

Abstract
N-methyl-D-aspartate (NMDA) receptor plays important roles in learning and memory. NMDA receptors are a tetramer that consists of two glycine-binding subunits GluN1, two glutamate-binding subunits (i.e., GluN2A, GluN2B, GluN2C, and GluN2D), a combination of a GluN2 subunit and glycine-binding GluN3 subunit (i.e., GluN3A or GluN3B), or two GluN3 subunits. Recent studies revealed that the specific expression and distribution of each subunit are deeply involved in neural excitability, plasticity, and synaptic deficits. The present article summarizes reports on the dysfunction of NMDA receptors and responsible subunits in various neurological and psychiatric disorders, including schizophrenia, autoimmune-induced glutamatergic receptor dysfunction, mood disorders, and autism. A key role for the GluN2D subunit in NMDA receptor antagonist-induced psychosis has been recently revealed.
Keywords: GluN1, GluN2D, knockout mice, NMDA receptor subtype, phencyclidine, psychiatric disorders
Introduction
Postsynaptic N-Methyl-D-aspartate (NMDA) receptor is ionotropic glutamate receptor that mediates excitatory signaling in the presence of glycine and glutamate. NMDA receptors are a tetramer that consists of two glycine-binding subunits GluN1, two glutamate-binding subunits (i.e., GluN2A, GluN2B, GluN2C, and GluN2D), a combination of a GluN2 subunit and glycine-binding GluN3 subunit (i.e., GluN3A or GluN3B), or two GluN3 subunits. Moriyoshi et al. first isolated the cDNA of the rat NMDA receptor [1]. Subsequently, each subunit of the NMDA receptor was successfully cloned, including GluN1 [2], GluN2A-2C [3, 4], GluN2D [5], splice variants of GluN1 [2, 6-9], and GluN3A-3B [10-15]. GluN2 subunits are differentially expressed throughout the central nervous system [16-18]. NMDA receptor ion channels allow Na+ and Ca2+ ionic flows into the cell and K+ ions from the cell to outside in a voltage-dependent manner. Therefore, single-channel conductance is calculated from the summation of these three ionic flows. Subunit composition influences single-channel conductance, Mg2+ blockade, and Ca2+ permeability [19-22]. The NMDA receptor channel containing the GluN1 subunit and a GluN2A or GluN2B subunit yields a single larger conductance level associated with higher permeability of Ca2+ ions and higher sensitivity to extracellular Mg2+ blockade. In contrast, the NMDA receptor channel containing GluN1 and the glutamate-binding subunit GluN2C shows two lower conductance sublevels with frequent direct transitions, reduced permeability of Ca2+ ions, and less sensitivity to extracellular Mg2+ blockade [19]. Receptor channels containing GluN1 and the glutamate-binding subunit GluN2D also have a low open probability, two conductance levels, reduced sensitivity to Mg2+ blockade, minimal desensitization, and a markedly slow deactivation time course [17, 23-29]. The deactivation time of the NMDA receptor channel composed of the GluN1 and GluN2D subunits is also affected by its splice variants GluN1-1a and GluN1-1b [30].
Each NMDA receptor subunit is composed of four discrete semiautonomous domains, including the extra-cellular amino-terminal domain (ATD), extracellular ligand-binding domain (LBD), transmembrane domain (TMD), and intracellular carboxyl-terminal domain (CTD; Fig. 1) [19]. The GluN2 ATD regulates agonist potency, the deactivation time course, the open probability, and the mean open/closed duration of different GluN2 subunits [25, 26]. Ryan et al. (2013) [31] found unique functions of the GluN2A CTD (i.e., the regulation of locomotor activity and impulsivity) and GluN2B CTD (i.e., the regulation of perceptual learning, anxiety, impulsivity, and motor coordination). In contrast, the GluN2A and GluN2B CTDs had similar functions in the regulation of reversal learning, associative learning, and motor learning. Corticostriatal or striatal GluN2B deletion and GluN2B antagonism in dorsal striatum impair choice learning, whereas cortical GluN2B deletion and GluN2B antagonism in orbitofrontal cortex impair shifting [32]. Although the GluN2B subunit has been implicated in both the acquisition and extinction of conditioned fear, GluN2C subunits in the amygdala are involved in the consolidation of learned fear responses. D-cycloserine selectively enhances the activity of NMDA receptors containing GluN2C subunit. The increased activity of GluN2C receptors may underlie the enhancement of fear extinction by D-cycloserine [33].
The expression of GluN3A protein is observed throughout the central nervous system in multiple neuronal cell types. GluN3A subunit expression is high in the postnatal week and then decreases in adult animals [34, 35]. The GluN3A subunit makes NMDA receptor ion channels less Ca2+-permeable and Mg2+-insensitive [36]. In the absence of GluN3A, the expression of markers of synaptic maturation is accelerated [37]. Enhanced responses of NMDA receptors and increased number of dendritic spines are observed in early stage of postnatal cerebrocortical neurons [38], and inducing long-term potentiation (LTP) at young synapses is easier [39]. GluN3A knockout (KO) mice showed impaired locomotor activity, increased sensitivity to acute and subacute inflammatory pain, and enhanced recognition, spatial learning, and memory function [40]. The overexpression of GluN3A retards synaptic maturation and attenuates LTP at adult synapses [39]. These data indicate that GluN3A acts as a molecular brake to limit the plasticity and maturation of excitatory synapses [34, 37] and may have a profound impact on several functional/behavioral activities in adult animals [40] (Table 1).
Table 1.
NEW IUPHAR nomenclature for glutamate ionotropic receptor subunits [http://www.iuphar-db.org/LGICNomenclature.jsp].
| NC-IUPHAR | Gene Name | Previous Nomenclatures | ||
|---|---|---|---|---|
|
NMDA Receptor Subunit Nomenclature |
Human | Rat and Mouse | Human | Rat and Mouse |
| GluN1 | GRIN1 | Grin1 | RP11-350O14.1, GluN1, MRD8, NMDA1, NMDAR1, NR1 | GluN1, NMDAR1, NR1 RP23-132N23.20-010, GluRξ1, M100174, Nmdar, Rgsc174 |
| GluN2A | GRIN2A | Grin2a | EPND, FESD, GluN2A, LKS, NMDAR2A, NR2A, RP11-297M9.2 | GluN2A, NMDAR2A, NR2A, GluRε1 |
| GluN2B | GRIN2B | Grin2b | GluN2B, MRD6, NMDAR2B, NR2B, hNR3 | AW490526, GluN2B, NR2B, Nmdar2b, GluRε2 |
| GluN2C | GRIN2C | Grin2c | GluN2C, NMDAR2C, NR2C | RP23-117K15.2, GluN2C, NMDAR2C, NR2C, GluRε3 |
| GluN2D | GRIN2D | Grin2d | EB11, GluN2D, NMDAR2D, NR2D | GluN2D, NMDAR2D, NR2D, GluRε4 |
| GluN3A | GRIN3A | Grin3a | GluN3A, NMDAR-L, NR3A | GluN3A, NR3, chi-1, mCG_120729, 6430537F04, A830097C19Rik, NMDAR-L, NR3A, mKIAA1973 |
| GluN3B | GRIN3B | Grin3b | GluN3B, NR3B | GluN3B, NMDAR3B, NR3B |
|
AMPA Receptor Subunit Nomenclature |
Human | Rat and Mouse | Human | Rat and Mouse |
| GluA1 | GRIA1 | Gria1 | GLUH1, GLUR1, GLURA, HBGR1 | RP23-102H8.1, 2900051M01Rik, Glr-1, Glr1, GluR-A, GluRA, Glur-1, Glur1, HIPA1, gluR-K1 |
| GluA2 | GRIA2 | Gria2 | GLUR2, GLURB, GluR-K2, HBGR2 | GluR-K2, GluR2, gluR-B GluR-B, Glur-2, Glur2 |
| GluA3 | GRIA3 | Gria3 | RP11-349N19.3, GLUR-C, GLUR-K3, GLUR3, GLURC, GluA3, MRX94 | GLUR3, GluR-3, GluR-C, GluR-K3 RP23-471M13.1, 2900064I19Rik, Glur-3, Glur3, Gluralpha3 |
| GluA4 | GRIA4 | Gria4 | GLUR4, GLUR4C, GLURD | GluR-D, GluR4, Glur-4, Glur4, Gluralpha4, spkw1 |
NMDA receptor dysfunction is involved in various disorders
NMDA receptors are implicated in neuronal development, synaptic plasticity, and learning and memory [19, 41]. NMDA receptor dysfunction is also involved in various psychiatric disorders, including schizophrenia, autoimmune-induced glutamatergic receptor dysfunction, mood disorders, autism, and drug-induced psychosis (Table 2).
Table 2.
