-amino acid residues, and the proteolysis experiment described above suggested that the region near Gln26-Gln27 may be particularly susceptible to proteolysis (Figure 5, Figure S4). Therefore, we examined α/β-peptides 22, α/β-VEGF-2, and 23, which are, respectively, the analogues of 20, α/β-VEGF-1, and 21, that contain the Gln26→Aib substitution (Figure 6).[34] The three new α/β-peptides each manifested substantial affinity for VEGF (Ki = 0.34–0.46 μM). These α/β-peptides bound slightly less tightly than did α/β-VEGF-1, but they were experimentally indistinguishable in terms of VEGF binding from the full-length α-peptide Z-VEGF and the parent two-helix α-peptide α-VEGF-2. As predicted, α/β-VEGF-2 manifested significantly reduced susceptibility to proteinase K digestion relative to other compounds evaluated in this series, with a half-life of 670 minutes, as previously reported.[26]
To determine whether the disulfide crosslink is important for binding of our α/β-peptides to VEGF we evaluated α/β-VEGF-1-LIN, the analogue of α/β-VEGF-1 that contains the native residue from Z-VEGF (His) in place of the Cys10 and lacks Cys39 (Figure 6). α/β-VEGF-1-LIN showed very little affinity for VEGF in the competition FP assay (Ki > 50 μM). The far UV-CD spectrum of α/β-VEGF-1-LIN in PBS, pH 7.5 showed a single minimum at ~206 nm, which is characteristic of helical secondary structure among α/β-peptides (Figure 7).[3, 36] However, the intensity of the normalized CD minimum for α/β-VEGF-1-LIN was weaker and blue-shifted relative to that of disulfide-crosslinked analogue α/β-VEGF-1 (Figure 7A). Changes in the CD spectrum of the type observed upon transformation of α/β-VEGF-1 to α/β-VEGF-1-LIN have previously been observed[3] for α/β-peptide modifications that appear to diminish the extent of helix formation. The weaker intensity of the CD minimum for α/β-VEGF-1-LIN relative to α/β-VEGF-1 suggests that the crosslink in α/β-VEGF-1 enhances the helicity of this α/β-peptide in aqueous solution. The intensity of the CD minimum for α/β-VEGF-1 in water changes little upon 10-fold dilution from 50 μM to 5 μM (Figure 7B), which suggests that the secondary structure of this α/β-peptide is not highly dependent on peptide aggregation.
Figure 7.
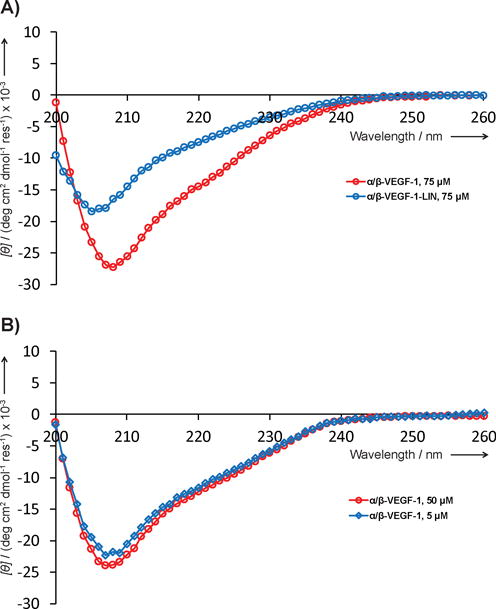
A) Far-UV CD spectrum for α/β-VEGF-1-LIN (blue) at 75 μM in PBS, pH 7.5. The previously reported spectrum for α/β-VEGF-1 (red) is shown for comparison.[26] B) Far-UV CD spectra for α/β-VEGF-1 at 50 μM (red) or 5 μM (blue) in water.
Discussion
The primary motivation for creating an α/β-peptide mimic of a protein-binding α-peptide is to retain high affinity for the target protein while significantly diminishing susceptibility to proteolysis. Most previous efforts of this type have begun with α-peptides that form a single, long α-helix, over most or all of the sequence, when bound to the target.[32] In several different systems, success has been achieved with α/β-peptides in which α→β replacement sites occur in simple patterns (αααβ or ααβαααβ) that provide 25–33% β residue content and disperse the β residues along the backbone.[5, 6, 9, 30, 31] α/β-Peptides with this β residue density typically display substantial resistance to proteolysis, particularly if cyclic β residues are employed. Furthermore, initial data suggest that such α/β-peptides may generally evade immunological recognition. Specifically, α/β-peptides containing multiple α→β replacements were not recognized by antibodies raised against the analogous α-peptides, and this type of α/β-peptide bound poorly to an MHC.[37] However, further studies are necessary to develop a more complete understanding of α/β-peptide interactions with the immune system.
