

Abstract
Alterations in dendritic spine morphology and postsynaptic structure are a hallmark of neurological disorders. Particularly spine pruning of striatal medium spiny neurons and aberrant rewiring of corticostriatal synapses have been associated with the pathology of Parkinson’s disease and L-DOPA induced dyskinesia, respectively. Owing to its low activation threshold the neuronal L-type calcium channel CaV1.3 is particularly critical in the control of neuronal excitability and thus in the calcium-dependent regulation of neuronal functions. CaV1.3 channels are located in dendritic spines and contain a C-terminal class 1 PDZ domain-binding sequence. Until today the postsynaptic PDZ domain proteins shank, densin-180, and erbin have been shown to interact with CaV1.3 channels and to modulate their current properties. Interestingly experimental evidence suggests an involvement of all three PDZ proteins as well as CaV1.3 itself in regulating dendritic and postsynaptic morphology. Here we briefly review the importance of CaV1.3 and its proposed interactions with PDZ proteins for the stability of dendritic spines. With a special focus on the pathology associated with Parkinson’s disease, we discuss the hypothesis that CaV1.3 L-type calcium channels may be critical modulators of dendritic spine stability.
Keywords: CACNA1D, PDZ domain, voltage-gated calcium channels, synaptic transmission, δ-catenin, synapse stability, Parkinson’s disease, autism spectrum disorders
Introduction
Voltage-gated calcium channels regulate a multitude of neuronal functions including presynaptic neurotransmitter release and the integration of postsynaptic signals leading to gene regulation and neuronal plasticity. In order to accomplish these tasks a remarkable functional heterogeneity of neuronal calcium channels exists. On the one hand, neurons express a number of channel isoforms displaying distinct gating and current properties. On the other hand, single channels may be functionally distinct with respect to differential targeting to specific neuronal compartments, their interactions with auxiliary calcium channel subunits, or by the formation of macro-molecular complexes with specific up- and downstream signaling proteins (for review see [1, 2]). L-type voltage-dependent calcium channels (LTCCs) occupy a key position in the activity-dependent regulation of neuronal development and thereby in mediating different forms of synaptic plasticity and in activity-induced regulation of gene expression. Calcium entering neurons through CaV1.2 and CaV1.3 calcium channels in response to membrane depolarization or synaptic activity contributes to synaptic plasticity [3], synaptic scaling [4], heterosynaptic molecular dynamics [5], and transcriptional regulation [6]. Thus, it is not surprising that a deficiency of LTCC channels or their increased activity leads to aberrant brain function and neurological disorders [3, 7-9]. Owing to its relatively low activation threshold the CaV1.3 isoform is particularly critical in the control of neuronal excitability and calcium-dependent regulation of neuronal development and disease [8, 10-12]. Disorders of the CNS are often accompanied by changes in the number and morphology of dendritic spines and thus the overall synaptic structure [13]. Particularly dendritic spine loss of striatal medium spiny neurons (MSN) has previously been shown to be involved in the pathology of Parkinson’s disease (PD). Furthermore, it has been hypothesized that the loss of MSN dendritic spines may underlie the development of L-DOPA induced dyskinesia [14-16]. Interestingly, there is evidence for a contribution and thus a therapeutic potential of LTCC in both PD as well as L-DOPA induced dyskinesia, although the underlying mechanisms have not yet been addressed.
Over the last years, distinct PDZ domain scaffold proteins have been identified as interaction partners and modulators of CaV1.3 channels. Three of these scaffold proteins and CaV1.3 modulators, namely shank1/3, densin-180, and erbin, are components of the excitatory postsynaptic compartment and have also been identified as regulators of dendritic morphology and postsynaptic structure. Here we review evidence in support of CaV1.3 regulation via its PDZ protein interaction partners. With a particular focus on postsynaptic adaptations observed in Parkinson’s disease, we discuss the hypothesis that CaV1.3 L-type calcium channels may be critical modulators of dendritic spine stability.
