

Abstract
Antioxidant glutathione (GSH) plays an important role in the regulation of immunity. However, little is known about its effects on humoral immunity, especially its action on effector molecules like antibody and complement. Given that these molecules contain abundant disulfide bonds, we speculated that GSH might influence the action of these proteins via its thiol function. Using a model of a glomerular mesangial cell (MC) lysis induced by antibodies plus complement, we addressed this hypothesis. Exposure of rat MCs to anti-Thy-1 antibody plus complement or anti-MC rabbit serum caused a complement-dependent cell lysis, which was completely blocked by GSH. Moreover, GSH potently prevented the antibody-mediated agglutination of red blood cells and aggregation of antibody-sensitized microspheres. Further analysis revealed that GSH inhibited antibody binding to antigens and promoted the conversion of the antibodies to its reduced forms. GSH also potently inhibited the formation and deposition of C5b-9 in MCs and suppressed both the classic and alternative complement activation pathway. Lastly, GSH attenuated P38 activation, an oxidative sensitive kinase that partially mediated the antibody- and complement-dependent MC lysis. Depletion of GSH via inhibiting gamma-glutamylcysteine synthetase or xCT transporter augmented P38 activation and sensitized MCs to the cell lysis. Collectively, our results indicate that GSH protects cells from immunological cell damage via mechanisms involving inhibition of antibody binding to the antigens, suppression of complement activation and augmentation of cellular defense mechanism. Our study provides novel mechanistic insights into the actions of GSH in the regulation of immune responses and suggests that GSH might be used to treat certain immune disorders.
Keywords: Glutathione, Antibody, Complement, Cell lysis, p38, Mesangial cells
Grapiical abstract
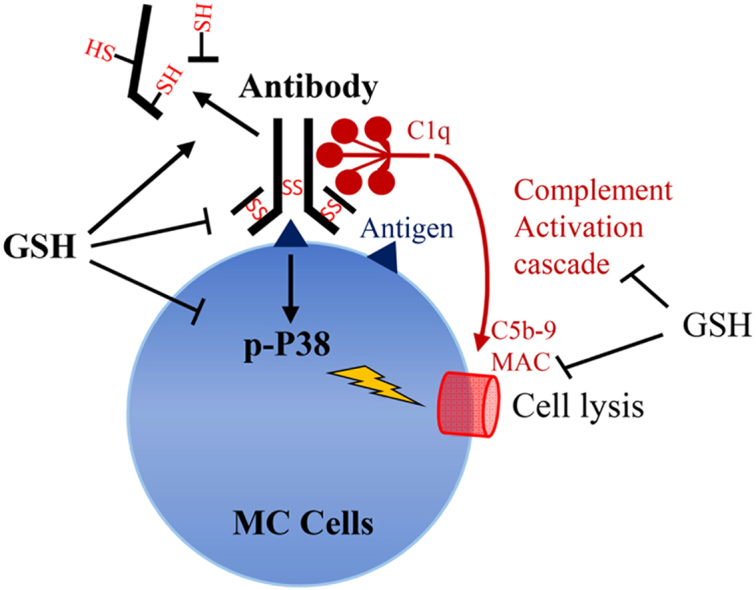
Highlights
-
•
Little information is available regarding the role of GSH on humoral immunity.
-
•
GSH inhibited antibody-triggered and complement-mediated immune responses.
-
•
GSH interfered with antibody binding to cell surface antigens via its thiol function.
-
•
GSH inhibited both the classic and alternative complement activation pathways.
-
•
GSH increased cell resistance to immunological injury via inhibition of P38.
1. Introduction
The tripeptide glutathione (γ-l-glutamyl-l-cysteinyl-glycine, GSH) is the most abundant low molecular weight thiol-containing peptide within the cells (intracellular concentration up to 10 ~ 15 mM). It exerts many biological functions. Most of them are attributable to its thiol function. GSH scavenges free radical and reactive oxygen species and facilitates the regeneration of other antioxidants. It is the major cellular defense mechanism against oxidative stress. Besides, GSH also serves as a key cofactor for many enzymes. Recently, GSH has been recognized to act not only as a major antioxidant within the cells, but also as a mediator of many other physiologic reactions, including metabolism of xenobiotics, thiol-disulfide exchange reactions, and cellular signaling. Dysfunction of GSH contributes to the initiation and development of many diseases [1].
GSH has been documented to regulate immune responses and has been shown to be critically implicated in the inflammatory diseases. GSH deficiency is one of the major pathogenic factors leading to the oxidative and immune diseases. Supplementation of GSH has been reported to be effective in prevention or alleviation of many immune-related diseases [1], [2]. GSH regulates several pivotal molecular events involved in immune responses. For examples, GSH affected the activation of NF-κB [3], [4], [5], [6], a transcription factor that plays a key role in the inflammation. It also potently suppressed inflammasome activation, a molecular event that links oxidative stress, inflammation and cell injury [7], [8], [9]. Furthermore, GSH modulated macrophage phagocytosis [10], [11] and T cell receptor-mediated signal transduction. It stimulated IL-2 production, promoted T cell proliferation, and enhanced T cell resistance to apoptosis [12], [13]. The mechanisms involved are thought to be related to its regulation on intracellular redox status and redox signaling.
Most of the studies regarding the regulatory effects of GSH on immunity have been limited to cell immunity. Our understanding about its roles on humoral immunity are still limited. In this aspect, GSH has been reported to selectively decrease IL-4-induced immunoglobulin (Ig) E and IgG4 production by blood mononuclear cells in vitro and decreased both IgE and IgG1 antibody production. In addition, in vivo administration of GSH precursor NAC decreased both IgE and IgG1 antibody responses to ovalbumin [14]. GSH has been reported to regulate IgE isotype switching via inhibition of NF-κB in cultured B cells [12]. NAC also significantly suppressed the specific antibody response via regulation of the functional molecules in B cells, as well as the production of IL-1 and IFN-γ [15]. Currently, few studies are available concerning the direct effect of GSH on soluble effectors of humoral immunity like antibodies and complement. Given that most of these proteins contain abundant disulfide bonds and that these bridges are indispensable for the maintenance of the normal structure and function [16], [17], we speculated that GSH might be a critically involved in the regulation of these molecules. This study was designed to address this hypothesis.
Using an in vitro model of antibody and complement-initiated cell lysis, we investigated the potential effects and mechanisms of GSH on antibody-initiated and complement-dependent cell injury. Here we present our results showing that GSH potently inhibited antibody- and complement-initiated immune responses through mechanisms involving its action on antibody, complement, and cell defense. Our study thus indicates that GSH potently regulates humoral immune responses and suggests that GSH has the potential to treat certain immune-mediated diseases.