Dysfunction of NMDA receptors and its experimental therapeutic treatments.
| Disorders | NMDAR-Subunit Related Alterations | Experimental Therapeutic Treatments |
|---|---|---|
| Schizophrenia | • Hypofunction of NMDA receptor on GABAergic neurons induce an imbalance in neural network activity [42]. • A majority of candidate genes associated with increasing risk for schizophrenia can modulate glutamate receptor functions or receptor-interacting proteins, and then affect signal transduction pathways [43-46]. • Two de novo mutations in Glun2A gene are reported in patients with sporadic schizophrenia [47]. • Expression of NMDA receptor subunit (GluN2A subunit in particular) is reduced in postmortem brain with schizophrenia [48]. |
• Agonists for glycine site (D-serine, glycine) or glycine transporter 1 inhibitors [49, 50]. • GluN2A-selective potentiators are of potential interest [51]. |
| Anti-NMDAR encephalitis | • Autoimmune-induced glutamatergic receptor dysfunctions. Anti-NMDA receptor antibodies reduced density of NMDA receptor and induced severe neurological symptoms including hallucinations, psychosis, and seizures [52]. | • Immunotherapy is effective for most patients with anti-NMDA receptor encephalitis [53]. |
| Depression | • Inhibitors of NMDA receptors, in particular GluN2B-containing receptors, result in fast and sustained therapeutic effects in depressive symptoms [54, 55]. | • Ketamine and GluN2B-selective antagonists [54, 55]. • NMDA receptor functional glycine site partial agonist [56]. |
| Autism spectrum disorders | • Alteration of GluN2B and SHANK3 are genetic risk factors. Reduced or enhanced functions of NMDA receptor are involved. Mechanisms are obscure [22, 47, 57-62]. | • Potential interest for NMDA receptor partial agonist [63]. |
| NMDA receptor antagonist-induced psychosis | • Administration of NMDA receptor antagonists induces abnormal behaviors in rodents and psychosis in humans [64-66]. • NMDA receptor antagonists induce increases in the cortical high frequency or gamma oscillations [67, 68]. • Effect of PCP is attenuated in GluN2D KO mice [69, 70]. |
• Potential interest for NMDA receptor co-agonists, glycine type I transport inhibitors, mGluR2/3 agonist or NMDA receptor potentiators [70-77]. • Prior embryonic medial ganglionic eminence cell transplantation into the medial prefrontal cortex [78]. |
Schizophrenia (NMDA Receptor Hypofunction)
Schizophrenia has been treated as a disease with hyperdopaminergic function, in which dopamine receptor antagonists effectively treat positive symptoms. However, dopamine antagonist treatment has not improved negative symptoms and/or cognitive deficits. Presently, the hypofunction theory of glutamatergic neurons in schizophrenia is also widely accepted. Furthermore, environmental factors and multiple genes that enhanced the risk for the onset of schizophrenia are being investigated [79]. As an example of research on environmental factors, a nationwide study of 2,486,646 Danish people reported familial and environmental risk factors for schizophrenia, rate ratios, population-attributable risks, and sex-specific cumulative incidences of several risk factors [80]. In parallel, multiple genetic loci have been reported by genetic linkage studies [81] and association studies [43, 44]. Genome-wide association studies (GWASs) investigate candidate genes, including voltage-gated calcium channel genes, miR-137 targets, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor genes (GRIA1, GRIA4), an NMDA receptor gene (GRIN2B), a metabotropic glutamate receptor gene (GRM5), an enzyme involved in glutamate metabolism GAD1, and a glutamate transporter SLC1A2. Gene expression data using human postmortem brain and human blood and relevant animal model data to identify candidate genes involved in schizophrenia integrate such studies [44-46]. In addition to the aforementioned candidate genes, genes with lower scores have also been reported, including an NMDA receptor gene (GRIN2A), a high-affinity sodium-coupled glutamate aspartate transporter GLAST-1 gene (SLC1A3), an AMPA receptor gene (GRIA3), a kainate receptor gene (GRIK4), and metabotropic glutamate receptor genes (GRM1, GRM4, and GRM7) [45]. Two de novo mutations in the GRIN2A gene, which encodes the NMDA receptor GluN2A subunit, have been reported in patients with sporadic schizophrenia [47]. In the Japanese population, the GluN2D subunit gene is also reported as a possible genome that is involved in schizophrenia susceptibility [82]. More precisely, the transcripts that encode the ionotropic glutamate receptor subunits GluN2D, GluA3, GluK2, and GluK3 and intracellular proteins GRIP1 and SynGAP1 are reduced in relay neurons in the medial dorsal thalamus in schizophrenia [83]. The susceptibility to schizophrenia is known to be increased by a small number of rare, recurrent genomic copy number variants (CNVs). Many small de novo mutations are found in the glutamatergic postsynaptic proteins, including NMDA receptor complex [84, 85]. These mutation-induced defects in glutamatergic transmission, especially via NMDA receptors, are consistent with the glutamatergic neuron hypofunction theory of schizophrenia.
NC-IUPHAR (the International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification). Greek symbols in NMDA receptor subunit names were applied to the mouse orthologue only. The protein name mirrors the gene name, with just the two letter code difference (i.e., GRIN1 translates to GluN1, Gria1 translates to GluA1).
Changes in binding of glutamate receptor, gene expression, and subunit protein expression, especially decreased GluN1 expression, in the prefrontal cortex [86-90], thalamus [91-94], hippocampus [95-99], and cerebellum [89] are shown using postmortem brain of patients with schizophrenia. The transcription of the GluN1 and GluN2B subunits of the NMDA receptor are regulated by nuclear respiratory factors 1 and 2 (NRF-1 and NRF-2). Specificity protein4 functionally regulates GluN1, GluN2A, and GluN2B in a complementary and concurrent/parallel manner with nuclear respiratory factors 1 and 2 (NRF-1 and NRF-2) [100]. One research group reported an increase in the expression of one spliced isoform, GluN1c2’, that was significantly increased in the anterior cingulate cortex in postmortem samples from aged schizophrenia patients [87]. With regard to the GluN1 subunit, GRIN1 (which encodes human GluN1) is not included in the candidate genes for schizophrenia, and complicated defects in glutamatergic postsynaptic signaling complexes, including NMDA receptors, AMPA receptors, and ARC, may induce schizophrenia symptoms.
Alterations in cortical inhibitory γ-aminobutyric acid (GABA) neurons have been demonstrated in many postmortem studies. For example, a subset of these neurons expressing the calcium-binding protein parvalbumin (PV) appear to have lower mRNA and protein levels of the glutamic acid decarboxylase (GAD67; 67kDa isozyme for GABA synthesis) and PV [101], indicating abnormal GABA neurotransmission by PV neurons in schizophrenia. Altered PV neuron function appears to underlie cognitive deficits in schizophrenia by disturbing the generation of cortical gamma-oscillations [101]. Interestingly, GluN2A mRNA expression is reduced to undetectable levels in approximately 50% of PV neurons in the prefrontal cortex in subjects with schizophrenia [48]. Genetically engineered mice, in which the GluN1 gene is eliminated during early postnatal development but not adulthood, replicated the reduced expression of both GAD67 and PV in cortical PV neurons [102]. These findings indicate that reduced transmission through NMDA receptors could be an upstream mechanism of altered PV neuron function in schizophrenia.
Numerous studies provided genetic, pharmaco-logical, and behavioral data to indicate that a reduction of glutamatergic function, especially NMDA receptors on inhibitory GABAergic interneurons, induces an imbalance between excitatory input and inhibitory input. These neural circuitry perturbations that underlie cognitive and executive dysfunctions lead to eventually psychosis [42].
Autoimmune-Induced Glutamatergic Receptor Dysfunction
Recently, autoimmune synaptic encephalitides are defined brain diseases in human that cause severe neurological symptoms, including hallucinations, psychosis, and seizures, by autoantibody reactions with brain tissue. Among autoimmune encephalitis patients, autoantibodies against the extracellular domains of the NMDA receptor are frequently detected and the concentrations of autoantibodies are relevant to the developing psychotic and neurological symptoms [52]. Notably, in addition to having autoimmune encephalitis, some patients also have anti-NMDA receptor antibodies. Using serum from 459 patients admitted with acute schizophrenia, major depression (MD), and borderline personality disorder (BLPD) and matched controls, diverse NMDA receptor antibodies are detected in 15 subjects [103]. They were classified primarily as an initial schizophrenia (9.9%), MD (2.8%) or BLPD (0%) and controls (0.4%). Among these patients, two young females with acute disorganized behavior or catatonia were diagnosed with NMDA receptor encephalitis according to specific immunoglobulin G (IgG) antibodies against GluN1a, which were also increased in cerebrospinal fluid (CSF) [103]. The other two patients had a different epitope of IgG NMDA receptor antibodies against GluN1a/GluN2b but not against GluN1a alone, and they had no immunoreactive antibodies in CSF [103]. IgA and IgM anti-NMDA receptor antibodies were present in all of the other seropositive cases. The presence of IgG GluN1 antibodies in CSF is important for the diagnosis of NMDA receptor encephalitis because antibodies in some patients are detected only in CSF [104, 105]. Kayser et al. (2013) reported the frequency, symptoms types, and outcome in patients with anti-NMDA receptor encephalitis and isolated psychiatric manifestations in 571 patients with IgG antibodies against the GluN1 subunit of the NMDA receptor [53]. Of these 571 patients, 23 (4%) developed isolated psychiatric episodes. Interestingly, for all 23 patients, age (median, 20 years), sex (91% female), and tumor association (43%; ovarian teratoma in all cases) were similar to the population at large. Predominant symptoms included delusional thinking (74%), mood disturbances (70%, usually manic), and aggression (57%). Brain magnetic resonance imaging findings were abnormal in 10 of 22 patients (45%), and CSF analysis showed pleocytosis in 17 of 22 patients (77%). Gresa-Arribas et al. (2014) [105] recently studied 250 patients with anti-NMDA receptor encephalitis and reported antibodies that targeted a main epitope region at GluN1 amino acid 369. The epitope repertoire did not differ between patients with different outcomes and did not change during relapses. Although autoimmune encephalitis is a rare disease, systematic checks of NMDA receptor antibodies are desired for patients with acute psychiatric symptoms, especially for young females. Patients with anti-NMDA receptor encephalitis mostly respond to immunotherapy [53]. When first-line treatments (e.g., steroids, intravenous immunoglobulin, and plasmapheresis) fail, second-line immunotherapy (e.g., rituximab and cyclophosphamide) is usually effective [104].