Dendritic spine pathology in neuronal disease
Dendritic spines are considered to be hotspots for neuronal plasticity. They bear the potential of transforming alterations in local synaptic strength into long-term memory manifested by morphological alterations (reviewed in [13]). This remarkable feature of dendritic spines was discovered 15 years ago by the observation that the local synaptic induction of LTP in cultured neurons can induce the outgrowth of dendritic filopodia [17, 18]. While not all studies over the last years on the role of dendritic spines are coherent and notable exceptions to the rule may exist (discussed in [13]), the current hypothesis on the role of dendritic spines is that induction of long-lasting memory, for example by LTP, is paralleled and manifested by an increased size and stability of dendritic spines, which in turn reflects the increased synaptic strength. Conversely, changes in neuronal signaling leading to long-term depression (LTD) may induce spine pruning and ultimately to the loss of synaptic connections. Due to the multitude of causes and the multifactorial pathology of neurological disorders it is often difficult to identify the underlying disease mechanisms. Yet common to a variety of neurological disorders including autism spectrum disorders, schizophrenia, intellectual disabilities, as well as neurodegenerative diseases such as Alzheimer’s or Parkinson’s, are alterations in dendritic spine morphology, which may underlie the pathological changes in memory and cognition. The pathological dendritic spine alterations range from local hyperconnectivity in autism spectrum disorders to reduced spine size and density in schizophrenia and spine loss in Alzheimer’s disease [19]. In Parkinson’s disease morphological changes of dendritic spines are a critical aspect of the disease etiology. In addition, they are associated with treatment-induced defects in the connectivity. A wealth of studies in mice, rats and patients have meanwhile established that in MSNs of the striatum, the GABAergic striatal projection neurons undergo spine pruning in PD and PD-like animal models (reviewed in [20]). This spine loss particularly leads to a decrease in the total number of striatal asymmetric glutamatergic synapses, which also receive input from midbrain dopaminergic neurons. Recent studies using BAC transgenic mice, in which the two distinct MSN populations can be selectively visualized, predominantly revealed the loss of glutamatergic synapses and spines of striatopallidal MSNs expressing the dopamine D2 receptor [16, 21]. GABAergic MSNs constitute 90–95% of all striatal neurons and can be classified based on two separate projection pathways: neurons of the direct pathway expressing D1 dopamine receptors, and neurons of the indirect pathway expressing D2 dopamine receptors [20, 22, 23]. The reduction of dopamine in PD has been suggested to increase the excitability of D2 receptor expressing MSNs of the indirect pathway. This altered activity subsequently induced the selective loss of dendritic spines with a subsequent downregulation of excitatory synapses [24]. Recent studies aiming at resolving the mechanisms underlying the major debilitating side effects in the treatment of PD, L-DOPA-induced dyskinesia, provide additional evidence for the selective involvement of D2 expressing MSNs. Dyskinesia induced by chronic L-DOPA treatment in animal models of dopamine depletion was associated with an abnormal restoration of dendritic spines in D2 receptor expressing neurons [14, 15, 25] as well as a concomitant reduction of spines in D1 receptor expressing MSNs [16]. Together this suggests that an aberrant rewiring of cortico-striatal synapses on MSNs may underlie the pathology of L-DOPA-induced dyskinesia.
Postsynaptic CAV1.3 signaling complexes
1. Functional Heterogeneity of CaV1.3 Splice Variants
CaV1.3 α1 subunits form slowly inactivating calcium channels activating at more negative voltages than CaV1.2 [26]. These properties allow CaV1.3 to be particularly involved in regulating neuronal excitability, which is indeed supported by observations in chromaffin cells (reviewed in [27]) and dopamine midbrain neurons [28]. The early characterization of neuronal LTCC proteins predicted the existence of long and short variants of class D (α1D, CaV1.3) L-type calcium channels [29]. Indeed, it was later revealed that alternative splicing of CaV1.3 gives rise to a long (CaV1.342 or CaV1.3L) and two short (CaV1.342A; CaV1.343S) C-terminal variants, which differ in their voltage-dependence of activation and calcium conductance due to accelerated calcium-dependent inactivation in the short forms [30]. More specifically the absence of a C-terminal modulator in short splice variants facilitates CaV1.3 channel activation at lower voltages [31] and more pronounced calcium-dependent inactivation. However, single channel open probabilities are much higher for both short splice variants when compared to CaV1.3L. Both short and long splice variants are expressed in striatal MSNs [32] and in the substantia nigra, where CaV1.342A accounts for approximately 30% of the total CaV1.3 transcripts [30].
2. PDZ-protein Interaction Partners of CaV1.3
The full length variant of CaV1.3 contains a class 1 PDZ domain-binding sequence [33]. PDZ domains, which were first identified in PSD-95, the Drosophila discs large 1 (Dlg-1), and the tight junction protein zonula occludens 1(ZO-1), are highly abundant in scaffold proteins regulating the trafficking and clustering of receptors and ion channels as well as the organization of macromolecular signaling complexes (reviewed in [34]). Postsynaptic adaptor proteins of the shank family were the first identified PDZ domain interaction partners of CaV1.3 [33]. Meanwhile CaV1.3 channels have been shown to also interact with densin-180, erbin, harmonin, and whirlin. Whirlin and harmonin are both scaffold proteins implicated in the human Usher syndrome (USH) [35, 36], the most frequent cause of combined deaf-blindness [37]. In case of harmonin the inner ear defect in Usher syndrome may depend on the aberrant regulation of CaV1.3, as it is involved in voltage-dependent facilitation and exocytosis in inner hair cells [38]. Because of their particular expression in the postsynaptic compartment of central glutamatergic synapses, here we focus on the interaction of CaV1.3 with the PDZ proteins shank, erbin, and densin-180.
Shank proteins: Shank proteins are a family of postsynaptic scaffold proteins containing a variety of protein-protein interactions domains [39, 40]. These include ankyrin repeats, an SH3 domain, a PDZ domain, a long proline-rich region, and a sterile alpha motif (SAM) domain. Shank has been demonstrated to promote the selective enlargement of spine heads via a mechanism that depends on the postsynaptic recruitment of homer [41]. The interaction with homer1a thereby seems to be critical for regulating the structure and function of synapses in an activity-dependent manner [42]. A strong scaffolding role of shank proteins is also suggested by the observation that shank3 overexpression can induce the formation of functional dendritic spine synapses in aspiny cerebellar neurons [43]. An important role of shank proteins in the stability of dendritic spines is also indicated by the observation that shank1-deficient mice displayed smaller dendritic spines. This neuronal phenotype was associated with enhanced performance in a spatial learning task but impaired long-term memory [44]. The C-terminal class 1 PDZ domain-binding sequence of full length CaV1.3 α1 subunits has been shown to bind shank3 and shank1 proteins [33]. In striatal MSNs D2 dopaminergic and M1 muscarinic receptors suppress L-type calcium currents by a selective modulation of CaV1.3 channels [32]. Most importantly, this study further showed that medium spiny neurons co-express the long CaV1.3 isoform and shank1/3. Moreover the G-protein-coupled receptor modulation of CaV1.3 was disrupted by intracellular dialysis of a peptide competing for the CaV1.3 PDZ ligand and by application of peptides targeting the shank interaction with homer. Together, these findings implicate an important role of shank proteins in the formation of a signaling complex containing shank, CaV1.3 and D2 dopaminergic receptors at cortico-striatal synapses (Fig. 1).