2. Materials and methods
2.1. Materials
Calcein AM was obtained from Invitrogen (Tokyo, Japan). Dynabeads protein A and protein G Mag Sepharose were from Novex in life technologies (Life Technologies-Novex, Carlsbad, CA) and GE Healthcare (UK), respectively. HRP-conjugated anti-rabbit or mouse IgG and anti-β-actin antibodies were purchased from Cell Signaling, Inc. (Beverly, MA). Anti-Thy-1 monoclonal antibody 1–22-3 was kindly gifted by Dr. Kawachi (Institute of Nephrology, Niigata University). BSO was obtained from Cayman Chemical (Michigan, USA). Rhodamine-conjugated anti-rabbit IgG antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-mouse red blood cell (RBC) antibody was purchased from Abgent (San Diego, CA). The Easy-Titer IgG Assay Kit was purchased from Thermoscientific (Rockford, IL). The Enzyme immunoassay for assessment of complement functional activity kit was purchased from Wieslab (Malmö, Sweden). GSH and all other reagents were bought from Sigma (Tokyo, Japan).
2.2. Rabbit serum
Rabbit blood was collected from ear aricular artery of the Japanese White Strain Rabbit. The blood was allowed to clot for 30 min at room temperature. The serum was collected and stored at −80 °C until use. For inactivation of complement, the serum was heated at 56 °C for 30 min. All animal experiments were approved by the animal experiment committee of the University of Yamanashi and performed following the relevant guidelines and regulations.
2.3. Cells
Rat glomerular mesangial cells (MCs) were obtained from the outgrowth of the isolated glomeruli, as described previously [18], [19]. Cells were cultured in DMEM (Gibco-BRL, Gaithersburg, MD, USA) supplemented with 5% FBS for passage and expansion. For experiments, cells were cultured with DMEM containing 1% FBS with or without the presence of various stimuli.
2.4. Calcein-AM and propidium Iodide (PI) staining
Cell viability was determined by the Calcein-AM/PI live/dead staining as described previously [18]. Living cells exhibit positive staining for calcein-AM activity (green fluorescence), whereas dead cells stain positive for PI (red fluorescence).
2.5. Precipitation of membrane-bound antibodies
Immunoglobulins (Igs) bound to the cell membrane were precipitated using a mixture of protein A and G beads as reported previously [18]. MCs were reacted with rabbit serum or antibodies for the indicated period. After washing out the unbound free Igs with PBS for two times, total cellular proteins were extracted by suspending the cells in RIPA lysis buffer. Lysates were incubated on ice for 15 min with intermittent mixing and then centrifuged at 15,350×g for 10 min at 4 °C. The supernatant was recovered and determined for protein concentration using the Micro BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). Same amount of lysate in 300 μl RIPA was incubated with a mixture of protein A and G beads in a rotator at 4 °C overnight. The pellet was washed with 1 ml RIPA for three times and resuspended in 50 μl 2.5 X SDS sample buffer containing five mM DTT. After heat treatment at 95–100 °C for 5 min, supernatants were collected and loaded on a 10% gel for SDS-PAGE. The separated proteins were transferred to PVDF membrane and immunoblotted for cell bound-Igs.
2.6. Lactate dehydrogenase (LDH) release assay
Cell viability was evaluated by the release of LDH using an LDH cytotoxicity detection kit (Takara Bio, Inc., Otsu, Shiga, Japan). Briefly, cells in 96-well culture plate were exposed to various stimuli for the indicated time intervals. Culture medium was collected and added to wells at the volume of 30 μl. After reaction with the same volume of assay solution, the optical absorbance of the red color formed in the assay was measured at a wavelength of 490 nm with a UV–VIS spectrophotometer. LDH activity was calculated and expressed as a percentage of 100% whole release as made by exposing cells to Triton X-100.
2.7. Assessment of cell viability with WST reagent
Cells were seeded into 96-well culture plates and exposed to various stimuli in the presence or absence of GSH. WST reagent was added into each well 2 h before measurement of OD with a spectrometer at the wavelength of 450 nm [20].
2.8. Immunofluorescence staining
For immunofluorescence staining of membrane-bound IgG, mesangial cells were pretreated with 1% heat-treated rabbit serum in the presence or absence of the indicated concentration of GSH for 1 h. The cells were then rinsed with PBS, fixed with 3% paraformaldehyde, and stained with tetramethy1 rhodamine B isothiocyanate-conjugated anti-rabbit immunoglobulin G for 1 h. After washed with PBS, cells were observed under IF microscopy and positive IF signals in MCs were captured using a CCD camera attached to an Olympus BX50 microscope. For assessment of C9 deposition, MCs were treated with 10 μg/ml Thy-1 plus 10% human serum as a source of complement in the presence or absence of 5 mM GSH for 30 min. After washing and fixation as described above, cells were incubated with an anti-human C9 antibody at room temperature for 2 h, followed by a further step of washing and incubation with tetramethy1 rhodamine B isothiocyanate-conjugated secondary anti-rabbit immunoglobulin G antibody for an additional 1 h.
2.9. Red blood cell (RBC) agglutination assay
Mouse whole blood in a volume of about 300 μl was collected in a plastic tube containing 200-μl 0.5 M 10% EDTA and washed twice with 0.9% sodium chloride. A 1% suspension of these cells was prepared in the saline and added to 96-well plate that contains a serial dilution of anti-mouse RBC antibodies for 60 min. The formation of RBC agglutination was captured using a CCD camera attached to an Olympus BX50 microscope. For determination of complement-dependent RBC lysis, human serum at the final concentration of 5% was added and allowed to react for 4.5 h. The supernatants were collected, transferred to 96-well ELISA plates and evaluated for RBC lysis by measurement of hemoglobin absorbance at 405 nm.
2.10. Easy-Titer IgG Assay
Easy-Titer Mouse IgG Assay Kit (Pierce) was used for evaluation of the effect of GSH on microagglutination of antibody-sensitized microbeads. The assay was done following the manufacturer's protocol. Briefly, 20 μl of the anti-IgG-sensitized beads was pipetted into a 96-well plate, followed by the addition of the same volume of the diluted samples or standard IgG. After vigorous mixing on a plate mixer, 100 μl of blocking reagent was added. After a further step of mixing, the OD at 405 nm was measured using a UV/VIS spectrometer. The formation of microagglutination of the microbeads was photographed using a CCD camera attached to an Olympus BX50 microscope. The anti-thy-1 monoclonal antibody was used as a standard control.
2.11. Enzyme immunoassay for assessment of complement activity
Total complement activity of the alternative pathway in healthy human serum was measured using an assay kit from Wieslab (Comp AP330 kit, Euro Diagnostica, Malmö, Sweden) following the manufacturer's instruction. Briefly, human sera from four healthy donors or positive control provided by the manufacturer were pretreated with or without the indicated concentrations of GSH for 60 min. The samples were then diluted with a diluent that contained specific blocker to ensure that only the alternative pathway was activated. After reaction with specific activator coated in the wells of the microtiter plate at 37 °C for 1 h, the wells were washed, and a specific alkaline phosphatase-labeled antibody against C5b-9 was added. After a further washing step, alkaline phosphatase substrate solution was added. The color intensity, which was correlated with the amount of complement activation, was read with a UV/VIS spectrometer at 405 nM. The total complement activity relative to positive control was calculated.