Mood Disorders and NMDA Receptor GluN2B Subunit
Major depressive disorder is considered a mood disorder caused by a malfunction of the monoaminergic systems, but the possibility of the involvement of the glutamatergic system has also been suggested [106]. Gene linkage analysis confirmed a role specifically for the GluN2B subunit of the NMDA receptor in bipolar disorder [107]. A recent study reported statistically significant differences in allele and genotype frequencies between treatment-resistant depression (TRD) and non-TRD groups for the rs1805502 polymorphism within the GRIN2B gene [108]. Interestingly, the nonselective NMDA receptor antagonist ketamine produces a fast and sustained reduction of depressive symptoms in patients with TRD, supporting this hypothesis [54, 55]. The antidepressant-like effect of ketamine is estimated to occur through the following pathways. The activity of cortical GABA interneurons is regulated by NMDA receptors. The basal activity of pyramidal neurons, in contrast, is not directly regulated by NMDA receptors [109]. Ketamine blocks NMDA receptors, and the subsequent suppression of tonic glutamate input to GABAergic interneurons results in the disinhibition of glutamatergic transmission to pyramidal neurons in the prefrontal cortex.
In animals, low doses of ketamine induce an increase in spine density, enhance mammalian target of rapamycin (mTOR) signaling, and increase protein synthesis in the prefrontal cortex, accompanied by an antidepressant effect [110]. This is consistent with reports of the reduced expression of mTOR and its downstream signaling targets in postmortem brain samples from depressed patients [111]. The GluN2B-specific antagonist Ro 25-6981 (Table 3) induced robust antidepressant-like effects [112]. GluN2B antagonists trigger their antidepressant effects by altering the activity of mTOR [110, 113], suggesting that inhibiting the activity of GluN2B subunit-containing NMDA receptors accounts for the majority of the antidepressant effects of ketamine. A recent study by Wang et al. [114] reported that replacing GluN2B with GluN2A in genetically modified mice enhanced the expression of synaptic AMPA receptors by activating mTOR signaling, which resembles ketamine-induced changes.
Table 3.
| Agonist and co-agonist | GluN2A | GluN2B | GluN2C | GluN2D | GluN3A | GluN3B |
| EC50 (µM) | ||||||
| Glutamate | 7.7 | 2.3 | 1 | 0.39 | ||
| Glycine | 1.2 | 0.38 | 0.32 | 0.12 | 57 | 95 |
| Competitive antagonists | GluN2A | GluN2B | GluN2C | GluN2D | GluN3A | GluN3B |
| Ki (µM) | ||||||
| (R)-AP5 | 0.28 | 0.46 | 1.6 | 3.7 | ||
| UBP141 | 14 | 19 | 4.2 | 2.8 | ||
| Channel blockers | GluN2A | GluN2B | GluN2C | GluN2D | GluN3A | GluN3B |
| IC50 (µM) | ||||||
| (+)MK801 | 0.015 | 0.0099 | 0.024 | 0.038 | ||
| Ketamine | 5.4 | 5.1 | 1.2 | 2.9 | ||
| Phencyclidine | 0.82 | 0.16 | 0.16 | 0.22 | ||
| Noncompetitive antagonists | GluN2A | GluN2B | GluN2C | GluN2D | GluN3A | GluN3B |
| IC50 (µM) | ||||||
| Ifenprodil | 39 | 0.15 | 29 | 76 | ||
| Ro25-6981 | 52 | 0.009 | ||||
| Allosteric potentiators | GluN2A | GluN2B | GluN2C | GluN2D | GluN3A | GluN3B |
| EC50 (µM) | ||||||
| CIQ | NE | NE | 2.8 | 3.0 |
NE, no detectable effect.
GLYX-13 is an amidated tetrapeptide (threonine-proline-proline-threonine) and glycine-site modulator at the NMDA receptor. It also preferentially modulates GluN2B subunit-containing NMDA receptors [115]. GLYX-13 and ketamine increased both GluN2B and GluR1 protein levels, but no changes in mRNA expression level [56]. GLYX-13 induces an antidepressant-like effect in the absence of the usual side effects associated with ketamine, at least partially by directly modulating GluN2B subunit-containing NMDA receptors in the medial prefrontal cortex [56]. Altogether, a direct or indirect antagonist that is selective for the GluN2B subunit would be a new-generation antidepressant candidate for the treatment of TRD.
Autism and Animal Models
In the Diagnostic and Statistical Manual of Mental Disorders, 5th edition (http://www.dsm5.org/proposedrevision/Pages/NeurodevelopmentalDisorders.aspx; acc-essed April 11, 2014), social communication impair-ments and restricted repetitive patterns of behavior are described as core symptoms of autism spectrum disorder (ASD). Many changes in ASD are intratelen-cephalic (IT)-related (for review, see [118]). The pathological changes in ASD patient are increased thickness of cortical tissue, abnormal interhemispheric and long-range cortico-cortical synchrony, resulting in a relative disconnection of IT neurons in the contralateral cortex, and reduced thickness of the corpus callosum. Causal genetic alterations have not yet been determined, but several genetic risk factors contribute to idiopathic ASD. Genetic variants of GluN2A and GluN2B in human have reportedly been associated with mental retardation, epilepsy, and autism [22, 57, 58]. One de novo mutation in GluN2B in a patient with ASD has been reported [47]. Using 151 Korean trios, a family-based association test (FBAT) provides a statistically significant association between ASD and GRIN2B haplotype [59]. GRIN2B gene alterations, including mutations and gene disruption by apparently balanced chromosomal rearrangements, have been described in patients with intellectual disability and ASD [119].
Rare mutations in SHANK3 have been reported in patients with idiopathic ASD [60, 61]. Shank family proteins are scaffolding proteins that organize a cytoskeleton-associated signaling complex at the postsynaptic density of excitatory synapses, including NMDA receptors. Mutation in SHANK3 induces ASD, however the population of SHANK3 mutations is small in ASD [62]. Similarly, mutations in SHANK2 and SHANK1 associate with idiopathic ASD [61]. A de novo deletion in SHANK1 is detected in an unrelated male individual with ASD with higher functioning [121]. A rare autosomal SHANK1 deletion that is limited to males provides a possible contributory model for elucidating the male gender bias in autism [120].
Behavioral analyses of Shank1 mutant mice have shown impairments in social interaction and communication, increased self-grooming, and repetitive behaviors [121, 122] but enhanced spatial learning and memory [123]. Shank2 mutant mice carry a mutation that is identical to the ASD-associated microdeletion of SHANK2 gene (exons 6 and 7) [124]. This mutation results in a markedly decrease in NMDA receptor complex function and ASD-like behavior, containing reduced social interaction and communication (reflected by ultrasonic vocalizations, USVs), and repetitive jumping [124]. The NMDA receptor-AMPA receptor ratio in the synapse of Shank2 mutant mice is reduced, and GluN2A and GluN2B subunit-containing NMDA receptors are equally affected in Shank2 mutant mice [124]. Shank2 mutant mice with exon 7 deletion showed similar behavioral abnormalities and NMDA receptor hyperfunction [125]. Several lines of Shank3 mutant mice shows reduced social interaction and affiliation behaviors [126-129]. These mice exhibited alterations in the levels of synaptic glutamate receptors, that is, GluA1 (AMPA receptor subunit) in the hippocampus, GluA2 (AMPA receptor subunit), GluN2A and GluN2B in the striatum. Shank3 knockdown with a small-interfering RNA (siRNA) caused significant reductions of ionic or synaptic currents via NMDA receptors and reduced surface GluR1 expressions in rat cortical cultures [130].
Neuroligins are neuronal postsynaptic cell adhesion molecules. Neuroligin-1 (NL-1) is preferentially distributed at excitatory synapses [132]. NL-1 KO mice are also known as an animal model of ASD that exhibits a marked increase in repetitive grooming behavior similar to increased repetitive behavior observed in autism. This repetitive grooming abnormality in NL1 KO mice is associated with decrease in the ratio of the NMDA to the AMPA at synapses in corticostriatal pathway [132].