Fig. (1).
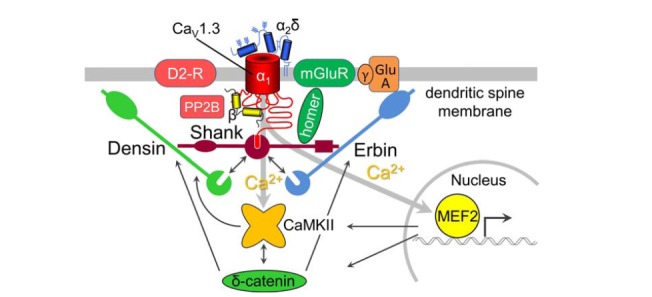
Model of the CaV1.3 signalosome in the membrane of dendritic spines: Shank proteins (shank1/3) bind to the distal C-terminus of CaV1.3 channels with their PDZ domain. Shank acts as scaffold for the association of metabotropic glutamate receptors via homer (mGluR, homer), AMPA receptors (GluA), and likely also for D2 dopamine receptors (D2-R) and calcineurin (PP2B). In striatal medium spiny neurons CaV1.3 activity is suppressed by D2-R mediated inhibition. In Parkinson’s disease dopamine depletion may thus increase CaV1.3 calcium influx, thereby destabilize the shank-CaV1.3 interaction and allow the PDZ domains of densin-180 and erbin to compete for binding to the CaV1.3 C-terminus. Subsequently erbin and densin induce alterations to dendritic spine morphology by contributing to the surface expression of AMPA receptors via the transmembrane AMPA-R regulatory proteins (TARP) γ2 (γ) and by regulating the postsynaptic localization of CaMKII and δ-catenin. Consequently increased calcium influx can activate the transcription factor myocyte enhancer factor 2 (MEF2) and regulate expression of genes involved in dendritic spine growth such as CaMKII and δ-catenin.
Densin-180: Densin was originally identified as a 180 kD (densin-180) protein constituent of the postsynaptic density fraction of rat forebrain [45]. Similar to other scaffold proteins densin-180 contains a number of protein-protein interaction motifs including leucine-rich repeats, a sialomucin domain, and a PDZ domain. Densin-180 has been shown to bind CaMKII and therefore is likely involved in anchoring CaMKII to the postsynaptic density, where it is ideally localized to respond to local calcium influx [46]. Indeed, later it was revealed that densin-180 binds the full length CaV1.3 α1 subunit via its PDZ domain and that both, densin and CaMKII, are required for calcium-dependent facilitation of CaV1.3 channels [47] (Fig. 1). Furthermore CaV1.3 and densin are both located in dendritic spines of cultured hippocampal neurons, where they may contribute to integrate postsynaptic signals [47]. Interestingly densin-180 also interacts with the shank proteins and overexpression of densin-180 in cultured hippocampal neurons has been shown to induce excessive branching of neuronal dendrites [48], which was antagonized by over-expression of shank3. Because shank blocked the binding of δ-catenin, which itself can induce excessive dendritic branching [49], the authors suggested that shank may inhibit the activation of a densin-180-dependent signaling pathway by δ-catenin.
Erbin: The PDZ protein erbin contains leucine-rich repeats and a PDZ domain [50] and is concentrated in postsynaptic membranes at the neuromuscular junction and in the central nervous system, where it also interacts with PSD-95 [51]. shRNA knockdown of erbin severely impaired dendritic morphogenesis [52]. Conversely, overexpression of erbin increased dendritic complexity. Similar to densin-180, erbin may act upstream of δ-catenin and regulate dendritic morphogenesis by maintaining appropriate localization of δ-catenin [52]. Most importantly, similar to densin-180 and shank1/3 also erbin can form a complex with the full length variant of CaV1.3 [53] (Fig. 1). Erbin co-immunprecipitated with CaV1.3 in heterologous expression systems as well as rat brain, showed somatodendritic colocalization in cultured neurons and augmented voltage-dependent facilitation (VDF) of CaV1.3 currents.