2.12. Western blot analysis
Total cellular protein was extracted by suspending the cells in SDS lysis buffer (62.5 mM Tris-HCl, 2% SDS, 10% glycerol) together with a freshly added proteinase inhibitor cocktail (Nakalai Tesque, Kyoto, Japan). Lysates were incubated on ice for 15 min with intermittent mixing and then centrifuged at 15,350×g for 10 min at 4 °C. The supernatant was recovered and determined for protein concentration using the Micro BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). Western blot was performed by the enhanced chemiluminescence system [18], [19], [20]. Briefly, extracted cellular proteins were separated by polyvinylidene difluoride membranes. After blocking with 3% BSA or milk in PBS, the membranes were either probed directly with HRP-conjugated anti-rabbit or mouse IgG (Cell Signaling; Beverly, MA, USA) for 2 h at room temperature or 4 °C overnight or with the primary antibodies and then with the HRP-labeled second antibodies. After washing, the bands were visualized by the enhanced chemiluminescence system (Nacalai Tesque). The chemiluminescent signal is captured with a Fujifilm image LAS-1000 analyzer (Fujifilm, Tokyo, Japan) and quantified with the NIH Image J software (http://rsb.info.nih.gov/ij). β-actin was probed to confirm equal loading of proteins.
2.13. Statistical analysis
All data were presented as mean ±SE. Differences between two groups were tested by Student's t-test. When more than two groups were compared, one-way ANOVA with Dunnett test was used. P values less than 0.05 were considered significant.
3. Results
3.1. Induction of antibody-triggered and complement-dependent glomerular MC lysis by a rabbit serum
We have recently reported that incubation of MCs with a rabbit serum from the Japanese White Strain Rabbit caused a complement-dependent MC lysis due to the presence of several antibodies against MCs [18]. In this study, we have used the same cell model. First, we confirmed our previous findings that incubation of MCs with the rabbit serum indeed caused cell damage, as evidenced by the appearance of cells with disrupted cell bodies (Fig. 1A), increased LDH release (Fig. 1B) and positive staining of PI (Fig. 1, C and D). The effect of the rabbit serum was comparable to that induced an anti-Thy-1 antibody plus complement, a well-reported model for induction of MC lysis [21], [22]. Also, the effect of the serum was completely abolished by treatment of the serum at 56 °C for 30 min, indicative of a mediating role of complement. Given that the serum provided both anti-MC antibodies and complement, and that it potently induced MC lysis in a way similar to Thy-1 antibody [18], we have used this model to evaluate the effects of GSH on antibody-initiated and complement-mediated cell injury.
Fig. 1.

Induction of rat MC lysis by rabbit serum. (A) Induction of MC lysis by rabbit serum. MCs were exposed to 10 μg/ml Thy-1 antibody in the presence of 5% native and heated-treated rabbit serum for 6 h. Cell morphology was photographed. (B) MCs were exposed to 10 μg/ml Thy-1 antibody in the presence of the indicated concentrations of rabbit serum for 6 h. Cellular supernatants were collected and assayed for LDH activities. Data shown are mean ±SE (n =4). ** P<0.01 vs. zero point control. (C) Calcein-AM/PI staining. Cells were either left untreated or treated with 5% rabbit serum in the absence or presence of 10 μg/ml anti-Thy-1 antibodies for 6 h. The viability of cells was determined using PI (red, dead cells) and calcein-AM (green, living cells) staining. (D) The percentage of PI-positive red cells per field in C. The number of PI-positive cells was counted. The data shown are mean ±SE (n =4). ** P<0.01 vs. zero control.
3.2. Suppressive effect of GSH on the rabbit serum-induced MC lysis
To determine the influence of GSH on antibody/complement-initiated cell injury, we determined the extent of cell death in the presence or absence of GSH. Fig. 2 shows that MC injury induced by the rabbit serum was completely blocked by GSH. GSH effectively prevented the appearance of PI-positive dead cells. Consistently, it blocked the loss of calcein-positive living cells (Fig. 2, A-C). The protective effect of GSH was also confirmed by WST assay. GSH significantly prevented the loss of cell viability caused by the rabbit serum (Fig. 2D). These results indicate that GSH potently prevents antibody/complement-elicited cell injury.
Fig. 2.

GSH suppresses the rabbit serum-induced MC cell lysis. (A) Calcein-AM/PI staining of rabbit serum-treated cells. Cells were treated with the indicated concentrations of rabbit serum in the presence or absence of 3 mM GSH for 6 h. The viability of cells was determined using calcein (green) and PI (red) staining. (B, C) The percentage of calcein- and PI-positive cells in A. The number of calcein-positive green and PI positive red cells was counted and expressed as percentage of the total cells. Data shown are mean ±SE (n =4). ## P<0.01 vs. zero control; ** P<0.01 vs. respective control. (D) Determination of cell viability using WST assay. Cells were treated the same as above. Cell viability was evaluated by WST assay. Data shown are mean ±SE (n =4). ## P<0.01 vs. zero control; ** P<0.01 vs. respective control.
3.3. Prevention of antibody binding to MC surface by GSH
Antibody-triggered and complement-dependent cell lysis involves many processes, including antibody binding to cell surface antigens, subsequent activation of complement via the classic pathway, formation of the “membrane attack complex”, and induction of cell lysis [23], [24]. To determine the mechanisms underlying the protective effects of GSH, we first examined the effect of GSH on the antibody binding to the antigens. For this purpose, we incubated MCs with rabbit serum and determined the amount of membrane-bound IgG using IF staining and immunoprecipitation, respectively. IF staining revealed that the rabbit serum contained antibodies against MC surface antigens. There was strong fluorescent signal at the cell membrane (Fig. 3A). In the presence of GSH, the signal was markedly reduced. To further confirm the result, MCs were incubated with rabbit serum or mouse anti-Thy-1 antibody and the membrane-bound IgG was precipitated using a mixture of protein-A and protein-G beads. In the presence of GSH, there was a concentration-dependent reduction in the level of the membrane-bound IgG (Figs. 3B and 3C).
Fig. 3.