Most α/β-peptides evaluated to date for inhibition of a protein-protein interaction mimic a single α-helix, but many protein-protein interfaces of interest are characterized by broader surfaces than can be effectively targeted by a single α-helix.[11] Complexes involving the receptor-recognition surfaces on the VEGF homodimer are representative of this type of expansive target. We sought to identify an α/β-peptide scaffold that would transcend an isolated helix.
The development described here of α/β-peptides that bind to the receptor-recognition surface of VEGF was inspired by the demonstration by Fedorova et al.[14] that the three-helix-bundle Z-domain scaffold could be used to generate α-peptides that target VEGF. The prospect of truncating from three to two helices[24, 25] provided further motivation because ~40-mers are significantly more tractable synthetically than are ~60-mers. In addition, a two-helix tertiary structure is more favorable than the three-helix tertiary structure in terms of maximizing the density of α→β substitutions. Multi-helix modules offer a greater challenge than does an isolated α-helix in terms of the placement of β residues or other non-proteinogenic residues, particularly if the new residues do not maintain the original side chain, as is the case with a cyclic β residue or Aib. Replacement sites in a multi-helix module must be chosen to avoid disrupting intermolecular contacts with the target protein, and also to avoid disrupting intramolecular contacts that are critical for folding. Starting from a full-length Z-domain derivative such as Z-VEGF, the most obvious positions for α→β substitution occur on the solvent-exposed side of helix 3. For helices 1 and 2, side chains from many of the solvent-exposed residues make contacts with VEGF, and replacements at these sites could therefore be deleterious to binding. For each of the three helices of Z-VEGF, α→β replacements at sites that pack against another helix in the native state could diminish tertiary structural stability. In contrast, a two-helix analogue presents a larger proportion of favorable opportunities for α→β substitution because both helices have a ‘back face’ that would be exposed to solvent in the VEGF-bound state.
The considerations outlined above led us to conclude that an iterative strategy would be necessary for identifying positions in a helix-loop-helix tertiary structure that could accommodate non-proteinogenic residues and for determining which type of non-proteinogenic residue could be accommodated (cyclic β vs. Aib). An iterative, site-by-site approach would contrast with the approach previously used to generate α/β-peptides that mimic isolated α-helices. This latter goal has been achieved by exploring small sets of α/β-peptides in which each registry of a repeating α/β pattern is evaluated, e.g., all four registries that are possible for the αααβ pattern, or all seven registries that are possible for the ααβαααβ pattern.[5, 6, 9, 30]
Having identified α-peptide 2 as a disulfide-crosslinked mini-Z-domain with high affinity for VEGF, we sought to identify sites that would tolerate β and/or Aib substitutions. Initial efforts focused on a relatively small number of positions identified that neither contact VEGF8-109 in the co-crystal structure with the full-length Z-domain nor are expected to be involved in intramolecular packing of the helices against one another. Affinity for VEGF was used to assess tolerance of substitution at the selected sites, only some of which proved amenable to substitution with a cyclic β and/or Aib residue. Once a set of candidate sites had been vetted in this way, the highest-affinity analogue was used as the starting point for evaluating a new set of potential substitution sites. At an intermediate stage in this iterative process (α/β-peptide 16, which contains four cyclic β residues and two Aib residues), we conducted a protease digestion assay to identify the regions of highest susceptibility to cleavage. As expected, proteolytic susceptibility was most pronounced in segments comprising multiple consecutive α residues. Placement of additional β and Aib residues in these segments was accomplished in a manner that largely preserved affinity for VEGF while enhancing resistance to degradation. This iterative development process ultimately provided α/β-peptides α/β-VEGF-1 and α/β-VEGF-2, high-affinity ligands for VEGF that are substantially less susceptible to proteinase K degradation relative to the parent α-peptide (α-VEGF-2) and even relative to the original three-helix α-peptide Z-VEGF.[26]
Although the positions of the non-proteinogenic residues for the iterative non-natural residue incorporation were selected based on the Z-VEGF+VEGF8-109 co-crystal structure,[14] our findings show that crystallographic information does not necessarily provide all of the insight a designer might require for rational design of high-affinity protein-binding peptides. For example, the lack of electron density for Z-VEGF residues 1–6 in the co-crystal structure led us to postulate that this segment was not important for binding to VEGF, but experimental results invalidated this hypothesis. In addition, the Z-VEGF+VEGF8-109 co-crystal structure suggested that the Ala30 side chain does not make contact with VEGF or participate in tertiary packing; however, placement of cyclic β residue ACPC at this position caused a substantial loss in affinity for VEGF. In contrast, Aib was tolerated at position 30. The apparent requirement for an α residue at this site was not readily deduced from the crystallographic data.