CAV1.3 as Potential Regulator of Post-synaptic Stability
1. Direct Evidence for CaV1.3 as Regulator of Dendritic Spine Morphology
As described above, all postsynaptic PDZ domain proteins interacting with CaV1.3L can alter dendritic spine morphology. This implicates a role of the calcium channel itself in regulating postsynaptic stability. However, in addition to this indirect evidence, CaV1.3 channels have also been associated with regulating dendritic spine stability in animal models of dopamine depletion, which induce a PD-like phenotype. One important piece of evidence comes from experiments with CaV1.3 knockout mice, which displayed a higher spine density in striatal MSNs that was associated with an increased mEPSC frequency [8]. In this study dopamine depletion caused a reduction in spine density in the D2 receptor-expressing MSNs and, most importantly, this reduction in spine density was not observed in the striatum of CaV1.3 knockout mice. Experiments on cultured D2 receptor expressing MSNs revealed that activity dependent spine pruning, which also involved activation of calcineurin and the transcription factor myocyte enhancer factor 2 (MEF2), was dependent on calcium influx via LTCCs [24]. However, in this case the effect was attributed to CaV1.2 channels, as spine pruning was not altered in cultures from CaV1.3 knockout mice. Nevertheless, it is conceivable that in this activity-dependent experimental paradigm (depolarization by 35 mM KCl) calcium influx through the more abundant CaV1.2 channels may be much stronger and thus sufficient to induce spine pruning even in the absence of CaV1.3. In a rodent model of L-DOPA induced dyskinesia following a nigrostriatal 6-hydroxydopamine (6-OHDA) lesion the chronic administration of L-type channel blockers reduced the development of disease as well as the associated abnormal dendritic spine formation [54]. Although a link to postsynaptic stability yet needs to be demonstrated, important insights into the potential role of CaV1.3 in neurological disease comes from recent discoveries. CACNA1D mutations associated with autism spectrum disorders [55, 56] and a severe congenital multiorgan syndrome with primary aldosteronism, seizures, and neurologic abnormalities [57, 58] suggest that CaV1.3 gain-of-function mutations may cause these diseases.
2. The Postsynaptic CaV1.3 Signalosome
The identified modulatory mechanisms of the three postsynaptic PDZ protein interaction partners of CaV1.3, densin, erbin, and shank, share a number of common principles: First, all three proteins have been demonstrated to interact with the C-terminal PDZ ligand (amino acid sequence ITTL) of the full length C-terminus of CaV1.3. Second, all three proteins are potential modulators of the stability of dendritic spines and thus synapse structure. Third, densin, erbin, and shank may act upstream of δ-catenin in regulating dendritic and postsynaptic morphogenesis. Fourth and most importantly, they all can, under certain physiological conditions, augment currents through CaV1.3 channels: densin-180, together with CaMKII, mediates calcium-dependent facilitation [47], erbin augments voltage-dependent facilitation [53], and shank mediates G-protein inhibition of L-type currents in striatal medium spiny neurons by D2 dopaminergic and M1 muscarinic receptors [32]. The latter modulation may provide a mechanism for synaptic plasticity associated with altered dendritic spine stability, namely by integrating glutamatergic signaling at corticostriatal synapses in relation to the strength of the dopaminergic input. Another link between CaV1.3 and D2 dopaminergic receptors was recently identified in substantia nigra dopamine neurons were CaV1.3 channels control the D2-autoreceptor responses [28]. Disruption of the above mentioned signaling pathways may thus contribute to a destabilization of dendritic spines, as frequently observed in neurological disease (see above). While SHANK proteins, especially SHANK3, are indeed well studied causative genes for autism spectrum disorders [40], a recent study also identified CACNA1D (CaV1.3) as risk gene [55]. Homozygous knock-out mice for densin-180 have been described to display symptoms related to schizophrenia and autism spectrum disorders associated with altered postsynaptic signaling in excitatory synapses [59]. In MSNs in Parkinson’s disease the loss of dopamine may lead to a reduction of CaV1.3 inhibition by G-protein coupled receptors. In fact D2 dopaminergic receptors and metabotropic glutamate receptors are tethered to CaV1.3 by binding via an interaction protein (e.g. homer) to shank. The aberrant regulation of this pathway will result in an increased calcium influx that can destabilize the shank-CaV1.3 interaction and favor competition of densin [48] or erbin for the PDZ ligand, which further augments calcium influx by calcium-dependent (densin [47]) or voltage-dependent (erbin [53]) facilitation. Thereby densin and erbin contribute to the postsynaptic localization of CaMKII and δ-catenin and thus changes in dendritic spine morphology. These modifications will be further enhanced by the activity-dependent activation of the transcription factor MEF2, which regulates the expression of genes involved in dendritic spine growth including CaMKII and δ-catenin. Furthermore, modulation of the surface expression of AMPA receptors by erbin binding to the transmembrane AMPA-R regulatory proteins (TARP) γ-2, which also provides another link to neuropsychiatric diseases [60], may contribute to synaptic and morphological adaptations. Alternatively also altered expression levels of PDZ domain proteins or CaV1.3 itself may affect the regulatory balance of the entire signaling complex, thereby enhancing or reducing calcium influx. Because the short splice variants of CaV1.3 lack the distal C-terminus and thus cannot bind PDZ domain proteins, changes in the expression of splice variants are also expected to influence CaV1.3 signaling. Although at this point signals and mechanisms regulating alternative splicing of CaV1.3 in neurons are elusive. An intriguing possibility for the regulation CaV1.3 calcium signaling is provided by RNA editing and by the observation that edited channels exhibit a strong reduction of calcium-dependent inactivation [61]. Taken together the available evidence indicates a role for CaV1.3 in regulating postsynaptic stability via signalling complexes established, maintained, and regulated by the C-terminal interaction partners densin, erbin, and shank.