GSH inhibits the binding of rabbit and mouse Igs to MC surface and promotes the cleavage of the disulfide bonds in Igs. (A-D) Effect of GSH on antibody binding to the cell surface. (A) Immunofluorescence staining of membrane-bound IgG. MCs were incubated with 3% rabbit serum for 2 h in the presence or absence of 5 mM GSH. The presence of rabbit Igs in cell surface was detected with rhodamine-conjugated anti-rabbit IgG antibody. Note the high red fluorescence in MCs and its prevention by GSH. (B, C) Precipitation of the cell membrane-bound Igs using protein A/G beads. MCs were exposed to 1% heat-treated rabbit serum (B) or 1.0 μg/ml anti-Thy-1 antibody (C) in the presence or absence of the indicated concentration of GSH for 30 min. After washing out the unbound free Igs, cells were lysed with SDS buffer. The cell-bound Igs were precipitated and subjected to Western blot analysis using an HRP-labeled anti-rabbit or anti–mouse antibody. (D) Western blot analysis of cell bound Igs. MCs were exposed to 1% heat-treated rabbit serum for the indicated time intervals in the presence or absence of 3 mM GSH. Cell bound Igs were determined using Western blot analysis. (E, F) Effect of GSH on the cleavage of disulfide bonds in Igs. Heat-treated rabbit serum (1%, E, F) or Thy-1 (1.0 μg/ml, G) was incubated with the indicated concentrations of GSH, NAC or 50 mM DTT for 60 min. After treatment, the samples were separated by SDS page and immunoblotted with an HRP-labeled anti-rabbit or anti–mouse antibody.
We also determined the amount of cell-bound IgG using Western blot analysis. MCs were incubated with the rabbit serum in the presence or absence of GSH for different intervals. After washing out the unbound IgG, the remaining IgG in the cellular lysates was detected using Western blot analysis. Fig. 3D shows that the rabbit IgG in Western blot was mainly localized at the location of 160 ~ 180 kDa, which corresponds to the predicted molecular mass of a homodimeric IgG consisting of two polypeptide chains. GSH treatment caused a reduction of membrane-bound IgG and a shift of IgG from 160 KDa to 50 kDa (which corresponds to the molecular weight of a single heavy chain), indicating a cleavage of the disulfide bonds among the heavy and light chains in rabbit IgG. This result was further confirmed by direct incubation of rabbit IgG or monoclonal Thy-1 antibody with GSH and its precursor NAC. Figs. 3E and 3F show that GSH and NAC treatment indeed caused a shift of rabbit IgG, an effect that is known to be inducible by the reducing agent DTT via splitting the disulfide bonds. GSH also caused a shift of monoclonal Thy-1 antibody from 160 kDa to 50 and 25 kDa (which corresponds to the molecular weight of the heavy and light chain, respectively). Of note, the antibody used for detection of rabbit IgG poorly reacted with IgG light chain. This explains why we only detected 50 kDa band in the case of rabbit IgG. Collectively, these observations indicate that GSH disrupts the disulfide bonds of IgG and reduces its binding to cell surface antigens.
3.4. Effect of GSH on antibody-mediated agglutination of RBCs and antibody-sensitized microbeads
To further confirm that GSH regulated the antibody-mediated immune responses, we examined the effect of GSH on antibody-initiated red blood cell agglutination. Fig. 4A shows that, in the presence of anti-mouse RBC antibody, agglutination of RBCs occurred. In the presence of GSH, however, the agglutination was inhibited. There was less number of RBCs in the cell aggregates (Fig. 4A). Consistent with the inhibitory effect of GSH on the rabbit serum-induced MC injury, GSH also significantly prevented RBC lysis induced by the antibody plus complement from the human serum (Fig. 4B). These observations thus indicate that GSH prevents antibody-dependent RBC agglutination and antibody/complement-mediated RBC lysis.
Fig. 4.

GSH prevents antibody-mediated agglutination of RBCs and antibody-sensitized microbeads. (A) Effect of GSH on antibody-mediated RBC agglutination. Mouse RBCs were exposed to the indicated dilutions of anti-mouse RBC antibody in the presence or absence of 5 mM GSH for 90 min. The formation of agglutinated RBCs was photographed. (B) Effect of GSH on antibody-initiated and complement-mediated RBC lysis. Mouse RBCs were exposed to the anti-mouse antibody (1 μg/ml) in the presence or absence of 5 mM GSH or NAC for 1.5 h. Afterward, 5% human serum were added as a source of complement. After reaction for 4.5 h, the supernatants were collected and measured for the absorbance at 405 nm. (C-G) The influence of GSH on the Easy-Titer IgG Assay. (C) Effect of GSH on antibody-elicited agglutination of the microbeads. The assay beads were incubated with the indicated concentrations of mouse anti-Thy-1 antibody that was pretreated with 3 mM GSH for 1 h. After reaction for 5 min, the agglutination of the microbeads was photographed. Note the obvious difference in the size of the aggregates between control and GSH-treated wells. (D) The standard curve generated from the OD obtained from a series of diluted mouse IgG in the presence or absence of 3 mM GSH. (E) The influence of GSH on the Easy-Titer IgG Assay. The same amount of anti-Thy-1 antibody in the presence or absence of 3 mM GSH was measured for their concentrations using the Easy-Titer IgG Assay Kit. Data shown are mean ±SE (n =4), ** P<0.01. (F) Effect of different concentrations of GSH on Easy-Titer IgG assay. The IgG assay was done in the presence of the indicated concentrations of GSH. Data shown are mean ±SE (n =4), * P<0.05; ** P<0.01. (G) Influence of NAC on Easy-Titer IgG assay. The IgG assay was done in the presence of 3 mM NAC. Data shown are mean ±SE (n =4). ** P<0.01.
Taking advantage of the Easy-Titer Mouse IgG Assay Kit, we also determined the effect of GSH on antibody-mediated agglutination of microbeads. The kit is used for accurate determination of IgG concentrations in various samples. It contains antibody-sensitized polystyrene beads. When reaction with IgG in the samples, the beads aggregate and result in a reduction of light absorption at 405 nm, which is proportional to the IgG concentration. Fig. 4C shows that addition of different concentrations of monoclonal antibody into the assay system caused agglutination of microbeads. Under the microscope, the size of the aggregates appeared to be in direct correlation with the concentration of the added IgG. GSH treatment blocked the formation of aggregates and shifted the standard curve downward (Fig. 4D). It significantly lowered the estimated concentration of the samples (Fig. 4E). This effect of GSH was concentration-dependent, being observable at the concentration as low as 10 μM (Fig. 4F). Moreover, GSH precursor NAC exerted the similar effect (Fig. 4G). These observations thus indicate that GSH prevents the interaction between antibody and antigen.
