

Calreticulin (CALR, also known as CRT) is a chaperone of the endoplasmic reticulum (ER) involved in the maintenance of reticular homeostasis and the presentation of MHC class I antigens.1,2 In addition, CRT is actively translocated to the outer leaflet of the plasma membrane by cancer cells that die in response to some (but not all) stimuli, including specific chemotherapeutics (e.g., doxorubicin, mitoxantrone, oxaliplatin, bortezomib), hypericin-based photodynamic therapy and ionizing irradiation.3-5 In all these settings, CRT exposure on the cell surface is required for dying cells to elicit an adaptive immune response specific for dead cell-associated antigens and associated with the development of immunological memory.6 Thus, wild-type murine cancer cells undergoing such an immunogenic variant of cellular demise – which is commonly known as “immunogenic cell death” (ICD) – can be used to efficiently vaccinate syngeneic immunocompetent mice against a subsequent challenge with live cells of the same type.7 Conversely, murine cancer cells that have been depleted of CRT by specific small-interfering RNAs and killed with ICD inducers are unable to elicit protective immunity upon inoculation into syngeneic immunocompetent hosts, a defect that can be corrected by the adsorption of recombinant CRT to the surface of dying cells.8 In line with this notion, spontaneous or therapy-driven CRT exposure by malignant cells has been linked with improved disease outcome in cohorts of acute myeloid leukemia (AML) and non-small cell lung carcinoma (NSCLC) patients.9-11 Of note, the immunostimulatory activity of CRT exposed on dying cancer cells has largely been attributed to its ability to operate as an “eat-me” signal,12 hence favoring the uptake of cell corpses by phagocytes expressing LDL receptor-related protein 1 (LRP1; also known as CD91).13,14 Until now, however, CRT exposure has been mostly studied in the context of ICD, which is associated with secretion of several other danger signals, including ATP, high-mobility group box 1 (HMGB1), annexin A1 (ANXA1), and type I interferon (IFN).6 To eliminate these potential confounders from the analysis, Chen and colleagues generated murine AML cells stably expressing CRT on the outer leaflet of the plasma membrane and compared them with WT cells for immunostimulatory and pathogenic potential in vivo.15 Unsuspectedly, the capacity of CRT-exposing AML cells to initiate a protective anticancer immune response did not correlate with increased phagocytosis but was linked to type I IFN signaling.15 These data highlight an interesting and unforeseen connection between CRT exposure in the absence of cell death and type I IFN signaling.
Chen and colleagues took advantage of four murine AML models: (1) WT C1498 cells; (2) C1498 cells engineered to constitutively expose CRT on their surface as a glycosylphosphatidylinisotol (GPI)-anchored protein (C1498.CRT cells), rather than as a fusion with a transmembrane domain (as previously done)16; (3) C1498 cells expressing the Kb-restricted SIY model antigen (C1498.SIY cells); and (4) C1498 cells co-expressing SIY and membrane-exposed CRT (C1498.SIY.CRT cells). Despite similar rates of proliferation and comparable ability to activate murine SIY-specific CD8+ 2C T cells in vitro (indicating no impairment in MHC class I presentation), C1498.CRT or C1498.SIY.CRT cells were less efficient than their control counterparts at generating highly progressive tumors upon subcutaneous or intravenous inoculation into immunocompetent WT C57BL/6 mice. Such a difference was abrogated in Rag2−/− mice (which are deficient in adaptive immunity), in Tcrbtm1MomTcrdtm1Mom mice (which lack αβ and γδ T cells) or in mice receiving CD4+- and CD8+-depleting antibodies. Importantly, mice that failed to develop tumors upon inoculation of C1498.CRT or C1498.SIY.CRT cells (70% and 90%, respectively) were fully protected upon a subsequent challenge with C1498 or C1498.SIY cells, respectively, suggesting that the constitutive exposure of CRT at the plasma membrane is sufficient to generate protective immunity against AML cells in most mice. In line with this notion, mice receiving intravenously C1498.SIY.CRT cells developed increased amounts of functionally superior SIY-specific CD8+ cytotoxic T lymphocytes in the spleen as compared with mice inoculated with C1498.SIY cells. Moreover, twice the amount of adoptively transferred SIY-specific CD8+ 2C T cells accumulated in mice bearing C1498.SIY.CRT cells than in mice receiving C1498.SIY cells, and such 2C cells had an improved secretory and lytic capacity.15
Of note, virtually none of splenic CD11b+ cells (including macrophages) took up fluorescently labeled C1498 or C1498.CRT cells upon intravenous administration. Conversely, a small population of CD11c+ cells (including dendritic cells and their precursors) stained positively for C1498 cell uptake in the same setting. However, this was only marginally improved when C1498.CRT cells were used, which could not explain the large differences in tumor progression and survival observed in previous experiments. Moreover, the intravenous inoculation of C1498.CRT cells did not promote superior dendritic cell activation as compared with the administration of C1498 cells, at least in terms of MHC class I and II levels on the cell surface, expression of co-stimulatory molecules, interleukin-12 (IL-12) production, and SIY-specific CD8+ 2C T-cell priming ex vivo.15 Nonetheless, CD8α+ dendritic cells were required for the immunostimulatory effects of CRT exposure, as demonstrated upon the diphtheria toxin-mediated removal of all dendritic cells in mice expressing the diphtheria toxin receptor under the CD11c promoter, as well as in Batf3−/− mice.15
Of note, a 2-fold increase in interferon, β (Ifnb) mRNA levels was detected in the spleen of mice 24 h upon intravenous inoculation of C1498.SIY.CRT versus C1498.SIY cells, an effect mainly attributed to CD11b+, rather than CD11c+, cells. Importantly, type I IFN signaling appeared to be critical for the immunostimulatory effects of constitutively exposed CRT as the survival advantage associated with the inoculation of C1498.SIY.CRT versus C1498.SIY cells was completely abrogated in Ifnar1−/− animals.15
Altogether, the findings by Chen and colleagues unveiled an unforeseen link between CRT exposure on living cells and pathologically relevant type I IFN signaling, at least in the setting of AML. However, several questions remain to be addressed. First, which are the molecular mechanisms linking CRT signaling to Ifnb upregulation in CD11b+ cells? Second, which cell population(s) are the actual target for type I IFN signaling in this setting? Third, do other danger signals involved in ICD participate in the elicitation of antitumor immune response to live cancer cells constitutively exposing CRT? Fourth, is some extent of cell death spontaneously occurring in vivo involved in this process, implying that danger signaling from living cells is intimately connected with ICD (Fig. 1)? Answering these questions will provide additional insights into the unsuspected capacity of surface-exposed CRT to initiate type I IFN-dependent anticancer immunity in vivo.