Conclusion
Accumulating evidence implicates the C-terminal modulation of CaV1.3 channels by PDZ proteins in regulating dendritic spine morphology and thereby postsynaptic stability. In the healthy brain these mechanism may contribute to synaptic plasticity associated with LTP or LTD or homeostatic scaling of the network excitability. However, aberrant regulation of CaV1.3 caused by mutations or deregulated expression of constituents of the putative CaV1.3 signalosome are likely causes of disease-associated alterations in synaptic structure. Particularly in Parkinson’s disease and L-DOPA-induced dyskinesia the gradual reduction in striatal dopaminergic signaling may deregulate CaV1.3 calcium handling. In theory the C-terminal modulation via PDZ protein interactions may provide a novel therapeutic target for specifically inhibiting CaV1.3 channels. In fact alternative strategies for therapeutically inhibiting LTCCs are becoming increasingly important considering the limitations of available drugs to specifically discriminate between the two major neuronal isoforms CaV1.2 and CaV1.3 [62, 63]. Yet, the next step will be testing whether and how the hypothesized modulatory interactions are relevant for disease-associated destabilization of synapses.
ACKNOWLEDGEMENTS
We thank our colleagues and collaborators for helpful discussions. The authors declare no competing financial interests. This work was supported by grants from the Austrian Science Fund (FWF): F4406 and F4415 and P24079.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Obermair G., Flucher B. In: Neuronal Functions of Auxiliary Calcium Channel Subunits, Modulation of Presynaptic Calcium Channels; Stephens, G.; Mochida, S. Netherlands S., editor. 2013. pp. 29–59. [Google Scholar]
- 2.Geisler S., Schopf C.L., Obermair G.J. Emerging evidence for specific neuronal functions of auxiliary calcium channel alpha2delta subunits. Gen. Physiol. Biophys. 2015;34(2):105–118. doi: 10.4149/gpb_2014037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moosmang S., Haider N., Klugbauer N., Adelsberger H., Langwieser N., Muller J., Stiess M., Marais E., Schulla V., Lacinova L., Goebbels S., Nave K.A., Storm D.R., Hofmann F., Kleppisch T. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J. Neurosci. 2005;25(43):9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ibata K., Sun Q., Turrigiano G.G. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57(6):819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 5.Rose J., Jin S.X., Craig A.M. Heterosynaptic molecular dynamics: locally induced propagating synaptic accumulation of CaM kinase II. Neuron. 2009;61(3):351–358. doi: 10.1016/j.neuron.2008.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greer P.L., Greenberg M.E. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59(6):846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Splawski I., Timothy K.W., Sharpe L.M., Decher N., Kumar P., Bloise R., Napolitano C., Schwartz P.J., Joseph R.M., Condouris K., Tager-Flusberg H., Priori S.G., Sanguinetti M.C., Keating M.T. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Day M., Wang Z., Ding J., An X., Ingham C.A., Shering A.F., Wokosin D., Ilijic E., Sun Z., Sampson A.R., Mugnaini E., Deutch A.Y., Sesack S.R., Arbuthnott G.W., Surmeier D.J. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 2006;9(2):251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- 9.Busquet P., Nguyen N.K., Schmid E., Tanimoto N., Seeliger M.W., Ben-Yosef T., Mizuno F., Akopian A., Striessnig J., Singewald N. CaV1.3 L-type Ca2+ channels modulate depression-like behaviour in mice independent of deaf phenotype. Int. J. Neuropsychopharmacol. 2010;13(4):499–513. doi: 10.1017/S1461145709990368. [DOI] [PubMed] [Google Scholar]
- 10.Singh A., Gebhart M., Fritsch R., Sinnegger-Brauns M.J., Poggiani C., Hoda J.C., Engel J., Romanin C., Striessnig J., Koschak A. Modulation of voltage- and Ca2+-dependent gating of CaV1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J. Biol. Chem. 2008;283(30):20733–20744. doi: 10.1074/jbc.M802254200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hasreiter J., Goldnagl L., Bohm S., Kubista H. Cav1.2 and Cav1.3 L-type calcium channels operate in a similar voltage range but show different coupling to Ca(2+)-dependent conductances in hippocampal neurons. Am. J. Physiol. Cell Physiol. 2014;306(12):C1200–C1213. doi: 10.1152/ajpcell.00329.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berger S.M., Bartsch D. The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res. 2014;357(2):463–476. doi: 10.1007/s00441-014-1936-3. [DOI] [PubMed] [Google Scholar]