3.5. Suppressive effect of GSH on complement activation
Antibody binding to antigens subsequently activates complement via the classic pathway and causes cell lysis via formation of the “membrane attack complex” of C5b-9 [23], [24]. To determine whether GSH also affected the complement activation, we examined the deposition of C9 after treatment of MCs with the Thy-1 antibody plus human serum (as a source of complement). IF staining revealed a deposition of C9 in Thy-1 treated cells, which was almost completely prevented in the presence of GSH (Fig. 5A). Consistent with this result, Western blot analysis of the cellular lysates revealed a significant reduction in C9 deposition in GSH-treated cells. Furthermore, the similar effect was also achieved by GSH precursor NAC (Fig. 5, B and C). Of note, the deposition of C9 was not detectable in the presence of heated human serum (Fig. 5B), suggesting that the deposition was the consequence of complement activation. Moreover, heat treatment itself did not affect C9 detection (Fig. 5D). These observations indicate that GSH prevents the antibody-mediated activation of the classic complement pathway.
Fig. 5.

GSH inhibits terminal attack complex formation induced by both classic and alternation pathway. (A-D) Effect of GSH on the cellular deposition of C9. (A) Immunofluorescent staining of C9. MCs were either left untreated (control) or treated with 10 μg/ml Thy-1 for 1 h, followed by incubation with 10% GSH-treated or untreated human serum for an additional 30 min. The cellular deposition of C9 was detected by IF staining. Note the deposition of C9 in Thy-1 plus human serum-treated cells and its prevention by GSH. (B) Western blot analysis of C9 in the cell lysate. Cells were treated with 10 μg/ml Thy-1 plus 10% native or heat-treated serum for 1 h in the presence or absence of 5 mM GSH or NAC. The cellular lysates were analyzed by native electrophoretic separation and immunoblotted for C9. (C) Densitometric analysis of the band intensity in (B). Data shown are mean ±SE (n =3). ** P<0.01. (D) Western blot analysis of C9 in native and heat-treated human serum. Note that the detection of C9 was not affected by heat treatment. (E-G) Effect of GSH on the alternative complement activation pathway. The complement activity induced by activation of the alternative pathway in the positive control (E) or 4 normal human sera (F) in the presence or absence of 3 mM GSH was determined via detection of the C5b-9 formation. (G) The influence of different concentrations of GSH on complement activity of a normal adult serum. Data shown are mean ±SE (n =4), ** P<0.01.
We also examined the effect of GSH on the alternative pathway. For this purpose, we have used a commercially available immunoassay kit. In this kit, the wells of microtiter strips are coated with specific activator of the alternative pathway. Activation of the pathway leads to the formation of C5b-9, the amount of which reflects the extent of the complement activation. Fig. 5 shows that GSH significantly inhibited the activation of the alternative pathway, as demonstrated by the suppression of complement activity in the positive control (Fig. 5E), as well as in human serum samples (Fig. 5F). This effect of GSH was concentration-dependent. This significant inhibition was achieved at the concentration as low as 100 μM (Fig. 5G). These observations indicate that GSH potently inhibits both classic and alternative complement activation pathways.
3.6. Suppressive effect of GSH on p38 activation
Apart from antibody and complement, the cellular defense against C5b-9-induced cell injury is another determinant of cell destiny. Previous studies have shown that the cell lysis induced by complement was partially mediated by induction of oxidative stress and activation of stress-sensitive kinases [25]. Under our experimental settings, we have demonstrated a partially mediating role of P38 [18]. As GSH is one of the major antioxidants against oxidative stress, we speculated that GSH might also work through modulation of the oxidative sensitive kinase. To test this hypothesis, we first confirmed our previous findings that inhibition of P38 with a specific chemical inhibitor SB2203580 significantly attenuated the rabbit serum-induced cell death (Fig. 6, A and B). Intriguingly, GSH also suppressed P38 activation under both basal and serum-stimulated conditions. To further establish the role of GSH, we used BSO (an inhibitor of gamma-glutamylcysteine synthetase) [26], and SSA (an inhibitor of the XCT transporter) [27] to deplete intracellular GSH. As shown in Figs. 6D to 6G, depletion of GSH with these agents promoted P38 activation and sensitized cells to the antibody/complement-initiated cell death. Of note, depletion of intracellular GSH with BSO and SSA did not greatly affect the cellular deposition of C9 (Fig. H), suggesting that these chemicals did not affect the formation of the membrane attack complex. Collectively, these observations indicate that GSH also exerts a protective role against antibody- and complement-initiated cell injury via modulation of oxidative sensitive P38.
Fig. 6.

Mediating role of p38 in complement-elicited cell death and its inhibition by GSH. (A, B) Effect of P38 in antibody/complement-initiated cell death. MCs preloaded with calcein-AM were exposed to 5% rabbit serum in the presence or absence of p38 inhibitor SB203580 (10 μM) for 6 h. Note the reduced number of PI-positive dead cells in SB203580-treated cells. The intensity of green fluorescence was determined with a fluorescence reader at 490–515 nm. The rate of cell survival relative to untreated control was calculated (Fig. 6B). # P<0.01 vs. control; ** P<0.01 vs. 5% rabbit serum control. (C) Induction of p38 phosphorylation by the rabbit serum and its prevention by GSH. MCs were incubated with 1% rabbit serum in the presence or absence of 2.5 mM GSH. The cellular lysates were subject to Western blot analysis of the level of phospho-p38. β-actin was used as loading control. (D-G) Depletion of intracellular GSH on antibody/complement-induced cell death and P38 activation. MCs were pretreated with the indicated concentration of BSO (D), or 500 μM SSA (E) for 6 h before exposing to rabbit serum for an additional 6 h. (F) MCs were pretreated with 10 mM BSO or 500 μM SSA before exposing to the indicated concentrations of rabbit serum for 15 min. The cellular lysates were subject to Western blot analysis of the level of phospho-p38. (G) Densitometric measurement of band intensity in F. Data shown are mean ±SE (n =4). # P<0.05 vs. zero control; * P<0.05 vs. respective control. H. Effect of GSH-modulating chemicals on cellular deposition of C9. Cells were pretreated with 5 mM BSO and 500 μM SSA for 6 h, followed by reaction with 10 μg/ml Thy-1 plus 10% native human serum for an additional 1 h. The cellular lysates were subjected to Western blot analysis of C9.
4. Discussion
In this study, we demonstrated that GSH prevented antibody-triggered and complement-dependent cell lysis. Furthermore, we provided evidence that GSH interfered with antibody-antigen binding, blunted complement activation and augmented cellular defense against complement-mediated cell injury. Our study thus characterized GSH as a potent inhibitor of antibody- and complement-initiated immune responses and suggested that GSH may have the therapeutic potential to treat certain immune-related diseases.
We have used an in vitro model of glomerular MC lysis induced by a rabbit serum to investigate the effect of GSH on immune responses. In our previous study, we have demonstrated that the serum contained antibodies against several different MC surface antigens and induced a complement-dependent MC lysis in a way similar to anti-Thy-1 antibody [18] (a well-used model for induction of mesangial cell injury both in vitro and in vivo [21], [22]). Given that the serum simultaneously provided antibodies and complement, it provided a cheap and convenient way to induce immunologic cell injury and to evaluate the effect of GSH on immune responses.