The strategy that culminated in development of α/β-VEGF-1 and α/β-VEGF-2, protease-resistant oligomers with an unnatural backbone that bind tightly to a protein target of high biomedical significance, is important because these α/β-peptides achieve an unprecedented molecular engineering goal. Previously described α/β-peptide antagonists have mimicked isolated α-helices, but the examples described here recapitulate a more complex folding pattern. Identifying replacement sites among the original proteinogenic residues has required us to accommodate not only points of contact with the target surface on VEGF but also intramolecular packing interactions. The difficulty of this engineering challenge is highlighted by a recently reported D-residue scan of a 33-residue peptide derived from the Z-domain.[27] This fundamental study revealed that substitution of a single L-α residue with its enantiomer at the majority of positions caused a profound decline in affinity for the target protein (an IgG fragment). The sites most tolerant of L→D residue replacement were mainly near the termini. No diastereomeric peptides containing more than one L→D replacement were reported, but the available data suggest that most multiple-replacement patterns would cause a substantial loss of affinity for the target and of the native helix-loop-helix tertiary structure. In light of these findings for heterochiral α-peptide analogues of Z-domain-derived motif, the ability we have demonstrated to generate helix-loop-helix analogues that contain multiple β-amino acid and Aib residues distributed across the sequence, and that retain high affinity for VEGF, for TNFα or for IgG,[26] demonstrates the potential power of this approach for engineering polypeptide binding agents that resist enzymatic degradation.
Although we were drawn to a Z-domain-derived tertiary structure as our starting point because of the copious α-helical secondary structure, the iterative replacement strategy illustrated here should, in principle, be applicable to poly-α-peptides with alternative tertiary structures. However, additional examples will be required in order to determine whether this strategy is truly general. It is encouraging that Horne et al.[38] have shown that non-natural residue substitutions can be tolerated within a tertiary structure that contains β-sheet in addition to α-helix secondary structure. Modules that have been used to develop binding agents for biomedically important proteins would be of particular interest for such efforts.[22]
Experimental Section
Materials
Fmoc-protected α-amino acids with compatible side-chain protecting groups and benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate (PyBOP) were purchased from Chem-Impex International and Novabiochem. Fmoc-protected β3-amino acids with compatible side-chain protecting groups were purchased from PepTech Corporation and Chem-Impex International. Fmoc-ACPC-OH was purchased from Chem-Impex International (15073). Fmoc-sAPC(OtBu)-OH was synthesized as previously described.[10b] Fmoc-APC(Boc)-OH was synthesized as previously described.[39]
Peptide synthesis and purification
α- and α/β-Peptides were synthesized on NovaPEG Rink Amide resin (Novabiochem, 855047) using microwave-assisted solid-phase conditions, as previously described,[26] or using a Symphony automated peptide synthesizer (Protein Technologies). During microwave synthesis, coupling reactions were carried out with a solution of Fmoc-protected amino acid activated with PyBOP, 0.1 M 1-hydoxybenzotriazole (HOBt), and N,N-diisopropylethylamine (DIEA) in 1-methyl-2-pyrrolidone (NMP). Fmoc deprotection reactions were carried using a solution of 5% piperazine, 0.1 M HOBt in N,N-dimethylformamide (DMF). After the final Fmoc deprotection reaction, α- and α/β-peptides were cleaved from the resin using a cleavage solution of 95% trifluoroacetic acid (TFA), 2.5% H2O, and 2.5% triisopropylsilane for oligomers with no cysteine residues and a cleavage solution of 94% TFA, 2.5% H2O, 2.5% ethanedithiol, and 1% triisopropylsilane for oligomers with cysteine residues. All peptides contain a free N-terminal amine. Cleavage solutions were concentrated under N2, and then peptides were precipitated with cold diethyl ether. For peptides with no disulfide bond, crude peptide mixtures were then subjected to purification by reverse-phase HPLC. For peptides with a disulfide bond, crude peptide mixtures were dissolved in a solution of dimethylsulfoxide (DMSO) (~10 mL DMSO for a 50 μmol resin loading) and ammonium hydroxide (~25 drops for a 50 μmol resin loading). These DMSO solutions were exposed to air to allow the peptides to oxidize over several days (4–7 days), followed by purification by reverse-phase HPLC. Alternatively, disulfide-containing peptides could also be purified as the bis-thiol immediately after cleavage (in the presence of reducing agent TCEP), subjected to oxidation using the above conditions, followed by repurification by HPLC.