Figure 1.
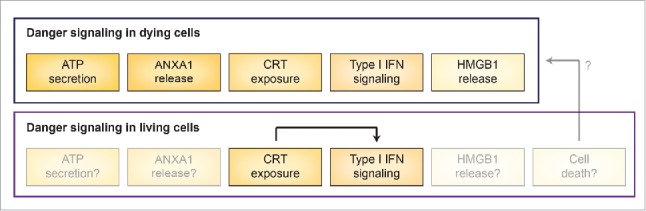
Danger signaling in dying and living cancer cells. Danger signaling in dying cells relies on exposure of calreticulin (CRT) on the outer leaflet of the plasma membrane, secretion of ATP, release of annexin A1 (ANXA1), synthesis of type I interferon (IFN), liberation of high mobility group box 1 (HMGB1), coupled with a robust cytotoxic response. Live cells appear to engage danger signaling as a consequence of CRT surface exposure resulting in type I IFN responses. It remains to be elucidated whether ATP secretion, ANXA1 production, HMGB1 release and/or some extent of cell death are also involved in this process.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 2003; 4:181-91; PMID:12612637; http://dx.doi.org/26137404 10.1038/nrm1052 [DOI] [PubMed] [Google Scholar]
- 2.Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 2011; 11:823-36; PMID:22076556; http://dx.doi.org/26137404 10.1038/nri3084 [DOI] [PubMed] [Google Scholar]
- 3.Pol J, Vacchelli E, Aranda F, Castoldi F, Eggermont A, Cremer I, Sautes-Fridman C, Fucikova J, Galon J, Spisek R et al.. Trial Watch: Immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology 2015; 4:e1008866; PMID:26137404; http://dx.doi.org/ 10.1080/2162402X.2015.1008866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adkins I, Fucikova J, Garg AD, Agostinis P, Spisek R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology 2014; 3:e968434; PMID:25964865; http://dx.doi.org/ 10.4161/21624011.2014.968434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 2014; 3:e28518; PMID:25071979; http://dx.doi.org/ 10.4161/onci.28518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2016; 17:97-111; PMID:27748397; http://dx.doi.org/25941621 10.1038/nri.2016.107 [DOI] [PubMed] [Google Scholar]
- 7.Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N et al.. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014; 3:e955691; PMID:25941621; http://dx.doi.org/ 10.4161/21624011.2014.955691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N et al.. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13:54-61; PMID:17187072; http://dx.doi.org/ 10.1038/nm1523 [DOI] [PubMed] [Google Scholar]
- 9.Fucikova J, Becht E, Iribarren K, Goc J, Remark R, Damotte D, Alifano M, Devi P, Biton J, Germain C et al.. Calreticulin expression in human non-small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res 2016; 76:1746-56; PMID:26842877; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-1142 [DOI] [PubMed] [Google Scholar]
- 10.Fucikova J, Truxova I, Hensler M, Becht E, Kasikova L, Moserova I, Vosahlikova S, Klouckova J, Church SE, Cremer I et al.. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood 2016; 128:3113-24; PMID:27802968; http://dx.doi.org/ 10.1182/blood-2016-08-731737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wemeau M, Kepp O, Tesniere A, Panaretakis T, Flament C, De Botton S, Zitvogel L, Kroemer G, Chaput N. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis 2010; 1:e104; PMID:21368877; http://dx.doi.org/ 10.1038/cddis.2010.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012; 12:860-75; PMID:23151605; http://dx.doi.org/ 10.1038/nrc3380 [DOI] [PubMed] [Google Scholar]
- 13.Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 2001; 14:303-13; PMID:11290339; http://dx.doi.org/ 10.1016/S1074-7613(01)00111-X [DOI] [PubMed] [Google Scholar]
- 14.Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, Henson PM. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med 2001; 194:781-95; PMID:11560994; http://dx.doi.org/ 10.1084/jem.194.6.781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X, Fosco D, Kline DE, Kline J. Calreticulin promotes immunity and type I interferon-dependent survival in mice with acute myeloid leukemia. Oncoimmunology 2017; In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M et al.. An immunosurveillance mechanism controls cancer cell ploidy. Science 2012; 337:1678-84; PMID:23019653; http://dx.doi.org/ 10.1126/science.1224922 [DOI] [PubMed] [Google Scholar]