Antibody-triggered complement-dependent cell lysis occurs in many pathological situations, including Thy-1 glomerulonephritis [21], [22]. The induction of cell lysis by antibody/complement involves many steps, including antibody binding to cell surface antigens, subsequent activation of complement via the classic pathway and formation of the “membrane attack complex” of C5b-9, and finally, induction of cell lysis [23], [24]. It is conceivable that the interruption of a single step of the cascade could greatly influence the cell fate. Apart from these molecular events, cell defense mechanism against C5b-9 is an additional factor governing cell destiny. In the current investigation, we demonstrated that GSH potently inhibited the antibody/complement-induced cell injury through mechanisms involving its action on the antibody, complement, and cellular defensive mechanisms.
The antibody is a tetrameric polypeptide structure, composed of two identical heavy chain and two identical light chain polypeptides, covalently linked by disulfide bridges. In Ig molecules, there are abundant disulfide bonds, which is indespensible for maintaining the normal structure and function [28]. In this study, we found that GSH and its precursor NAC induced cleavage of disulfide bonds in IgG. This effect of GSH destroyed the antigen binding structure and obliterated the antigen-binding function, as evidenced by the reduction in IgG binding to cell surface antigens and prevention of antibody-mediated formation of RBC cell agglutination, as well as aggregation of antibody-sensitized microbeads. Our results appeared to be in accordance with a report by Rogers et al. They found that thiol-reactive compound GSH prevented nonspecific antibody binding in immunohistochemistry. Concomitantly, GSH also reduced the specific signal. They speculated that it could be related to GSH binding to antibody SH residues and subsequent prevention of the unwanted disulfide bridges with substrate thiols [29]. Based on the results of this study, we speculated that the reduction of disulfide bonds in antibodies by GSH could also contributed to the reduced target signal.
Of note, the isotype of the antibody mediating RBC agglutination was IgM. This suppression of RBC agglutination by GSH indicated that the effect of GSH was not antibody isotype-specific. Previous studies have shown that incubation of IgM with DTT, a reducing chemical, inactivated IgM [30]. Also, a study by Roche, et al. demonstrated that treatment with IgG with DTT caused a preferential reduction of the hinge region disulfide bonds. The reduced IgG (rIgG, or half IgG) generated following DTT treatment was monovalent and lost its ability to agglutinate its target bacterial [31]. These data are in support of the view that the reduction of the disulfide bond in Igs by GSH could be responsible for the reduced agglutination of RBCs and microbeads in this study.
Besides its action on antibodies, GSH also potently inhibited complement activation. It inhibited Thy-1 antibody-induced deposition of human C9 in MCs. It also inhibited formation of C5b-9 by the activator of the alternative pathway. These observations indicate that GSH interfered with both classic and alternative complement activation pathways. Currently, the molecular mechanisms involved in the action of GSH are unclear. It could also be related to the reduction of disulfide bonds in complement components. Indeed, the existence of disulfide bridges complement components has been reported [17], [32]. In this study, we have found the amount of cell-bound C9 was reduced in the presence of GSH. As a final component of the complement activation cascade, the change of C9 may well reflected that activity of the complement system. C9 serves an ideal parameter for assessment of complement activation. Furthermore, C9 plays a critical role in determining the cytolytic efficiency of the C5b-9 complex [33], [34], [35]. In the absence of C9, the C5b-8 complex is ineffective in inducing membrane damage. Intriguingly, the membrane attack complex of complement (the dimeric C5b-9 complex) is held together by noncovalent forces [36], [37], [38]. C9 in the C5b-9 dimer was in a disulfide-linked dimeric form. The quaternary structure of the C5b-9 dimer is stabilized by the disulfide-linking of C9 [37], [38]. It is possible that C9 itself could be the target of GSH in the current investigation. In support of this notion, previous studies have shown that there are abundant disulfide bridges in C9 and C9 molecules can be covalently crosslinked (disulfide-linked) by GSH [17]. The function of C9 is sustained by disulfide bond-dependent conformational motifs. Disruption of intra-chain disulfide linkages in C9 by the reducing agent DTT has been reported to cause a loss of C9 function [39]. Thus, it is highly likely that disruption of the disulfide bond in C9 by GSH could be a potential mechanism by which GSH suppresses complement activation.
Another factor determining cell fate to antibody/complement-initiated cell injury is cell defense mechanisms. The insertion of C5b-9 (membrane attack) complexes into cell membranes cause many cell responses, including oxidative stress and activation of oxidative sensitive kinases. Several previous studies have demonstrated that a mediating role of stress-activated kinases in mediating C5b-9-induced cell injury [25], [40], [41]. GSH, as a major antioxidant, could protect the cells via modulation of intracellular redox status. Indeed, in a study demonstrating an implication of free radical in complement-induced cell injury, GSH has been shown to suppress complement-induced podocyte injury through inhibition of JNK [41]. Under our experimental setting, we have documented a pivotal role of P38, another oxidative sensitive MAPK in complement-induced MC lysis [18]. In this study, we observed that GSH affected both basal and the rabbit serum-initiated elevation in P38. Furthermore, depletion of intracellular GSH with BSO (an inhibitor of gamma-glutamylcysteine synthetase) [26], and SSA, which blocks the XCT transporter subunit and reduces cystine uptake [27], promoted P38 activation and sensitized cells to the antibody/complement-initiated cell death. These observations indicate that endogenous GSH exerted a protective role against antibody- and complement-initiated cell injury.
Our findings could have a great clinic and basic relevance. First, our study provided novel mechanistic insights into the suppressive effect of GSH on inflammatory cell injury. Apart from the well-documented action of GSH on ROS, GSH also directly suppressed antibody binding to cell surface antigens and inhibited both classic and alternative complement activation pathway through the direct cleavage of the disulfide bonds of the key molecules in the immunologic responses. The effects of GSH on antibody, complement and cell defense may contribute to the reported protective effect of GSH on mesangial cell lysis in an in vivo model of glomerular nephritis induced by anti-Thy-1 antibody [42], and on anti-FAS antibody-induced neutrophil apoptosis [43], [44]. Second, our study indicates that GSH might be used to modulate immune responses and prevent cell injury elicited by antibody and complement complex, such as transfusion reactions, autoimmune hemolytic anemia, erythroblastosis fetalis, goodpasture's syndrome, and antibody-dependent cell-mediated cytotoxicity (ADCC), etc. Third, dietary supplementation of GSH or its precursor NAC has been suggested as a potential way to prevent the oxidative diseases and aging. Given its strong suppressive action on humoral immunity, which is required for normal defense against pathogens, the supplementation of GSH should be taken with care. In fact, GSH has been reported to inhibit phagocytosis and enhance the mortality of bacterial-induced peritonitis [11]. Lastly, our study indicates that in the assay system involving antibody-antigen interaction, such as antibody-mediated RBC agglutination and Easy-Titer IgG Assay, special cautions should be paid to exclude the thiol-reactive chemicals in the assay system.