Autism has been reliably associated with electrophysiological endophenotypes that may be caused by NMDA receptor disruption on parvalbumin (PV)-containing interneurons [133]. In human, M1 (N1 event-related potential as measured by magneto-encephalography) latency is shifted by approximately 10% in ASD patients [134]. The N1 event-related potential is a negative peak approximately 100 ms after sensory stimulation that is linked to early attention [135]. In PV containing cell-type selective GluN1 KO mice, delayed N1 event-related potential latency, reduced sociability, and impairment of mating-related USVs are observed [133]. In mice, social behavior is known to be expressed as social investigation, intermale aggression, sexual behavior, and parental behavior. When aggressive behavior or sexual motivation of the test mouse toward the stimulus mouse is low in the social interaction test, a significant correlation is found between delayed N1 latency and reduced sociability (i.e., social approach with same-sex gonadectomized mouse) but not between N1 latency and premating USV power (i.e., social approach with female mouse) or T-maze performance [133]. Reduced sociability is one of core symptoms in the patients of ASD [134]. Poor USV emission mimics the social communication impairments in ASD. T-maze learning involves finding a food reward in one of two available locations at opposite ends of a T-shaped apparatus. Reversal requires the mouse to extinguish the location of the reinforcer in one arm and learn a new location of the reinforcer in the other arm. Failure to switch to the new position may be analogous to the inflexibility of routines that is characteristic of autism [136]. Therefore, the increased N1 latency, impairments of sociability and USVs in PV-selective GluR1 KO mice have some analogy with characteristic of autism [133]. Treatment with the NMDA receptor functional glycine site partial agonist GLYX-13 rescued the USV deficits [63]. Considering that GLYX-13 rescued the pro-social USV deficits, the reduction of NMDA receptor function may be an important therapeutic target for autism. Further investigation of the precise mechanisms that modulate the reduction of sociability, poor USV, and inflexible reversal learning is required.
Genetic alterations of GluN2A or 2B subunits are reported in humans with autism. As animal models of ASD, Shank mutations or NL1 KO alter NMDA receptor function and induce ASD-like behavior, including reduced sociability and stereotyped grooming behavior. PV-selective GluN1 KO mice exhibited delayed N1 latency, reduced sociability, impaired mating-related USVs, and inflexible T-maze learning. Various animal studies of ASD revealed that reduced NMDA receptor function in corticostriatal PV-containing neurons is involved in ASD-like behavior. The improvement of USV deficits by the partial agonist GLYX-13 in animal models might be extended to the treatment of autism in humans.
NMDA Receptor Antagonist-Induced Psychosis
NMDA receptor antagonists, including PCP, ketamine, and MK-801, induce experimentally abnormal behavior in rodents and transient psychosis in humans. NMDA receptor antagonists overstimulate neurons, but locally administered PCP does not induce excitation. The mechanism of NMDA receptor antagonist-induced excitation has been hypothesized to involve the NMDA receptor-modulated inhibition of GABA interneurons and disinhibition of pyramidal neurons, consistent with reductions of extracellular GABA levels and elevations of extracellular glutamate levels [64-66].
NMDA receptor antagonists result in an increase of cortical high-frequency or gamma oscillations in animals, and these phenomena might be involved in psychosis [67, 68]. Systemic administration of an NMDA receptor antagonist increased the power of high-frequency oscillations. Tetrodotoxin infusion directly into the nucleus accumbens immediately and markedly reduced the power of accumbal high-frequency oscillations, that is correlated with changes in high-frequency oscillations recorded in distant cortical sites, suggesting that the nucleus accumbens is an important neural generator of oscillations [137].
Ketamine induces negative symptoms which are related to the degree of its occupancy of NMDA receptors [138]. Neuroimaging data suggest that the production of schizophrenia-like symptoms by ketamine is associated with frontal cortical activation, reflected by increases in frontal cortical perfusion [139-141], cortical glucose metabolism [142, 143], and glutamate levels [144]. Acute or chronic administration of ketamine both differentially and brain region-specifically modulates acetylcholine, dopamine, serotonin, and norepinephrine levels. Additionally, chronic administration of ketamine markedly reduces the glycine levels and induced gene expression changes of important neurotransmitter receptor systems, e.g. some members of the dopamine and serotonin receptor families [145]. Chronic administration of ketamine but not MK-801 elicited a reduction of the peak oscillatory frequency of kainic acid-elicited oscillations in ex vivo slices [146]. de Bartolomeisa et al. [147] show that ketamine and MK-801 induce the expression of Homer1a and Arc early genes in cortical regions, whereas ketamine and MK-801 reduce Homer1b and PSD-95 expression in cortical and striatal regions. These findings suggest that ketamine and MK-801 might have slightly different mechanisms of action in the induction of experimental psychotic-like behavior in rodents.
Systemic PCP also produces long-lasting excitation in the prefrontal cortex along with the enhanced locomotor activity and behavioral stereotypies in rats [148]. Comparisons of the effects of acute ketamine and PCP administration generally show that PCP increases impulsive responding. Ketamine does not have the same effect like PCP on impulsive responding and consequently produces more subtle cognitive deficits in attentional set-shifting [149]. Interestingly, ketamine, which is more frequently used in clinical settings, does not result in extensive cognitive deficits induced by using PCP administration [149]. As previously mentioned, inhibition of the activity of GluN2B-containing NMDA receptors can be responsible for the majority of the antidepressant effects of ketamine. Although ketamine can act on the GluN2B subunit, the influence of GluN2B antagonism by ketamine may differ between wildtype and disease-model mice. Acute application of Ro 25-6981, a GluN2B-selective antagonist, rescues LTP and gamma oscillation deficits in slices derived from Ts65Dn mice (i.e., a Down syndrome model mice associated with cognitive impairment), whereas prolonged treatment induces the persistent rescue of LTP [150]. In contrast, Ro 25-6981 has no effect on LTP in wildtype mice but reduces gamma oscillations both acutely and following prolonged treatment [150].
PCP is not known to have antidepressive actions. Chronic PCP exposure intensely affects the relative immunoreactivity of GluN2 subunits in the frontal cortex. PCP induces significant increases in GluN2D immunoreactivity and protein expression, and a shift to a predominance of the GluN2D subunit in the frontal cortex [151]. Recently, we showed that the action of PCP involves the GluN2D subunit. In wildtype and GluN2A KO mice, PCP increases locomotor activity and extracellular dopamine levels in the striatum and prefrontal cortex, but not in GluN2D KO mice [69] (Table 4). Acute or repeated administration of PCP is not able to increase locomotor activity in GluN2D KO mice [69] (Table 4). These results indicate that acting of PCP at the GluN2D subunit induces enhanced dopaminergic transmission and increases in locomotor activity. To determine the precise mechanism of action of PCP, the effects of a GluN2C/2D antagonist and GluN2C/2D potentiator were investigated in wildtype and GluN2D KO mice. PCP and UBP141 (a GluN2C/2D antagonist) induced potent motor impairment in wildtype mice but not in GluN2D KO mice [70] (Table 4). In contrast, CIQ, a GluN2C/2D potentiator, induced severe motor impairment in GluN2D KO mice but not in wildtype mice [70] (Table 4), suggesting that the GluN2D subunit plays an essential role in the effects of PCP and UBP141, and an appropriate balance between the GluN2C and GluN2D subunits might be needed for appropriate motor performance. GluN2D subunits mainly exist in brainstem structures, the globus pallidus, the thalamus, and the subthalamic nucleus. The c-fos gene is differentially and PCP-dependently expressed in wildtype and GluN2D KO mice, and the number of Fos-positive cells increases after PCP administration in the basal ganglia motor circuit in wildtype mice but not in GluN2D KO mice [70] (Table 4). These results suggest that the GluN2D subunit within motor circuitry is a key subunit for PCP-induced motor impairment, which requires an intricate balance between GluN2C- and GluN2D-mediated excitatory outputs.
Table 4.
Differential effects of antagonists or potentiators between GluN2 subunit genotypes.
| Treatment | WT | GluN2A KO | GluN2D KO | Ref. No. | |
|---|---|---|---|---|---|
| Dopamine release (in vivo brain dialysis) | PCP (3 mg/kg, s.c.) | ↑↑ | ↑↑ | → | [69] |
| Locomotor activity | PCP (3 mg/kg, s.c.) | ↑↑ | ↑↑ | → | [69] |
| Sensitization | Subchronic PCP (3 mg/kg, s.c.) | ↑↑ | → | → | [69] |
| Motor performance (rotarod test) | PCP (3 or 5 mg/kg, s.c.) | ↓↓ | n.d. | ↓ | [70] |
| UBP141 (3 mM, 20 µl, i.c.) | ↓↓ | n.d. | → | [70] | |
| CIQ (20 mg/kg, i.p.) | → | n.d. | ↓↓ | [70] | |
| c-fos expression | PCP (10 mg/kg, s.c.) | ↑↑↑ | n.d. | ↑ or → | [70] |
| UBP141 (3 mM, 20 µl, i.c.) | ↑ | n.d. | → | [70] | |
| CIQ (20 mg/kg, i.p.) | ↑↑↑ | n.d. | ↑↑↑ | [70] |
n.d., not determined.