Protein expression
VEGF165 was expressed and purified as previously reported.[29]
VEGF fluorescence polarization assays
Direct binding and competition fluorescence polarization assays were performed as previously described using the previously reported tracer, BODIPY-v114*ΔNT (Sequence = (BODIPYTMR)-X-CDIHV(Nle)WEWECFERL-NH2, where X = [2-(2-amino-ethoxy)-ethoxy]acetic acid, (Nle) = norleucine, and cysteines are linked in intramolecular disulfide, reported Kd = 21 nM).[19, 26, 29] To account for slight variations in the activity of protein solutions resulting from different VEGF165 expressions, the Kd for the tracer was experimentally determined for each new VEGF165 stock (using direct-binding mode),[29] and this Kd used to determine Ki values in the resulting competition FP assays using that VEGF165 expression. Briefly, DMSO stock solutions of potential inhibitors were prepared and their concentration determined by UV absorbance at 280 nm.[40] Competition FP assays were performed by adding 2 μL of potential inhibitor in DMSO to 48 μL of a solution containing VEGF165 and tracer (Final conditions = 40 nM VEGF165, 10 nM tracer in 50 mM NaCl, 16.2 mM Na2HPO4, 3.8 mM KH2PO4, 0.15 mM NaN3, 0.15 mM EDTA, 0.5 mg/mL Pluronic-F68, pH 7.5). Plates were covered from light and incubated at room temperature for 5–7 hours, and then read on either an Envision 2100 or a BioTek Synergy 2 microplate reader. Inhibition constants (Ki) were calculated from FP data using a competitive binding model[41] in GraphPad Prism 4.0. See Figure S1 for all FP data, fitted Ki values, and a discussion of Ki fitting and an estimation of experimental uncertainty.
Circular Dichroism (CD)
CD experiments were carried out on a Aviv Model 420 CD Spectrometer. Solutions of peptide were prepared in PBS, pH 7.5 or pure water at approximately 75 μM or 50 μM, with the exact concentration determined by UV-absorbance.[40] Spectra were collected in a 0.1 cm quartz cuvette, a 1 nm wavelength step size, a 10 second averaging time, and at 20 °C. For measurement at 5 μM, the 50 μM solution was diluted 10-fold in water and a spectrum was collected in a 1 cm quartz cuvette. Spectra were normalized to units of [θ] (deg cm2 dmol−1 res−1) × 10−3. Spectra for Z-VEGF and α/β-VEGF-1 were previously reported in the Supporting Information of Checco et al.,[26] but are displayed here for comparison.
Proteinase K stability assay
Proteinase K digestions were performed as previously described, with slight modifications.[26] Briefly, peptide stocks were prepared in TBS, pH 7.5 with 10% DMSO at 80 μM, as determined by UV absorbance.[40] A stock solution of 50 μg/mL proteinase K (Novagen 70663-4) was prepared in TBS, pH 7.5 (without DMSO). Peptide stocks were diluted and proteinase K stock solution was added to give final concentrations of 40 μM peptide, 10 μg/mL proteinase K, 5% DMSO in TBS, pH 7.5 and the reaction was allowed to proceed at room temperature. Reactions were run in duplicate. At each time point, 50 μL of the protease reaction was quenched with 100 μL of 50% acetonitrile in water with 1% TFA, and 125 μL of this quenched solution was injected onto analytical scale reverse-phase HPLC. The amount of peptide remaining, relative to the “0 minute” time point, was determined by integration of the peak at 220 nm in the resulting chromatogram corresponding to the intact peptide. Half-lives were determined by fitting the time course of degradation of intact peptide to an exponential decay model in GraphPad Prism 4.0.