- 13.Sala C., Segal M. Dendritic spines: the locus of structural and functional plasticity. Physiol. Rev. 2014;94(1):141–188. doi: 10.1152/physrev.00012.2013. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y., Meredith G.E., Mendoza-Elias N., Rademacher D.J., Tseng K.Y., Steece-Collier K. Aberrant restoration of spines and their synapses in L-DOPA-induced dyskinesia: involvement of corticostriatal but not thalamostriatal synapses. J. Neurosci. 2013;33(28):11655–11667. doi: 10.1523/JNEUROSCI.0288-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishijima H., Suzuki S., Kon T., Funamizu Y., Ueno T., Haga R., Suzuki C., Arai A., Kimura T., Suzuki C., Meguro R., Miki Y., Yamada J., Migita K., Ichinohe N., Ueno S., Baba M., Tomiyama M. Morphologic changes of dendritic spines of striatal neurons in the levodopa-induced dyskinesia model. Mov. Disord. 2014;29(3):336–343. doi: 10.1002/mds.25826. [DOI] [PubMed] [Google Scholar]
- 16.Fieblinger T., Graves S.M., Sebel L.E., Alcacer C., Plotkin J.L., Gertler T.S., Chan C.S., Heiman M., Greengard P., Cenci M.A., Surmeier D.J. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat. Commun. 2014;5:5316. doi: 10.1038/ncomms6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maletic-Savatic M., Malinow R., Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283(5409):1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- 18.Engert F., Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399(6731):66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- 19.Penzes P., Vanleeuwen J.E. Impaired regulation of synaptic actin cytoskeleton in Alzheimer's disease. Brain Res. Brain Res. Rev. 2011;67(1-2):184–192. doi: 10.1016/j.brainresrev.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villalba R.M., Smith Y. Differential striatal spine pathology in Parkinson's disease and cocaine addiction: a key role of dopamine? Neuroscience. 2013;251:2–20. doi: 10.1016/j.neuroscience.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Surmeier D.J., Ding J., Day M., Wang Z., Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30(5):228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Surmeier D.J., Graves S.M., Shen W. Dopaminergic modulation of striatal networks in health and Parkinson's disease. Curr. Opin. Neurobiol. 2014;29c:109–117. doi: 10.1016/j.conb.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fieblinger T., Sebastianutto I., Alcacer C., Bimpisidis Z., Maslava N., Sandberg S., Engblom D., Cenci M.A. Mechanisms of dopamine D1 receptor-mediated ERK1/2 activation in the parkinsonian striatum and their modulation by metabotropic glutamate receptor type 5. J. Neurosci. 2014;34(13):4728–4740. doi: 10.1523/JNEUROSCI.2702-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian X., Kai L., Hockberger P.E., Wokosin D.L., Surmeier D.J. MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Mol. Cell. Neurosci. 2010;44(1):94–108. doi: 10.1016/j.mcn.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suarez L.M., Solis O., Carames J.M., Taravini I.R., Solis J.M., Murer M.G., Moratalla R. L-DOPA treatment selectively restores spine density in dopamine receptor D2-expressing projection neurons in dyskinetic mice. Biol. Psychiatry. 2014;75(9):711–722. doi: 10.1016/j.biopsych.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 26.Koschak A., Reimer D., Huber I., Grabner M., Glossmann H., Engel J., Striessnig J. alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J. Biol. Chem. 2001;276(25):22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- 27.Mahapatra S., Calorio C., Vandael D.H., Marcantoni A., Carabelli V., Carbone E. Calcium channel types contributing to chromaffin cell excitability, exocytosis and endocytosis. Cell Calcium. 2012;51(3-4):321–330. doi: 10.1016/j.ceca.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 28.Dragicevic E., Poetschke C., Duda J., Schlaudraff F., Lammel S., Schiemann J., Fauler M., Hetzel A., Watanabe M., Lujan R., Malenka R.C., Striessnig J., Liss B. Cav1.3 channels control D2-autoreceptor responses via NCS-1 in substantia nigra dopamine neurons. Brain. 2014;137(Pt 8):2287–2302. doi: 10.1093/brain/awu131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hell J.W., Westenbroek R.E., Warner C., Ahlijanian M.K., Prystay W., Gilbert M.M., Snutch T.P., Catterall W.A. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J. Cell Biol. 1993;123(4):949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bock G., Gebhart M., Scharinger A., Jangsangthong W., Busquet P., Poggiani C., Sartori S., Mangoni M.E., Sinnegger-Brauns M.J., Herzig S., Striessnig J., Koschak A. Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J. Biol. Chem. 2011;286(49):42736–42748. doi: 10.1074/jbc.M111.269951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lieb A., Ortner N., Striessnig J. C-terminal modulatory domain controls coupling of voltage-sensing to pore opening in Cav1.3 L-type Ca(2+) channels. Biophys. J. 2014;106(7):1467–1475. doi: 10.1016/j.bpj.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olson P.A., Tkatch T., Hernandez-Lopez S., Ulrich S., Ilijic E., Mugnaini E., Zhang H., Bezprozvanny I., Surmeier D.J. G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J. Neurosci. 2005;25(5):1050–1062. doi: 10.1523/JNEUROSCI.3327-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H., Maximov A., Fu Y., Xu F., Tang T.S., Tkatch T., Surmeier D.J., Bezprozvanny I. Association of CaV1.3 L-type calcium channels with Shank. J. Neurosci. 2005;25(5):1037–1049. doi: 10.1523/JNEUROSCI.4554-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye F., Zhang M. Structures and target recognition modes of PDZ domains: recurring themes and emerging pictures. Biochem. J. 2013;455(1):1–14. doi: 10.1042/BJ20130783. [DOI] [PubMed] [Google Scholar]