PCP and ketamine are used as experimental psychosis-inducing drugs in rodents. Prenatal treatment with PCP causes cognitive impairment of memory, PCP sensitization, and deficits of sensorimotor gating [152, 153]. These behavioral deficits may be caused by impairments in the neuronal progenitor proliferations and decreased densities of glutamatergic neurons in the prefrontal cortex following prenatal PCP treatment [154]. Prenatally PCP-administered mice display behavioral deficits in cognitive memory and sensorimotor gating until adulthood, and they are used as an experimental model of schizophrenia. The administration of D-serine improved the PCP-induced reduction of prepulse inhibition and cognitive deficits [71, 72]. Glycine type I (GlyT1) transport inhibitors (GTIs) are applied for PCP-induced deficits and have been proven effective (for review, see [73]). Furthermore, the cognitive impairment and behavioral effects caused by NMDA receptor antagonists may be reduced by facilitating GABAergic neurotransmission [155], administering metabotropic glutamate 2/3 receptor agonists [74-76], or prior embryonic medial ganglionic eminence cell transplantation in the medial prefrontal cortex that many of these cells differentiate into cortical GABAergic interneurons [78]. Systemic administration of CIQ (a positive allosteric modulator selective for GluN2C/GluN2D-containing NMDA receptors) reversed deficits induced by MK-801 in prepulse inhibition in mice [77].
Concluding remarks
NMDA receptors have been implicated in physiological processes, and the dysfunction of NMDA receptors is known to be involved in various psychiatric disorders. The hypofunction of NMDA receptors might be a common mechanism that underlies schizophrenia and anti-NMDA receptor encephalitis. Unique findings related to the physiological functions of the GluN2B subunit in mood disorders provide important information that may have wide clinical applications. Our finding that PCP preferentially acts on the GluN2D subunit in vivo suggests the possibility that the GluN2D molecule might be a main target related to PCP-induced psychosis. Subunit-selective treatments may have important clinical implications.
Fig. (1).
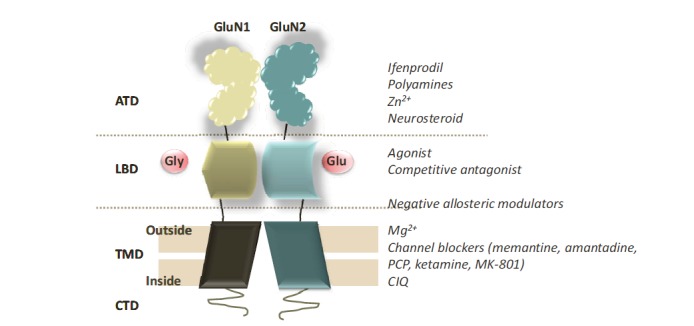
Model of the structure of NMDA receptors. NMDA receptors are glutamate-activated ion channels in the presence of glycine expressed throughout the central nervous system. The full NMDA receptor is a tetramer, but only a GluN1/GluN2 dimer is shown. These receptors are mainly composed of two glycine (Gly)-binding GluN1 subunits and two glutamate (Glu)-binding GluN2 subunits. Each subunit is organized into four distinct domains: an extracellular N-terminal domain (ATD), a ligand-binding domain (LBD), a transmembrane pore-forming domain (TMD), and an intracellular C-terminal domain (CTD) [19].
ACKNOWLEDGEMENTS
We acknowledge Mr. Michael Arends for assistance with editing the manuscript. This work was supported by grants from the Ministry of Health, Labour and Welfare of Japan (H22-Iyaku-015 and H25-Iyaku-020), JSPS KAKENHI (grant no. 23390377 and 24659549), MEXT KAKENHI (grant no. 25116532), the Smoking Research Foundation, the Naito Foundation, and the Astellas Foundation for Research on Metabolic Disorders.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Moriyoshi K., Masu M., Ishii T., et al. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- 2.Yamazaki M., Mori H., Araki K., Mori K.J., Mishina M. Cloning, expression and modulation of a mouse NMDA receptor subunit. FEBS Lett. 1992;300:39–45. doi: 10.1016/0014-5793(92)80160-i. [DOI] [PubMed] [Google Scholar]
- 3.Monyer H., Sprengel R., Schoepfer R., et al. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 4.Kutsuwada T., Kashiwabuchi N., Mori H., et al. Molecular diversity of the NMDA receptor channel. Nature. 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- 5.Ikeda K., Nagasawa M., Mori H., et al. Cloning and expression of the epsilon 4 subunit of the NMDA receptor channel. FEBS Lett. 1992;313:34–38. doi: 10.1016/0014-5793(92)81178-o. [DOI] [PubMed] [Google Scholar]
- 6.Sugihara H., Moriyoshi K., Ishii T., Masu M., Nakanishi S. Structures and properties of seven isoforms of the NMDA receptor generated by alternative splicing. Biochem. Biophys. Res. Commun. 1992;185:826–832. doi: 10.1016/0006-291x(92)91701-q. [DOI] [PubMed] [Google Scholar]
- 7.Nakanishi N., Axel R., Shneider N.A. Alternative splicing generates functionally distinct N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. USA. 1992;89:8552–8556. doi: 10.1073/pnas.89.18.8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durand G.M., Gregor P., Zheng X., et al. Cloning of an apparent splice variant of the rat N-methyl-D-aspartate receptor NMDAR1 with altered sensitivity to polyamines and activators of protein kinase C. Proc. Natl. Acad. Sci. USA. 1992;89:9359–9363. doi: 10.1073/pnas.89.19.9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anantharam V., Panchal R.G., Wilson A., et al. Combinatorial RNA splicing alters the surface charge on the NMDA receptor. FEBS Lett. 1992;305:27–30. doi: 10.1016/0014-5793(92)80648-z. [DOI] [PubMed] [Google Scholar]
- 10.Ciabarra A.M., Sullivan J.M., Gahn L.G., et al. Cloning and characterization of chi-1: a developmentally regulated member of a novel class of the ionotropic glutamate receptor family. J. Neurosci. 1995;15:6498–6508. doi: 10.1523/JNEUROSCI.15-10-06498.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sucher N.J., Akbarian S., Chi C.L., et al. Developmental and regional expression pattern of a novel NMDA receptor-like subunit (NMDAR-L) in the rodent brain. J. Neurosci. 1995;15:6509–6520. doi: 10.1523/JNEUROSCI.15-10-06509.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersson O., Stenqvist A., Attersand A., von Euler G. Nucleotide sequence, genomic organization, and chromosomal localization of genes encoding the human NMDA receptor subunits NR3A and NR3B. Genomics. 2001;78:178–184. doi: 10.1006/geno.2001.6666. [DOI] [PubMed] [Google Scholar]
- 13.Nishi M., Hinds H., Lu H.P., Kawata M., Hayashi Y. Motoneuron-specific expression of NR3B, a novel NMDA-type glutamate receptor subunit that works in a dominant-negative manner. J. Neurosci. 2001;21:RC185. doi: 10.1523/JNEUROSCI.21-23-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chatterton J.E., Awobuluyi M., Premkumar L.S., et al. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature. 2002;415:793–798. doi: 10.1038/nature715. [DOI] [PubMed] [Google Scholar]
- 15.Matsuda K., Kamiya Y., Matsuda S., Yuzaki M. Cloning and characterization of a novel NMDA receptor subunit NR3B: a dominant subunit that reduces calcium permeability. Brain Res. Mol. Brain Res. 2002;100:43–52. doi: 10.1016/s0169-328x(02)00173-0. [DOI] [PubMed] [Google Scholar]
- 16.Akazawa C., Shigemoto R., Bessho Y., Nakanishi S., Mizuno N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol. 1994;347:150–160. doi: 10.1002/cne.903470112. [DOI] [PubMed] [Google Scholar]
- 17.Monyer H., Burnashev N., Laurie D.J., Sakmann B., Seeburg P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe M., Inoue Y., Sakimura K., Mishina M. Developmental changes in distribution of NMDA receptor channel subunit mRNAs. Neuroreport. 1992;3:1138–1140. doi: 10.1097/00001756-199212000-00027. [DOI] [PubMed] [Google Scholar]
- 19.Traynelis S.F., Wollmuth L.P., McBain C.J., et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. 2004. [DOI] [PubMed]
- 21.Paoletti P. Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci. 2011;33:1351–1365. doi: 10.1111/j.1460-9568.2011.07628.x. [DOI] [PubMed] [Google Scholar]
- 22.Endele S., Rosenberger G., Geider K., et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet. 2010;42:1021–1026. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- 23.Wyllie D.J., Behe P., Nassar M., Schoepfer R., Colquhoun D. Single-channel currents from recombinant NMDA NR1a/NR2D receptors expressed in Xenopus oocytes. Proc. Biol. Sci. 1996;263:1079–1086. doi: 10.1098/rspb.1996.0159. [DOI] [PubMed] [Google Scholar]