Supplementary Material
Acknowledgments
This research was supported by NIGMS (GM056414, to S.H.G.). J.W.C. was supported in part by a Biotechnology Training Grant (NIH Grant T32 GM008349).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Arkin MR, Tang YY, Wells JA. Chem Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 2.a) Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Drug Discovery Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]; b) Steer DL, Lew RA, Perlmutter P, Smith AI, Aguilar MI. Curr Med Chem. 2002;9:811–822. doi: 10.2174/0929867024606759. [DOI] [PubMed] [Google Scholar]
- 3.Horne WS, Price JL, Gellman SH. Proc Natl Acad Sci U S A. 2008;105:9151–9156. doi: 10.1073/pnas.0801135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Sadowsky JD, Schmitt MA, Lee HS, Umezawa N, Wang SM, Tomita Y, Gellman SH. J Am Chem Soc. 2005;127:11966–11968. doi: 10.1021/ja053678t. [DOI] [PubMed] [Google Scholar]; b) Sadowsky JD, Fairlie WD, Hadley EB, Lee HS, Umezawa N, Nikolovska-Coleska Z, Wang SM, Huang DCS, Tomita Y, Gellman SH. J Am Chem Soc. 2007;129:139–154. doi: 10.1021/ja0662523. [DOI] [PubMed] [Google Scholar]
- 5.Horne WS, Johnson LM, Ketas TJ, Klasse PJ, Lu M, Moore JP, Gellman SH. Proc Natl Acad Sci U S A. 2009;106:14751–14756. doi: 10.1073/pnas.0902663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boersma MD, Haase HS, Peterson-Kaufman KJ, Lee EF, Clarke OB, Colman PM, Smith BJ, Horne WS, Fairlie WD, Gellman SH. J Am Chem Soc. 2012;134:315–323. doi: 10.1021/ja207148m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith BJ, Lee EF, Checco JW, Evangelista M, Gellman SH, Fairlie WD. ChemBioChem. 2013;14:1564–1572. doi: 10.1002/cbic.201300351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson LM, Mortenson DE, Yun HG, Horne WS, Ketas TJ, Lu M, Moore JP, Gellman SH. J Am Chem Soc. 2012;134:7317–7320. doi: 10.1021/ja302428d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horne WS, Boersma MD, Windsor MA, Gellman SH. Angew Chem Int Ed. 2008;47:2853–2856. doi: 10.1002/anie.200705315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Johnson LM, Horne WS, Gellman SH. J Am Chem Soc. 2011;133:10038–10041. doi: 10.1021/ja203358t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Peterson-Kaufman KJ, Haase HS, Boersma MD, Lee EF, Fairlie WD, Gellman SH. ACS Chem Biol. 2015;10:1667–1675. doi: 10.1021/acschembio.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Jones S, Thornton JM. Proc Natl Acad Sci U S A. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lo Conte L, Chothia C, Janin J. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]; c) Keskin O, Gursoy A, Ma B, Nussinov R. Chem Rev. 2008;108:1225–1244. doi: 10.1021/cr040409x. [DOI] [PubMed] [Google Scholar]
- 12.Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA, deVos AM. Cell. 1997;91:695–704. doi: 10.1016/s0092-8674(00)80456-0. [DOI] [PubMed] [Google Scholar]