5. Conclusion
In summary, we demonstrated, for the first time, that GSH protected cells from antibody/complement-induced lysis through multiple mechanisms, including inhibition of antibody binding to antigens, disruption of complement activation and augmentation of cellular defense against oxidative stress. Our study thus provided novel mechanistic insights into the immunoregulatory action of GSH and suggested that GSH may be exploited to treat certain immune-associated disorders.
Conflict of interest statement
The authors confirm that they have no competing interests.
Acknowledgements
We thank Dr. Hiroshi Kawachi (Department of Cell Biology, Institute of Nephrology, Niigata University) for supplying us anti-Thy-1 monoclonal antibodies used in this study. This work was supported by a grant from the Ministry of Education, Culture, Sports, Science and Technology, Japan (26461219 and 26462438 to J. Y).
References
- 1.Rahman I., MacNee W. Regulation of redox glutathione levels and gene transcription in lung inflammation: therapeutic approaches. Free Radic. Biol. Med. 2000;28(9):1405–1420. doi: 10.1016/s0891-5849(00)00215-x. [DOI] [PubMed] [Google Scholar]
- 2.Ghezzi P. Role of glutathione in immunity and inflammation in the lung. Int J. Gen. Med. 2011;4:105–113. doi: 10.2147/IJGM.S15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lou H., Kaplowitz N. Glutathione depletion down-regulates tumor necrosis factor alpha-induced NF-kappa B activity via I kappa B kinase-dependent and -independent mechanisms. J. Biol. Chem. 2007;282(40):29470–29481. doi: 10.1074/jbc.M706145200. [DOI] [PubMed] [Google Scholar]
- 4.Lou H., Kaplowitz N. Glutathione depletion down-regulates tumor necrosis factor (TNF) alpha-induced NF-kappa B activity via I kappa B kinase (IKK)-independent and -dependent mechanisms. J. Biol. Chem. 2007;46(4):281a–282a. doi: 10.1074/jbc.M706145200. [DOI] [PubMed] [Google Scholar]
- 5.Liao B.C., Hsieh C.W., Lin Y.C., Wung B.S. The glutaredoxin/glutathione system modulates NF-kappaB activity by glutathionylation of p65 in cinnamaldehyde-treated endothelial cells. Toxicol. Sci. 2010;116(1):151–163. doi: 10.1093/toxsci/kfq098. [DOI] [PubMed] [Google Scholar]
- 6.Jeon K.-I., Jeong J.-Y., Jue D.-M. Thiol-reactive metal compounds inhibit NF-κB activation by blocking IκB kinase. J. Immunol. 2000;164(11):5981–5989. doi: 10.4049/jimmunol.164.11.5981. [DOI] [PubMed] [Google Scholar]
- 7.Braga T.T., Forni M.F., Correa-Costa M., Ramos R.N., Barbuto J.A., Branco P., Castoldi A., Hiyane M.I., Davanso M.R., Latz E., Franklin B.S., Kowaltowski A.J., Camara N.O. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017;7:39884. doi: 10.1038/srep39884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim H.Y., Kim S.J., Lee S.M. Activation of NLRP3 and AIM2 inflammasomes in Kupffer cells in hepatic ischemia/reperfusion. FEBS J. 2015;282(2):259–270. doi: 10.1111/febs.13123. [DOI] [PubMed] [Google Scholar]
- 9.Shin J.N., Fattah E.A., Bhattacharya A., Ko S., Eissa N.T. Inflammasome activation by altered proteostasis. J. Biol. Chem. 2013;288(50):35886–35895. doi: 10.1074/jbc.M113.514919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J.M., Kim H., Kwon S.B., Lee S.Y., Chung S.C., Jeong D.W., Min B.M. Intracellular glutathione status regulates mouse bone marrow monocyte-derived macrophage differentiation and phagocytic activity. Biochem Biophys. Res Commun. 2004;325(1):101–108. doi: 10.1016/j.bbrc.2004.09.220. [DOI] [PubMed] [Google Scholar]
- 11.Goswami M., Sharma D., Khan N.M., Checker R., Sandur S.K., Jawali N. Antioxidant supplementation enhances bacterial peritonitis in mice by inhibiting phagocytosis. J. Med Microbiol. 2014;63(Pt 3):355–366. doi: 10.1099/jmm.0.067173-0. [DOI] [PubMed] [Google Scholar]
- 12.Eylar E., Rivera-Quinones C., Molina C., Baez I., Molina F., Mercado C.M. N-acetylcysteine enhances T cell functions and T cell growth in culture. Int Immunol. 1993;5(1):97–101. doi: 10.1093/intimm/5.1.97. [DOI] [PubMed] [Google Scholar]
- 13.Suthanthiran M., Anderson M.E., Sharma V.K., Meister A. Glutathione regulates activation-dependent DNA-synthesis in highly purified normal human lymphocytes-T stimulated via the Cd2-antigen and Cd3-antigen. Proc. Natl. Acad. Sci. USA. 1990;87(9):3343–3347. doi: 10.1073/pnas.87.9.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeannin P., Delneste Y., Lecoanet-Henchoz S., Gauchat J.F., Life P., Holmes D., Bonnefoy J.Y. Thiols decrease human interleukin (IL) 4 production and IL-4-induced immunoglobulin synthesis. J. Exp. Med. 1995;182(6):1785–1792. doi: 10.1084/jem.182.6.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giordani L., Quaranta M.G., Malorni W., Boccanera M., Giacomini E., Viora M. N-acetylcysteine inhibits the induction of an antigen-specific antibody response down-regulating CD40 and CD27 co-stimulatory molecules. Clin. Exp. Immunol. 2002;129(2):254–264. doi: 10.1046/j.1365-2249.2002.01897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schur P.H., Christian G.D. The role of disulfide bonds in the complement-fixing and precipitating properties of 7S rabbit and sheep antibodies. J. Exp. Med. 1964;120(4):531. doi: 10.1084/jem.120.4.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lengweiler S., Schaller J., Rickli E.E. Identification of disulfide bonds in the ninth component (C9) of human complement. FEBS Lett. 1996;380(1–2):8–12. doi: 10.1016/0014-5793(95)01541-8. [DOI] [PubMed] [Google Scholar]
- 18.Piao H., Chi Y., Zhang X., Zhang Z., Gao K., Niimi M., Kamiyama M., Zhang J., Takeda M., Yao J. Suramin inhibits antibody binding to cell surface antigens and disrupts complement-mediated mesangial cell lysis. J. Pharmacol. Sci. 2016;132(4):224–234. doi: 10.1016/j.jphs.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 19.Yao J., Hiramatsu N., Zhu Y., Morioka T., Takeda M., Oite T., Kitamura M. Nitric oxide-mediated regulation of connexin43 expression and gap junctional intercellular communication in mesangial cells. J. Am. Soc. Nephrol. 2005;16(1):58–67. doi: 10.1681/ASN.2004060453. [DOI] [PubMed] [Google Scholar]