- 24.Clarke R.J., Johnson J.W. NMDA receptor NR2 subunit dependence of the slow component of magnesium unblock. J. Neurosci. 2006;26:5825–5834. doi: 10.1523/JNEUROSCI.0577-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gielen M., Siegler Retchless B., Mony L., Johnson J.W., Paoletti P. Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature. 2009;459:703–707. doi: 10.1038/nature07993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan H., Hansen K.B., Vance K.M., Ogden K.K., Traynelis S.F. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J. Neurosci. 2009;29:12045–12058. doi: 10.1523/JNEUROSCI.1365-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vicini S., Wang J.F., Li J.H., et al. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J. Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- 28.Wyllie D.J., Behe P., Colquhoun D. Single-channel activations and concentration jumps: comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. J. Physiol. 1998;510(Pt 1):1–18. doi: 10.1111/j.1469-7793.1998.001bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vance K.M., Simorowski N., Traynelis S.F., Furukawa H. Ligand-specific deactivation time course of GluN1/GluN2D NMDA receptors. Nat. Commun. 2011;2:294. doi: 10.1038/ncomms1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vance K.M., Hansen K.B., Traynelis S.F. GluN1 splice variant control of GluN1/GluN2D NMDA receptors. J. Physiol. 2012;590:3857–3875. doi: 10.1113/jphysiol.2012.234062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan T.J., Kopanitsa M.V., Indersmitten T., et al. Evolution of GluN2A/B cytoplasmic domains diversified vertebrate synaptic plasticity and behavior. Nat. Neurosci. 2013;16:25–32. doi: 10.1038/nn.3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brigman J.L., Daut R.A., Wright T., et al. GluN2B in corticostriatal circuits governs choice learning and choice shifting. Nat. Neurosci. 2013;16:1101–1110. doi: 10.1038/nn.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogden K.K., Khatri A., Traynelis S.F., Heldt S.A. Potentiation of GluN2C/D NMDA Receptor Subtypes in the Amygdala Facilitates the Retention of Fear and Extinction Learning in Mice. Neuropsychopharmacology. 2014;39:625–637. doi: 10.1038/npp.2013.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henson M.A., Roberts A.C., Perez-Otano I., Philpot B.D. Influence of the NR3A subunit on NMDA receptor functions. Prog. Neurobiol. 2010;91:23–37. doi: 10.1016/j.pneurobio.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong H.K., Liu X.B., Matos M.F., et al. Temporal and regional expression of NMDA receptor subunit NR3A in the mammalian brain. J. Comp. Neurol. 2002;450:303–317. doi: 10.1002/cne.10314. [DOI] [PubMed] [Google Scholar]
- 36.Tong G., Takahashi H., Tu S., et al. Modulation of NMDA receptor properties and synaptic transmission by the NR3A subunit in mouse hippocampal and cerebrocortical neurons. J. Neurophysiol. 2008;99:122–132. doi: 10.1152/jn.01044.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henson M.A., Larsen R.S., Lawson S.N., et al. Genetic deletion of NR3A accelerates glutamatergic synapse maturation. PLoS One. 2012;7:e42327. doi: 10.1371/journal.pone.0042327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das S., Sasaki Y.F., Rothe T., et al. Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature. 1998;393:377–381. doi: 10.1038/30748. [DOI] [PubMed] [Google Scholar]
- 39.Roberts A.C., Diez-Garcia J., Rodriguiz R.M., et al. Downregulation of NR3A-containing NMDARs is required for synapse maturation and memory consolidation. Neuron. 2009;63:342–356. doi: 10.1016/j.neuron.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohamad O., Song M., Wei L., Yu S.P. Regulatory roles of the NMDA receptor GluN3A subunit in locomotion, pain perception and cognitive functions in adult mice. J. Physiol. 2013;591:149–168. doi: 10.1113/jphysiol.2012.239251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Citri A., Malenka R.C. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- 42.Moghaddam B., Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37:4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stefansson H., Ophoff R.A., Steinberg S., et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schizophrenia Psychiatric Genome-Wide Association Study C. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ayalew M., Le-Niculescu H., Levey D.F., et al. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol. Psychiatry. 2012;17:887–905. doi: 10.1038/mp.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cross-Disorder Group of the Psychiatric Genomics C Genetic Risk Outcome of Psychosis C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarabeux J., Kebir O., Gauthier J., et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry. 2011;1:e55. doi: 10.1038/tp.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bitanihirwe B.K., Lim M.P., Kelley J.F., Kaneko T., Woo T.U. Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry. 2009;9:71. doi: 10.1186/1471-244X-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tuominen H.J., Tiihonen J., Wahlbeck K. Glutamatergic drugs for schizophrenia: a systematic review and meta-analysis. Schizophr. Res. 2005;72:225–234. doi: 10.1016/j.schres.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 50.Javitt D.C. Glycine transport inhibitors in the treatment of schizophrenia. Handbook Exp. Pharmacol. 2012;213:367–399. doi: 10.1007/978-3-642-25758-2_12. [DOI] [PubMed] [Google Scholar]
- 51.Costa B.M., Irvine M.W., Fang G., et al. A novel family of negative and positive allosteric modulators of NMDA receptors. J. Pharmacol. Exp. Ther. 2010;335:614–621. doi: 10.1124/jpet.110.174144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mikasova L., De Rossi P., Bouchet D., et al. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain. 2012;135:1606–1621. doi: 10.1093/brain/aws092. [DOI] [PubMed] [Google Scholar]
- 53.Kayser M.S., Titulaer M.J., Gresa-Arribas N., Dalmau J. Frequency and characteristics of isolated psychiatric episodes in anti-N-methyl-d-aspartate receptor encephalitis. JAMA Neurol. 2013;70:1133–1139. doi: 10.1001/jamaneurol.2013.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berman R.M., Cappiello A., Anand A., et al. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 55.Zarate C.A., Jr, Singh J., Manji H.K. Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol. Psychiatry. 2006;59:1006–1020. doi: 10.1016/j.biopsych.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 56.Burgdorf J., Zhang X.L., Nicholson K.L., et al. GLYX-13, a NMDA receptor glycine-site functional partial agonist, induces antidepressant-like effects without ketamine-like side effects. Neuropsychopharmacology. 2013;38:729–742. doi: 10.1038/npp.2012.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O'Roak B.J., Vives L., Girirajan S., et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Talkowski M.E., Rosenfeld J.A., Blumenthal I., et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoo H.J., Cho I.H., Park M., Yang S.Y., Kim S.A. Family based association of GRIN2A and GRIN2B with Korean autism spectrum disorders. Neurosci. Lett. 2012;512:89–93. doi: 10.1016/j.neulet.2012.01.061. [DOI] [PubMed] [Google Scholar]
- 60.Durand C.M., Betancur C., Boeckers T.M., et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang Y.H., Ehlers M.D. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78:8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moessner R., Marshall C.R., Sutcliffe J.S., et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burgdorf J., Moskal J.R., Brudzynski S.M., Panksepp J. Rats selectively bred for low levels of play-induced 50 kHz vocalizations as a model for autism spectrum disorders: a role for NMDA receptors. Behav. Brain Res. 2013;251:18–24. doi: 10.1016/j.bbr.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yonezawa Y., Kuroki T., Kawahara T., Tashiro N., Uchimura H. Involvement of gamma-aminobutyric acid neurotransmission in phencyclidine-induced dopamine release in the medial prefrontal cortex. Eur. J. Pharmacol. 1998;341:45–56. doi: 10.1016/s0014-2999(97)01435-0. [DOI] [PubMed] [Google Scholar]
- 65.Moghaddam B., Adams B., Verma A., Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Amitai N., Kuczenski R., Behrens M.M., Markou A. Repeated phencyclidine administration alters glutamate release and decreases GABA markers in the prefrontal cortex of rats. Neuropharmacology. 2012;62:1422–1431. doi: 10.1016/j.neuropharm.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pinault D. N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol. Psychiatry. 2008;63:730–735. doi: 10.1016/j.biopsych.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 68.Wood J., Kim Y., Moghaddam B. Disruption of prefrontal cortex large scale neuronal activity by different classes of psychotomimetic drugs. J. Neurosci. 2012;32:3022–3031. doi: 10.1523/JNEUROSCI.6377-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hagino Y., Kasai S., Han W., et al. Essential role of NMDA receptor channel epsilon4 subunit (GluN2D) in the effects of phencyclidine, but not methamphetamine. PLoS One. 2010;5:e13722. doi: 10.1371/journal.pone.0013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamamoto H., Kamegaya E., Sawada W., et al. Involvement of the N-methyl-D-aspartate receptor GluN2D subunit in phencyclidine-induced motor impairment, gene expression, and increased Fos immunoreactivity. Mol. Brain. 2013;6:56. doi: 10.1186/1756-6606-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lipina T., Labrie V., Weiner I., Roder J. Modulators of the glycine site on NMDA receptors, D-serine and ALX 5407, display similar beneficial effects to clozapine in mouse models of schizophrenia. Psychopharmacology (Berl.) 2005;179:54–67. doi: 10.1007/s00213-005-2210-x. [DOI] [PubMed] [Google Scholar]
- 72.Hashimoto K., Fujita Y., Ishima T., Chaki S., Iyo M. Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of the glycine transporter-1 inhibitor NFPS and D-serine. Eur. Neuropsychopharmacol. 2008;18:414–421. doi: 10.1016/j.euroneuro.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 73.Javitt D.C. Glutamatergic theories of schizophrenia. Isr. J. Psychiatry Relat. Sci. 2010;47:4–16. [PubMed] [Google Scholar]
- 74.Homayoun H., Jackson M.E., Moghaddam B. Activation of metabotropic glutamate 2/3 receptors reverses the effects of NMDA receptor hypofunction on prefrontal cortex unit activity in awake rats. J. Neurophysiol. 2005;93:1989–2001. doi: 10.1152/jn.00875.2004. [DOI] [PubMed] [Google Scholar]