- 35.Kersten F.F., van Wijk E., van Reeuwijk J., van der Zwaag B., Marker T., Peters T.A., Katsanis N., Wolfrum U., Keunen J.E., Roepman R., Kremer H. Association of whirlin with Cav1.3 (alpha1D) channels in photoreceptors, defining a novel member of the usher protein network. Invest. Ophthalmol. Vis. Sci. 2010;51(5):2338–2346. doi: 10.1167/iovs.09-4650. [DOI] [PubMed] [Google Scholar]
- 36.Gregory F.D., Bryan K.E., Pangrsic T., Calin-Jageman I.E., Moser T., Lee A. Harmonin inhibits presynaptic Cav1.3 Ca(2)(+) channels in mouse inner hair cells. Nat. Neurosci. 2011;14(9):1109–1111. doi: 10.1038/nn.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mathur P., Yang J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta. 2014 doi: 10.1016/j.bbadis.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gregory F.D., Pangrsic T., Calin-Jageman I.E., Moser T., Lee A. Harmonin enhances voltage-dependent facilitation of Cav1.3 channels and synchronous exocytosis in mouse inner hair cells. J. Physiol. 2013;591(Pt 13):3253–3269. doi: 10.1113/jphysiol.2013.254367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheng M., Kim E. The Shank family of scaffold proteins. J. Cell Sci. 2000;113(Pt 11):1851–1856. doi: 10.1242/jcs.113.11.1851. [DOI] [PubMed] [Google Scholar]
- 40.Jiang Y.H., Ehlers M.D. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78(1):8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sala C., Piech V., Wilson N.R., Passafaro M., Liu G., Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31(1):115–130. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- 42.Sala C., Futai K., Yamamoto K., Worley P.F., Hayashi Y., Sheng M. Inhibition of dendritic spine morphogenesis and synaptic transmission by activity-inducible protein Homer1a. J. Neurosci. 2003;23(15):6327–6337. doi: 10.1523/JNEUROSCI.23-15-06327.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roussignol G., Ango F., Romorini S., Tu J.C., Sala C., Worley P.F., Bockaert J., Fagni L. Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J. Neurosci. 2005;25(14):3560–3570. doi: 10.1523/JNEUROSCI.4354-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hung A.Y., Futai K., Sala C., Valtschanoff J.G., Ryu J., Woodworth M.A., Kidd F.L., Sung C.C., Miyakawa T., Bear M.F., Weinberg R.J., Sheng M. Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 2008;28(7):1697–1708. doi: 10.1523/JNEUROSCI.3032-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Apperson M.L., Moon I.S., Kennedy M.B. Characterization of densin-180, a new brain-specific synaptic protein of the O-sialoglycoprotein family. J. Neurosci. 1996;16(21):6839–6852. doi: 10.1523/JNEUROSCI.16-21-06839.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strack S., Robison A.J., Bass M.A., Colbran R.J. Association of calcium/calmodulin-dependent kinase II with developmentally regulated splice variants of the postsynaptic density protein densin-180. J. Biol. Chem. 2000;275(33):25061–25064. doi: 10.1074/jbc.C000319200. [DOI] [PubMed] [Google Scholar]
- 47.Jenkins M.A., Christel C.J., Jiao Y., Abiria S., Kim K.Y., Usachev Y.M., Obermair G.J., Colbran R.J., Lee A. Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. 2010;30(15):5125–5135. doi: 10.1523/JNEUROSCI.4367-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quitsch A., Berhorster K., Liew C.W., Richter D., Kreienkamp H.J. Postsynaptic shank antagonizes dendrite branching induced by the leucine-rich repeat protein Densin-180. J. Neurosci. 2005;25(2):479–487. doi: 10.1523/JNEUROSCI.2699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim K., Sirota A., Chen Yh Y.H., Jones S.B., Dudek R., Lanford G.W., Thakore C., Lu Q. Dendrite-like process formation and cytoskeletal remodeling regulated by delta-catenin expression. Exp. Cell Res. 2002;275(2):171–184. doi: 10.1006/excr.2002.5503. [DOI] [PubMed] [Google Scholar]
- 50.Borg J.P., Marchetto S., Le Bivic A., Ollendorff V., Jaulin-Bastard F., Saito H., Fournier E., Adelaide J., Margolis B., Birnbaum D. ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nat. Cell Biol. 2000;2(7):407–414. doi: 10.1038/35017038. [DOI] [PubMed] [Google Scholar]
- 51.Huang Y.Z., Wang Q., Xiong W.C., Mei L. Erbin is a protein concentrated at postsynaptic membranes that interacts with PSD-95. J. Biol. Chem. 2001;276(22):19318–19326. doi: 10.1074/jbc.M100494200. [DOI] [PubMed] [Google Scholar]