- 20.Yan Q., Gao K., Chi Y., Li K., Zhu Y., Wan Y., Sun W., Matsue H., Kitamura M., Yao J. NADPH oxidase-mediated upregulation of connexin43 contributes to podocyte injury. Free Radic. Biol. Med. 2012;53(6):1286–1297. doi: 10.1016/j.freeradbiomed.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto T., Wilson C.B. Complement dependence of antibody-induced mesangial cell injury in the rat. J. Immunol. 1987;138(11):3758–3765. [PubMed] [Google Scholar]
- 22.Hori Y., Yamada K., Hanafusa N., Okuda T., Okada N., Miyata T., Couser W.G., Kurokawa K., Fujita T., Nangaku M. Crry, a complement regulatory protein, modulates renal interstitial disease induced by proteinuria. Kidney Int. 1999;56(6):2096–2106. doi: 10.1046/j.1523-1755.1999.00765.x. [DOI] [PubMed] [Google Scholar]
- 23.Dunkelberger J.R., Song W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20(1):34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- 24.Noris M., Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33(6):479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren G., Huynh C., Bijian K., Cybulsky A.V. Role of apoptosis signal-regulating kinase 1 in complement-mediated glomerular epithelial cell injury. Mol. Immunol. 2008;45(8):2236–2246. doi: 10.1016/j.molimm.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 26.Drew R., Miners J.O. The effects of buthionine sulphoximine (BSO) on glutathione depletion and xenobiotic biotransformation. Biochem Pharmacol. 1984;33(19):2989–2994. doi: 10.1016/0006-2952(84)90598-7. [DOI] [PubMed] [Google Scholar]
- 27.Gout P.W., Buckley A.R., Simms C.R., Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15(10):1633–1640. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- 28.Liu H., May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs. 2012;4(1):17–23. doi: 10.4161/mabs.4.1.18347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rogers A.B., Cormier K.S., Fox J.G. Thiol-reactive compounds prevent nonspecific antibody binding in immunohistochemistry. Lab Invest. 2006;86(5):526–533. doi: 10.1038/labinvest.3700407. [DOI] [PubMed] [Google Scholar]
- 30.Okuno T., Kondelis N. Evaluation of dithiothreitol (DTT) for inactivation of IgM antibodies. J. Clin. Pathol. 1978;31(12):1152–1155. doi: 10.1136/jcp.31.12.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roche A.M., Richard A.L., Rahkola J.T., Janoff E.N., Weiser J.N. Antibody blocks acquisition of bacterial colonization through agglutination. Mucosal Immunol. 2015;8(1):176–185. doi: 10.1038/mi.2014.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sahu A., Lambris J.D. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 2001;180:35–48. doi: 10.1034/j.1600-065x.2001.1800103.x. [DOI] [PubMed] [Google Scholar]
- 33.Stolfi R.L. Immune lytic transformation: a state of irreversible damage generated as a result of the reaction of the eighth component in the guinea pig complement system. J. Immunol. 1968;100(1):46–54. [PubMed] [Google Scholar]
- 34.Yamamoto K.I. Lytic activitylytic activity of C5-9 complexes for erythrocytes from the species other than sheep: C9 rather than C8-dependent variation in lytic activity. J. Immunol. 1977;119(4):1482–1485. [PubMed] [Google Scholar]
- 35.Yamamoto K., Kawashima T., Migita S. Glutathione-catalyzed disulfide-linking of C9 in the membrane attack complex of complement. J. Biol. Chem. 1982;257(15):8573–8576. [PubMed] [Google Scholar]
- 36.Stanley K.K. The molecular mechanism of complement C9 insertion and polymerisation in biological membranes. Curr. Top. Microbiol Immunol. 1989;140:49–65. doi: 10.1007/978-3-642-73911-8_5. [DOI] [PubMed] [Google Scholar]
- 37.Ware C.F., Kolb W.P. Assembly of the functional membrane attack complex of human complement: formation of disulfide-linked C9 dimers. Proc. Natl. Acad. Sci. USA. 1981;78(10):6426–6430. doi: 10.1073/pnas.78.10.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto K., Migita S. Mechanisms for the spontaneous formation of covalently linked polymers of the terminal membranolytic complement protein (C9) J. Biol. Chem. 1983;258(13):7887–7889. [PubMed] [Google Scholar]
- 39.Hatanaka M., Seya T., Inai S., Shimizu A. The functions of the ninth component of human complement are sustained by disulfide bonds with different susceptibilities to reduction. Biochim Biophys. Acta. 1994;1209(1):117–122. doi: 10.1016/0167-4838(94)90146-5. [DOI] [PubMed] [Google Scholar]
- 40.Cybulsky A.V., Takano T., Papillon J., Bijian K., Guillemette J. Activation of the extracellular signal-regulated kinase by complement C5b-9. Am. J. Physiol. Ren. Physiol. 2005;289(3):F593–F603. doi: 10.1152/ajprenal.00066.2005. [DOI] [PubMed] [Google Scholar]
- 41.Peng H., Takano T., Papillon J., Bijian K., Khadir A., Cybulsky A.V. Complement activates the c-Jun N-terminal kinase/stress-activated protein kinase in glomerular epithelial cells. J. Immunol. 2002;169(5):2594–2601. doi: 10.4049/jimmunol.169.5.2594. [DOI] [PubMed] [Google Scholar]
- 42.Mosley K., Waddington S.N., Ebrahim H., Cook T., Cattell V. Inducible nitric oxide synthase induction in Thy 1 glomerulonephritis is complement and reactive oxygen species dependent. Exp. Nephrol. 1999;7(1):26–34. doi: 10.1159/000020581. [DOI] [PubMed] [Google Scholar]
- 43.Deas O., Dumont C., Mollereau B., Metivier D., Pasquier C., Bernard-Pomier G., Hirsch F., Charpentier B., Senik A. Thiol-mediated inhibition of FAS and CD2 apoptotic signaling in activated human peripheral T cells. Int Immunol. 1997;9(1):117–125. doi: 10.1093/intimm/9.1.117. [DOI] [PubMed] [Google Scholar]
- 44.Watson R.W., Rotstein O.D., Jimenez M., Parodo J., Marshall J.C. Augmented intracellular glutathione inhibits Fas-triggered apoptosis of activated human neutrophils. Blood. 1997;89(11):4175–4181. [PubMed] [Google Scholar]