- 75.Moghaddam B., Adams B.W. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- 76.Krystal J.H., Abi-Saab W., Perry E., et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology (Berl.) 2005;179:303–309. doi: 10.1007/s00213-004-1982-8. [DOI] [PubMed] [Google Scholar]
- 77.Suryavanshi P.S., Ugale R.R., Yilmazer-Hanke D., Stairs D.J., Dravid S.M. GluN2C/GluN2D subunit-selective NMDA receptor potentiator CIQ reverses MK-801-induced impairment in prepulse inhibition and working memory in Y-maze test in mice. Br. J. Pharmacol. 2014;171:799–809. doi: 10.1111/bph.12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tanaka D.H., Toriumi K., Kubo K., Nabeshima T., Nakajima K. GABAergic precursor transplantation into the prefrontal cortex prevents phencyclidine-induced cognitive deficits. J. Neurosci. 2011;31:14116–14125. doi: 10.1523/JNEUROSCI.2786-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gejman P.V., Sanders A.R., Kendler K.S. Genetics of schizophrenia: new findings and challenges. Annu. Rev. Genomics Hum. Genet. 2011;12:121–144. doi: 10.1146/annurev-genom-082410-101459. [DOI] [PubMed] [Google Scholar]
- 80.Sorensen H.J., Nielsen P.R., Pedersen C.B., et al. Population impact of familial and environmental risk factors for schizophrenia: A nationwide study. Schizophr. Res. 2014;153:214–219. doi: 10.1016/j.schres.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 81.Ng M.Y., Levinson D.F., Faraone S.V., et al. Meta-analysis of 32 genome-wide linkage studies of schizophrenia. Mol. Psychiatry. 2009;14:774–785. doi: 10.1038/mp.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Makino C., Shibata H., Ninomiya H., Tashiro N., Fukumaki Y. Identification of single-nucleotide polymorphisms in the human N-methyl-D-aspartate receptor subunit NR2D gene, GRIN2D, and association study with schizophrenia. Psychiatr. Genet. 2005;15:215–221. doi: 10.1097/00041444-200509000-00014. [DOI] [PubMed] [Google Scholar]
- 83.Sodhi M.S., Simmons M., McCullumsmith R., Haroutunian V., Meador-Woodruff J.H. Glutamatergic gene expression is specifically reduced in thalamocortical projecting relay neurons in schizophrenia. Biol. Psychiatry. 2011;70:646–654. doi: 10.1016/j.biopsych.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kirov G., Pocklington A.J., Holmans P., et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fromer M., Pocklington A.J., Kavanagh D.H., et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Akbarian S., Sucher N.J., Bradley D., et al. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 1996;16:19–30. doi: 10.1523/JNEUROSCI.16-01-00019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kristiansen LV, Beneyto M, Haroutunian V, Meador-Woodruff JH. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. 2006. [DOI] [PubMed]
- 88.Beneyto M., Meador-Woodruff J.H. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology. 2008;33:2175–2186. doi: 10.1038/sj.npp.1301604. [DOI] [PubMed] [Google Scholar]
- 89.Pinacho R., Villalmanzo N., Roca M., et al. Analysis of Sp transcription factors in the postmortem brain of chronic schizophrenia: a pilot study of relationship to negative symptoms. J. Psychiatr. Res. 2013;47:926–934. doi: 10.1016/j.jpsychires.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 90.Weickert C.S., Fung S.J., Catts V.S., et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol. Psychiatry. 2013;18:1185–1192. doi: 10.1038/mp.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ibrahim H.M., Hogg A.J., Jr, Healy D.J., et al. Ionotropic glutamate receptor binding and subunit mRNA expression in thalamic nuclei in schizophrenia. Am. J. Psychiatry. 2000;157:1811–1823. doi: 10.1176/appi.ajp.157.11.1811. [DOI] [PubMed] [Google Scholar]
- 92.Clinton S.M., Meador-Woodruff J.H. Abnormalities of the NMDA Receptor and Associated Intracellular Molecules in the Thalamus in Schizophrenia and Bipolar Disorder. Neuropsychopharmacology. 2004;29:1353–1362. doi: 10.1038/sj.npp.1300451. [DOI] [PubMed] [Google Scholar]
- 93.Clinton S.M., Haroutunian V., Meador-Woodruff J.H. Up-regulation of NMDA receptor subunit and post-synaptic density protein expression in the thalamus of elderly patients with schizophrenia. J. Neurochem. 2006;98:1114–1125. doi: 10.1111/j.1471-4159.2006.03954.x. [DOI] [PubMed] [Google Scholar]
- 94.Dracheva S., Byne W., Chin B., Haroutunian V. Ionotropic glutamate receptor mRNA expression in the human thalamus: absence of change in schizophrenia. Brain Res. 2008;1214:23–34. doi: 10.1016/j.brainres.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gao X.M., Sakai K., Roberts R.C., et al. Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am. J. Psychiatry. 2000;157:1141–1149. doi: 10.1176/appi.ajp.157.7.1141. [DOI] [PubMed] [Google Scholar]
- 96.Beneyto M., Kristiansen L.V., Oni-Orisan A., McCullumsmith R.E., Meador-Woodruff J.H. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology. 2007;32:1888–1902. doi: 10.1038/sj.npp.1301312. [DOI] [PubMed] [Google Scholar]
- 97.McCullumsmith R.E., Kristiansen L.V., Beneyto M., et al. Decreased NR1, NR2A, and SAP102 transcript expression in the hippocampus in bipolar disorder. Brain Res. 2007;1127:108–118. doi: 10.1016/j.brainres.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pilowsky L.S., Bressan R.A., Stone J.M., et al. First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol. Psychiatry. 2006;11:118–119. doi: 10.1038/sj.mp.4001751. [DOI] [PubMed] [Google Scholar]
- 99.Vrajova M., Stastny F., Horacek J., et al. Expression of the hippocampal NMDA receptor GluN1 subunit and its splicing isoforms in schizophrenia: postmortem study. Neurochem. Res. 2010;35:994–1002. doi: 10.1007/s11064-010-0145-z. [DOI] [PubMed] [Google Scholar]
- 100.Priya A, Johar K, Wong-Riley MT. Specificity protein 4 functionally regulates the transcription of NMDA receptor subunits GluN1, GluN2A, and GluN2B. Biochim Biophys Acta. 1833;1833:2745–56. doi: 10.1016/j.bbamcr.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lewis D.A. Inhibitory neurons in human cortical circuits: substrate for cognitive dysfunction in schizophrenia. Curr. Opin. Neurobiol. 2014;26:22–26. doi: 10.1016/j.conb.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Belforte J.E., Zsiros V., Sklar E.R., et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Steiner J., Walter M., Glanz W., et al. Increased prevalence of diverse N-methyl-D-aspartate glutamate receptor antibodies in patients with an initial diagnosis of schizophrenia: specific relevance of IgG NR1a antibodies for distinction from N-methyl-D-aspartate glutamate receptor encephalitis. JAMA Psychiatry. 2013;70:271–278. doi: 10.1001/2013.jamapsychiatry.86. [DOI] [PubMed] [Google Scholar]
- 104.Titulaer M.J., McCracken L., Gabilondo I., et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12:157–165. doi: 10.1016/S1474-4422(12)70310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gresa-Arribas N., Titulaer M.J., Torrents A., et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13:167–177. doi: 10.1016/S1474-4422(13)70282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hashimoto K., Sawa A., Iyo M. Increased levels of glutamate in brains from patients with mood disorders. Biol. Psychiatry. 2007;62:1310–1316. doi: 10.1016/j.biopsych.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 107.Martucci L., Wong A.H., De Luca V., et al. N-methyl-D-aspartate receptor NR2B subunit gene GRIN2B in schizophrenia and bipolar disorder: Polymorphisms and mRNA levels. Schizophr. Res. 2006;84:214–221. doi: 10.1016/j.schres.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 108.Zhang C., Li Z., Wu Z., et al. A study of N-methyl-D-aspartate receptor gene (GRIN2B) variants as predictors of treatment-resistant major depression. Psychopharmacology (Berl.) 2014;231:685–693. doi: 10.1007/s00213-013-3297-0. [DOI] [PubMed] [Google Scholar]
- 109.Homayoun H., Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li N., Lee B., Liu R.J., et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jernigan C.S., Goswami D.B., Austin M.C., et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2011;35:1774–1779. doi: 10.1016/j.pnpbp.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lima-Ojeda J.M., Vogt M.A., Pfeiffer N., et al. Pharmacological blockade of GluN2B-containing NMDA receptors induces antidepressant-like effects lacking psychotomimetic action and neurotoxicity in the perinatal and adult rodent brain. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2013;45:28–33. doi: 10.1016/j.pnpbp.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 113.Li N., Liu R.J., Dwyer J.M., et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol. Psychiatry. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang C.C., Held R.G., Chang S.C., et al. A critical role for GluN2B-containing NMDA receptors in cortical development and function. Neuron. 2011;72:789–805. doi: 10.1016/j.neuron.2011.09.023. [DOI] [PubMed] [Google Scholar]