- 13.a) Muller YA, Li B, Christinger HW, Wells JA, Cunningham BC, DeVos AM. Proc Natl Acad Sci U S A. 1997;94:7192–7197. doi: 10.1073/pnas.94.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fuh G, Wu P, Liang WC, Ultsch M, Lee CV, Moffat B, Wiesmann C. J Biol Chem. 2006;281:6625–6631. doi: 10.1074/jbc.M507783200. [DOI] [PubMed] [Google Scholar]; c) Chen Y, Wiesmann C, Fuh G, Li B, Christinger HW, McKay P, de Vos AM, Lowman HB. J Mol Biol. 1999;293:865–881. doi: 10.1006/jmbi.1999.3192. [DOI] [PubMed] [Google Scholar]; d) Bostrom J, Yu SF, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C, Fuh G. Science. 2009;323:1610–1614. doi: 10.1126/science.1165480. [DOI] [PubMed] [Google Scholar]; e) Fellouse FA, Wiesmann C, Sidhu SS. Proc Natl Acad Sci U S A. 2004;101:12467–12472. doi: 10.1073/pnas.0401786101. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, Koide S, Sidhu SS. J Mol Biol. 2007;373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]; g) Mandal K, Uppalapati M, Ault-Riche D, Kenney J, Lowitz J, Sidhu SS, Kent SBH. Proc Natl Acad Sci U S A. 2012;109:14779–14784. doi: 10.1073/pnas.1210483109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fedorova A, Zobel K, Gill HS, Ogasawara A, Flores JE, Tinianow JN, Vanderbilt AN, Wu P, Meng YG, Williams SP, Wiesmann C, Murray J, Marik J, Deshayes K. Chem Biol. 2011;18:839–845. doi: 10.1016/j.chembiol.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 15.Pan B, Li B, Russell SJ, Tom JYK, Cochran AG, Fairbrother WJ. J Mol Biol. 2002;316:769–787. doi: 10.1006/jmbi.2001.5370. [DOI] [PubMed] [Google Scholar]
- 16.a) Ferrara N, Gerber HP, LeCouter J. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]; b) Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 17.a) Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N. Nature. 1993;362(6423):841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]; b) Ferrara N, Hillan KJ, Gerber HP, Novotny W. Nat Rev Drug Discovery. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]; c) Carmeliet P, Jain RK. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Grothey A, Galanis E. Nat Rev Clin Oncol. 2009;6:507–518. doi: 10.1038/nrclinonc.2009.110. [DOI] [PubMed] [Google Scholar]
- 18.Fairbrother WJ, Christinger HW, Cochran AG, Fuh C, Keenan CJ, Quan C, Shriver SK, Tom JYK, Wells JA, Cunningham BC. Biochemistry. 1998;37:17754–17764. doi: 10.1021/bi981931e. [DOI] [PubMed] [Google Scholar]
- 19.Haase HS, Peterson-Kaufman KJ, Levengood SKL, Checco JW, Murphy WL, Gellman SH. J Am Chem Soc. 2012;134:7652–7655. doi: 10.1021/ja302469a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nilsson B, Moks T, Jansson B, Abrahmsen L, Elmblad A, Holmgren E, Henrichson C, Jones TA, Uhlen M. Protein Eng. 1987;1:107–113. doi: 10.1093/protein/1.2.107. [DOI] [PubMed] [Google Scholar]
- 21.a) Nord K, Gunneriusson E, Ringdahl J, Stahl S, Uhlen M, Nygren PA. Nat Biotechnol. 1997;15:772–777. doi: 10.1038/nbt0897-772. [DOI] [PubMed] [Google Scholar]; b) Lofblom J, Feldwisch J, Tolmachev V, Carlsson J, Stahl S, Frejd FY. FEBS Lett. 2010;584:2670–2680. doi: 10.1016/j.febslet.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 22.a) Binz HK, Amstutz P, Pluckthun A. Nat Biotechnol. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]; Skerra A. Curr Opin Biotechnol. 2007;18:295–304. doi: 10.1016/j.copbio.2007.04.010. [DOI] [PubMed] [Google Scholar]; b) Gronwall C, Stahl S. J Biotechnol. 2009;140:254–269. doi: 10.1016/j.jbiotec.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 23.a) Baum RP, Prasad V, Muller D, Schuchardt C, Orlova A, Wennborg A, Tolmachev V, Feldwisch J. J Nucl Med. 2010;51:892–897. doi: 10.2967/jnumed.109.073239. [DOI] [PubMed] [Google Scholar]; b) Sorensen J, Sandberg D, Sandstrom M, Wennborg A, Feldwisch J, Tolmachev V, Astrom G, Lubberink M, Garske-Roman U, Carlsson J, Lindman H. J Nucl Med. 2014;55:730–735. doi: 10.2967/jnumed.113.131243. [DOI] [PubMed] [Google Scholar]