- 52.Arikkath J., Israely I., Tao Y., Mei L., Liu X., Reichardt L.F. Erbin controls dendritic morphogenesis by regulating localization of delta-catenin. J. Neurosci. 2008;28(28):7047–7056. doi: 10.1523/JNEUROSCI.0451-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calin-Jageman I., Yu K., Hall R.A., Mei L., Lee A. Erbin enhances voltage-dependent facilitation of Ca(v)1.3 Ca2+ channels through relief of an autoinhibitory domain in the Ca(v)1.3 alpha1 subunit. J. Neurosci. 2007;27(6):1374–1385. doi: 10.1523/JNEUROSCI.5191-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schuster S., Doudnikoff E., Rylander D., Berthet A., Aubert I., Ittrich C., Bloch B., Cenci M.A., Surmeier D.J., Hengerer B., Bezard E. Antagonizing L-type Ca2+ channel reduces development of abnormal involuntary movement in the rat model of L-3,4-dihydroxyphenylalanine-induced dyskinesia. Biol. Psychiatry. 2009;65(6):518–526. doi: 10.1016/j.biopsych.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 55.De Rubeis S., He X., Goldberg A.P., Poultney C.S., Samocha K., Cicek A.E., Kou Y., Liu L., Fromer M., Walker S., Singh T., Klei L., Kosmicki J., Shih-Chen F., Aleksic B., Biscaldi M., Bolton P.F., Brownfeld J.M., Cai J., Campbell N.G., Carracedo A., Chahrour M.H., Chiocchetti A.G., Coon H., Crawford E.L., Curran S.R., Dawson G., Duketis E., Fernandez B.A., Gallagher L., Geller E., Guter S.J., Hill R.S., Ionita-Laza J., Jimenz Gonzalez P., Kilpinen H., Klauck S.M., Kolevzon A., Lee I., Lei I., Lei J., Lehtimaki T., Lin C.F., Ma'ayan A., Marshall C.R., McInnes A.L., Neale B., Owen M.J., Ozaki N., Parellada M., Parr J.R., Purcell S., Puura K., Rajagopalan D., Rehnstrom K., Reichenberg A., Sabo A., Sachse M., Sanders S.J., Schafer C., Schulte-Ruther M., Skuse D., Stevens C., Szatmari P., Tammimies K., Valladares O., Voran A., Li-San W., Weiss L.A., Willsey A.J., Yu T.W., Yuen R.K., Cook E.H., Freitag C.M., Gill M., Hultman C.M., Lehner T., Palotie A., Schellenberg G.D., Sklar P., State M.W., Sutcliffe J.S., Walsh C.A., Scherer S.W., Zwick M.E., Barett J.C., Cutler D.J., Roeder K., Devlin B., Daly M.J., Buxbaum J.D. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pinggera A., Lieb A., Benedetti B., Lampert M., Monteleone S., Liedl K.R., Tuluc P., Striessnig J. CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type Ca2+ channels. Biol. Psychiatry. 2015;77(9):816–822. doi: 10.1016/j.biopsych.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Azizan E.A., Poulsen H., Tuluc P., Zhou J., Clausen M.V., Lieb A., Maniero C., Garg S., Bochukova E.G., Zhao W., Shaikh L.H., Brighton C.A., Teo A.E., Davenport A.P., Dekkers T., Tops B., Kusters B., Ceral J., Yeo G.S., Neogi S.G., McFarlane I., Rosenfeld N., Marass F., Hadfield J., Margas W., Chaggar K., Solar M., Deinum J., Dolphin A.C., Farooqi I.S., Striessnig J., Nissen P., Brown M.J. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet. 2013;45(9):1055–1060. doi: 10.1038/ng.2716. [DOI] [PubMed] [Google Scholar]
- 58.Scholl U.I., Goh G., Stolting G., de Oliveira R.C., Choi M., Overton J.D., Fonseca A.L., Korah R., Starker L.F., Kunstman J.W., Prasad M.L., Hartung E.A., Mauras N., Benson M.R., Brady T., Shapiro J.R., Loring E., Nelson-Williams C., Libutti S.K., Mane S., Hellman P., Westin G., Akerstrom G., Bjorklund P., Carling T., Fahlke C., Hidalgo P., Lifton R.P. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013;45(9):1050–1054. doi: 10.1038/ng.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carlisle H.J., Luong T.N., Medina-Marino A., Schenker L., Khorosheva E., Indersmitten T., Gunapala K.M., Steele A.D., O'Dell T.J., Patterson P.H., Kennedy M.B. Deletion of densin-180 results in abnormal behaviors associated with mental illness and reduces mGluR5 and DISC1 in the postsynaptic density fraction. J. Neurosci. 2011;31(45):16194–16207. doi: 10.1523/JNEUROSCI.5877-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tao Y., Chen Y.J., Shen C., Luo Z., Bates C.R., Lee D., Marchetto S., Gao T.M., Borg J.P., Xiong W.C., Mei L. Erbin interacts with TARP gamma-2 for surface expression of AMPA receptors in cortical interneurons. Nat. Neurosci. 2013;16(3):290–299. doi: 10.1038/nn.3320. [DOI] [PubMed] [Google Scholar]
- 61.Huang H., Tan B.Z., Shen Y., Tao J., Jiang F., Sung Y.Y., Ng C.K., Raida M., Kohr G., Higuchi M., Fatemi-Shariatpanahi H., Harden B., Yue D.T., Soong T.W. RNA editing of the IQ domain in Ca(v)1.3 channels modulates their Ca(2)(+)-dependent inactivation. Neuron. 2012;73(2):304–316. doi: 10.1016/j.neuron.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sinnegger-Brauns M.J., Huber I.G., Koschak A., Wild C., Obermair G.J., Einzinger U., Hoda J.C., Sartori S.B., Striessnig J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol. Pharmacol. 2009;75(2):407–414. doi: 10.1124/mol.108.049981. [DOI] [PubMed] [Google Scholar]
- 63.Ortner N.J., Bock G., Vandael D.H., Mauersberger R., Draheim H.J., Gust R., Carbone E., Tuluc P., Striessnig J. Pyrimidine-2,4,6-triones are a new class of voltage-gated L-type Ca2+ channel activators. Nat. Commun. 2014;5:3897. doi: 10.1038/ncomms4897. [DOI] [PMC free article] [PubMed] [Google Scholar]