- 115.Zhang X.L., Sullivan J.A., Moskal J.R., Stanton P.K. A NMDA receptor glycine site partial agonist, GLYX-13, simultaneously enhances LTP and reduces LTD at Schaffer collateral-CA1 synapses in hippocampus. Neuropharmacology. 2008;55:1238–1250. doi: 10.1016/j.neuropharm.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ogden K.K., Traynelis S.F. New advances in NMDA receptor pharmacology. Trends Pharmacol. Sci. 2011;32:726–733. doi: 10.1016/j.tips.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kvist T., Steffensen T.B., Greenwood J.R., et al. Crystal Structure and Pharmacological Characterization of a Novel NMDA Receptor Antagonist at the GluN1 Glycine Binding Site. J. Biol. Chem. 2013;288:33124–33135. doi: 10.1074/jbc.M113.480210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shepherd G.M. Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 2013;14:278–291. doi: 10.1038/nrn3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dimassi S., Andrieux J., Labalme A., et al. Interstitial 12p13.1 deletion involving GRIN2B in three patients with intellectual disability. Am. J. Med. Genet. A. 2013;161:2564–2569. doi: 10.1002/ajmg.a.36079. [DOI] [PubMed] [Google Scholar]
- 120.Sato D., Lionel A.C., Leblond C.S., et al. SHANK1 Deletions in Males with Autism Spectrum Disorder. Am. J. Hum. Genet. 2012;90:879–887. doi: 10.1016/j.ajhg.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Silverman J.L., Turner S.M., Barkan C.L., et al. Sociability and motor functions in Shank1 mutant mice. Brain Res. 2011;1380:120–137. doi: 10.1016/j.brainres.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wohr M., Roullet F.I., Hung A.Y., Sheng M., Crawley J.N. Communication impairments in mice lacking Shank1: reduced levels of ultrasonic vocalizations and scent marking behavior. PLoS One. 2011;6:e20631. doi: 10.1371/journal.pone.0020631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hung A.Y., Futai K., Sala C., et al. Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 2008;28:1697–1708. doi: 10.1523/JNEUROSCI.3032-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Won H., Lee H.R., Gee H.Y., et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature. 2012;486:261–265. doi: 10.1038/nature11208. [DOI] [PubMed] [Google Scholar]
- 125.Schmeisser M.J., Ey E., Wegener S., et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature. 2012;486:256–260. doi: 10.1038/nature11015. [DOI] [PubMed] [Google Scholar]
- 126.Bozdagi O., Sakurai T., Papapetrou D., et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism. 2010;1:15. doi: 10.1186/2040-2392-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yang M., Bozdagi O., Scattoni M.L., et al. Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. J. Neurosci. 2012;32:6525–6541. doi: 10.1523/JNEUROSCI.6107-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang X., McCoy P.A., Rodriguiz R.M., et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 2011;20:3093–3108. doi: 10.1093/hmg/ddr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Peca J., Feliciano C., Ting J.T., et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Duffney L.J., Wei J., Cheng J., et al. Shank3 deficiency induces NMDA receptor hypofunction via an actin-dependent mechanism. J. Neurosci. 2013;33:15767–15778. doi: 10.1523/JNEUROSCI.1175-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Song J.Y., Ichtchenko K., Sudhof T.C., Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. USA. 1999;96:1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Blundell J., Blaiss C.A., Etherton M.R., et al. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 2010;30:2115–2129. doi: 10.1523/JNEUROSCI.4517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Saunders J.A., Tatard-Leitman V.M., Suh J., et al. Knockout of NMDA receptors in parvalbumin interneurons recreates autism-like phenotypes. Autism Res. 2013;6:69–77. doi: 10.1002/aur.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Roberts T.P., Khan S.Y., Rey M., et al. MEG detection of delayed auditory evoked responses in autism spectrum disorders: towards an imaging biomarker for autism. Autism Res. 2010;3:8–18. doi: 10.1002/aur.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Naatanen R., Picton T. The N1 wave of the human electric and magnetic response to sound: a review and an analysis of the component structure. Psychophysiology. 1987;24:375–425. doi: 10.1111/j.1469-8986.1987.tb00311.x. [DOI] [PubMed] [Google Scholar]
- 136.Crawley J.N. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol. 2007;17:448–459. doi: 10.1111/j.1750-3639.2007.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Olszewski M., Dolowa W., Matulewicz P., Kasicki S., Hunt M.J. NMDA receptor antagonist-enhanced high frequency oscillations: Are they generated broadly or regionally specific? Eur. Neuropsychopharmacol. 2013;23:1795–1805. doi: 10.1016/j.euroneuro.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 138.Stone J.M., Erlandsson K., Arstad E., et al. Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy: a [(123)I]CNS-1261 SPET study. Psychopharmacology (Berl.) 2008;197:401–408. doi: 10.1007/s00213-007-1047-x. [DOI] [PubMed] [Google Scholar]
- 139.Holcomb H.H., Lahti A.C., Medoff D.R., Cullen T., Tamminga C.A. Effects of noncompetitive NMDA receptor blockade on anterior cingulate cerebral blood flow in volunteers with schizophrenia. Neuropsychopharmacology. 2005;30:2275–2282. doi: 10.1038/sj.npp.1300824. [DOI] [PubMed] [Google Scholar]
- 140.Holcomb H.H., Lahti A.C., Medoff D.R., Weiler M., Tamminga C.A. Sequential regional cerebral blood flow brain scans using PET with H2(15)O demonstrate ketamine actions in CNS dynamically. Neuropsychopharmacology. 2001;25:165–172. doi: 10.1016/S0893-133X(01)00229-9. [DOI] [PubMed] [Google Scholar]
- 141.Lahti A.C., Holcomb H.H., Medoff D.R., Tamminga C.A. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport. 1995;6:869–872. doi: 10.1097/00001756-199504190-00011. [DOI] [PubMed] [Google Scholar]
- 142.Vollenweider F.X., Leenders K.L., Scharfetter C., et al. Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). Eur. Neuropsychopharmacol. 1997;7:9–24. doi: 10.1016/s0924-977x(96)00039-9. [DOI] [PubMed] [Google Scholar]
- 143.Breier A., Malhotra A.K., Pinals D.A., Weisenfeld N.I., Pickar D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am. J. Psychiatry. 1997;154:805–811. doi: 10.1176/ajp.154.6.805. [DOI] [PubMed] [Google Scholar]
- 144.Stone J.M., Dietrich C., Edden R., et al. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol. Psychiatry. 2012;17:664–665. doi: 10.1038/mp.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chatterjee M., Verma R., Ganguly S., Palit G. Neurochemical and molecular characterization of ketamine-induced experimental psychosis model in mice. Neuropharmacology. 2012;63:1161–1171. doi: 10.1016/j.neuropharm.2012.05.041. [DOI] [PubMed] [Google Scholar]
- 146.McNally J.M., McCarley R.W., Brown R.E. Chronic Ketamine Reduces the Peak Frequency of Gamma Oscillations in Mouse Prefrontal Cortex Ex vivo. Front. Psychiatry. 2013;4:106. doi: 10.3389/fpsyt.2013.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.de Bartolomeis A., Sarappa C., Buonaguro E.F., et al. Different effects of the NMDA receptor antagonists ketamine, MK-801, and memantine on postsynaptic density transcripts and their topography: Role of Homer signaling, and implications for novel antipsychotic and pro-cognitive targets in psychosis. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2013;46C:1–12. doi: 10.1016/j.pnpbp.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 148.Jodo E. The role of the hippocampo-prefrontal cortex system in phencyclidine-induced psychosis: A model for schizophrenia. J. Physiol. Paris. 2013;107:434–440. doi: 10.1016/j.jphysparis.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 149.Gastambide F., Mitchell S.N., Robbins T.W., Tricklebank M.D., Gilmour G. Temporally distinct cognitive effects following acute administration of ketamine and phencyclidine in the rat. Eur. Neuropsychopharmacol. 2013;23:1414–1422. doi: 10.1016/j.euroneuro.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 150.Hanson J.E., Weber M., Meilandt W.J., et al. GluN2B antagonism affects interneurons and leads to immediate and persistent changes in synaptic plasticity, oscillations, and behavior. Neuropsychopharmacology. 2013;38:1221–1233. doi: 10.1038/npp.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lindahl J.S., Keifer J. Glutamate receptor subunits are altered in forebrain and cerebellum in rats chronically exposed to the NMDA receptor antagonist phencyclidine. Neuropsychopharmacology. 2004;29:2065–2073. doi: 10.1038/sj.npp.1300485. [DOI] [PubMed] [Google Scholar]
- 152.Lu L., Mamiya T., Lu P., et al. Prenatal exposure to phencyclidine produces abnormal behaviour and NMDA receptor expression in postpubertal mice. Int. J. Neuropsychopharmacol. 2010;13:877–889. doi: 10.1017/S1461145709990757. [DOI] [PubMed] [Google Scholar]
- 153.Lu L., Mamiya T., Lu P., et al. Prenatal exposure to PCP produces behavioral deficits accompanied by the overexpression of GLAST in the prefrontal cortex of postpubertal mice. Behav. Brain Res. 2011;220:132–139. doi: 10.1016/j.bbr.2011.01.035. [DOI] [PubMed] [Google Scholar]
- 154.Toriumi K., Mouri A., Narusawa S., et al. Prenatal NMDA receptor antagonism impaired proliferation of neuronal progenitor, leading to fewer glutamatergic neurons in the prefrontal cortex. Neuropsychopharmacology. 2012;37:1387–1396. doi: 10.1038/npp.2011.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Castner S.A., Arriza J.L., Roberts J.C., et al. Reversal of ketamine-induced working memory impairments by the GABAAalpha2/3 agonist TPA023. Biol. Psychiatry. 2010;67:998–1001. doi: 10.1016/j.biopsych.2010.01.001. [DOI] [PubMed] [Google Scholar]