- 24.Braisted AC, Wells JA. Proc Natl Acad Sci U S A. 1996;93:5688–5692. doi: 10.1073/pnas.93.12.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Starovasnik MA, Braisted AC, Wells JA. Proc Natl Acad Sci U S A. 1997;94:10080–10085. doi: 10.1073/pnas.94.19.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Checco JW, Kreitler DF, Thomas NC, Belair DG, Rettko NJ, Murphy WL, Forest KT, Gellman SH. Proc Natl Acad Sci U S A. 2015;112:4552–4557. doi: 10.1073/pnas.1420380112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simon MD, Maki Y, Vinogradov AA, Yu H, Lin Y, Kajihara Y, Pentelute BL. J Am Chem Soc. 2016;138:12099–12111. doi: 10.1021/jacs.6b03765. [DOI] [PubMed] [Google Scholar]
- 28.Webster JM, Zhang R, Gambhir SS, Cheng Z, Syud FA. ChemBioChem. 2009;10:1293–1296. doi: 10.1002/cbic.200900062. [DOI] [PubMed] [Google Scholar]
- 29.Peterson KJ, Sadowsky JD, Scheef EA, Pal S, Kourentzi KD, Willson RC, Bresnick EH, Sheibani N, Gellman SH. Anal Biochem. 2008;378:8–14. doi: 10.1016/j.ab.2008.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheloha RW, Maeda A, Dean T, Gardella TJ, Gellman SH. Nat Biotechnol. 2014;3:653–655. doi: 10.1038/nbt.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson LM, Barrick S, Hager MV, McFedries A, Homan EA, Rabaglia ME, Keller MP, Attie AD, Saghatelian A, Bisello A, Gellman SH. J Am Chem Soc. 2014;136:12848–12851. doi: 10.1021/ja507168t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson LM, Gellman SH. Methods Enzymol. 2013;523:407–429. doi: 10.1016/B978-0-12-394292-0.00019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karle IL, Balaram P. Biochemistry. 1990;29:6747–6756. doi: 10.1021/bi00481a001. [DOI] [PubMed] [Google Scholar]
- 34.Yamaguchi H, Kodama H, Osada S, Kato F, Jelokhani-Niaraki M, Kondo M. Biosci Biotechnol Biochem. 2003;67:2269–2272. doi: 10.1271/bbb.67.2269. [DOI] [PubMed] [Google Scholar]
- 35.Ebeling W, Hennrich N, Klockow M, Metz H, Orth HD, Lang H. Eur J Biochem. 1974;47:91–97. doi: 10.1111/j.1432-1033.1974.tb03671.x. [DOI] [PubMed] [Google Scholar]
- 36.Horne WS, Price JL, Keck JL, Gellman SH. J Am Chem Soc. 2007;129:4178–4180. doi: 10.1021/ja070396f. [DOI] [PubMed] [Google Scholar]
- 37.Cheloha RW, Sullivan JA, Wang T, Sand JM, Sidney J, Sette A, Cook ME, Suresh M, Gellman SH. ACS Chem Biol. 2015;10:844–854. doi: 10.1021/cb500888q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.a) Reinert ZE, Lengyel GA, Horne WS. J Am Chem Soc. 2013;135:12528–12531. doi: 10.1021/ja405422v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lengyel GA, Reinert ZE, Griffith BD, Horne WS. Org Biomol Chem. 2014;12:5375–5381. doi: 10.1039/c4ob00886c. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Reinert ZE, Horne WS. Org Biomol Chem. 2014;12:8796–8802. doi: 10.1039/c4ob01769b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee HS, LePlae PR, Porter EA, Gellman SH. J Org Chem. 2001;66:3597–3599. doi: 10.1021/jo001534l. [DOI] [PubMed] [Google Scholar]
- 40.Gill SC, Von Hippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 41.a) Roehrl MHA, Wang JY, Wagner G. Biochemistry. 2004;43:16056–16066. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]; b) Boersma MD, Sadowsky JD, Tomita YA, Gellman SH. Protein Sci. 2008;17:1232–1240. doi: 10.1110/ps.032896.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.