

 Dehydroformylation, a challenging, underexplored reaction, can be performed under mild conditions using a designed cooperative base metal catalyst system.
Dehydroformylation, a challenging, underexplored reaction, can be performed under mild conditions using a designed cooperative base metal catalyst system.
Abstract
Dehydroformylation, or the reaction of aldehydes to produce alkenes, hydrogen gas, and carbon monoxide, is a powerful transformation that is underdeveloped despite the high industrial importance of the reverse reaction, hydroformylation. Interestingly, nature routinely performs a related transformation, oxidative dehydroformylation, in the biosynthesis of cholesterol and related sterols under mild conditions using base-metal catalysts. In contrast, chemists have recently developed a non-oxidative dehydroformylation method; however, it requires high temperatures and a precious-metal catalyst. Careful study of both approaches has informed our efforts to design a base-metal catalyzed, mild dehydroformylation method that incorporates benefits from each while avoiding several of their respective disadvantages. Importantly, we show that cooperative base metal catalysis presents a powerful, mechanistically unique approach to reactions which are difficult to achieve using conventional catalyst design.
Introduction
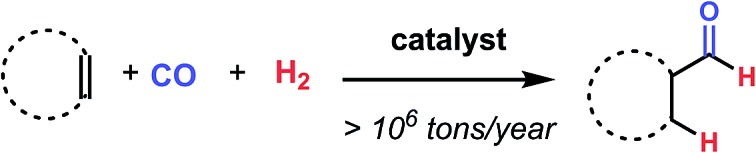
Hydroformylation, the addition of a unit of H2 and CO across an olefin to form a branched or linear aldehyde (Fig. 1), is a reaction that has been known and well-studied since its serendipitous discovery in 1938 by Otto Roelen.1 This “oxo process” is an important method of synthesis with industrial output of >10 000 000 tons per year (Fig. 1).2 So called “oxo chemicals”, either aldehydes or alcohols and olefins derived from further hydrogenation and manipulation, are important products for a multitude of industrial applications and processes. Stemming from this importance, the reaction has remained the subject of significant scientific efforts with advances continuing to be made.1b,3
Fig. 1. Hydroformylation is a highly important industrial reaction.
By contrast, the reverse reaction, dehydroformylation, or the formal loss of a unit of H2 and CO to form olefins from aldehydes, has been relatively unexplored in the literature until recently.4 Reports of the reaction do exist as early as the 1960's, often using rhodium, ruthenium, or palladium, and usually at high temperature and/or with low selectivity for the olefin over other products.5–8 While the rarity of this reaction might seem surprising given the ubiquity of hydroformylation, this can be ascribed to the presence of several challenges, including inherent endothermicity and potential for side reactivity such as hydrogenation of the product alkenes.9 Another barrier is provided by examining the microscopic reverse of the hydroformylation mechanism. Namely, a low valent, electron rich metal complex is desirable for oxidative addition into C–H bonds, such as the acyl-hydride bond.10 However, upon elimination of CO, an electron-deficient ligand is introduced into the system, which can bind the coordinatively unsaturated intermediate and hinder its insertion into another C–H bond (Fig. 2). Many previously reported dehydroformylation methods require forcing conditions to either scavenge or expel carbon monoxide from solution, suggesting that this consideration is a serious limiter of reaction efficiency.4,11 Similarly, studies on aldehyde isomerization, which proceeds through the reversible, final CO insertion/C–H reductive elimination steps of hydroformylation, have identified carbonyl-bound catalyst as a key deactivation pathway.12 Recognizing this as a significant opportunity for catalyst design, we set out to identify current progress towards dehydroformylation to inform our own efforts in the area.
Fig. 2. One challenge for achieving catalytic dehydroformylation through an organometallic mechanism is the coexistence of a low-valent, electron-rich intermediate [M] and carbon monoxide in the same reaction, leading to sequestration of the catalytically-active species as an off-cycle carbonyl adduct.

During 2015, two notable advances were made toward an efficient method for dehydroformylation, both of which deal with the problematic nature of CO in distinct ways. Dong and coworkers pursued a “transfer hydroformylation” strategy using Rh(xantphos) and a strained formyl acceptor to provide a system wherein CO is immediately sequestered by a favorable, forward hydroformylation event (Fig. 3).13 While such reactivity had been observed previously, this catalyst system represents a significant improvement over the earlier methods, where yields were low, ester side products were observed, and the reaction had to be performed at high temperature and high pressure of carbon monoxide.8 In the wake of that development, Nozaki and coworkers described a method of acceptorless dehydroformylation using a carefully engineered iridium complex to produce olefins with H2 and CO as the sole by-products.14 The optimized catalyst, a well-defined, bulky N-heterocyclic carbene (NHC) hydroxytetraphenylcyclopentadienyl iridium species (1), provides unprecedented selectivity for dehydroformylation over other potential side processes and good thermal stability (Fig. 3). This stability proved essential for its compatibility with carbon monoxide, as the elimination of it and dihydrogen require high temperatures (>160 °C) to proceed efficiently. While enabling, long-chain aldehydes often produced internal olefin isomers through undesired isomerization activity, and decarbonylation and hydrogenation side reactions can compete under some circumstances.
Fig. 3. Two recent advances towards the dehydroformylation reaction were provided in 2015. Both of these methods use noble metal catalysts and proceed through inner-sphere, organometallic mechanisms.

Interestingly, nature also utilizes a formal dehydroformylation process as a step in methyl group eliminations using a number of cytochrome P450 enzymes (Fig. 4). In this biological reaction, carbon monoxide is never formed and the iron center is never bound directly to the substrate.15,16 This “outer sphere” dehydroformylation process, which proceeds at physiological temperatures and displays excellent selectivity, has inspired the development of biomimetic transformations featuring synthetic iron porphyrin compounds.17,18
Fig. 4. Nature also pursues dehydroformylation of molecules, albeit through an oxidative, outer-sphere, stepwise process. The oxidative demethylation of lanosterol mediated by CYP51 contains an oxidative dehydroxymethylation sequence, itself containing an oxidative dehydroformylation step.

Our laboratory was drawn to aspects of each of these reported methods. The low temperature operation of the Dong transfer hydroformylation and P450-mediated reactions is appealing from an operational standpoint; however, both of these methods require sacrificial reagents in stoichiometric quantities. Conversely, the Nozaki dehydroformylation requires high temperatures for efficient reactions. Finally, the fact that the enzymatic process utilizes a base metal (iron) challenged us to restrict our thinking to earth-abundant metal strategies. With all of these considerations in mind, we set out to design a catalyst system that could capture all of these desirable characteristics, while effectively dealing with the carbon monoxide inhibition problem.
The early stages of our planning, like that of others, considered the problematic coexistence of CO and H2 in the presence of a low-valent, electron rich metal species. Inspiration for overcoming this hurdle was provided by the enzymatic dehydroformylation, a process where a high-valent, base metal species is used which never coordinates CO. Additionally, the stepwise, outer-sphere nature of this transformation suggested that we might be able to design a system wherein the substrate is never ligated directly to the metal center, freeing us from the constraint that a single catalyst must be able to effect the transformation. We wondered whether such a decoupling of the dehydrogenation and decarbonylation steps could be used to permit the use of cooperative, base metal catalysis.
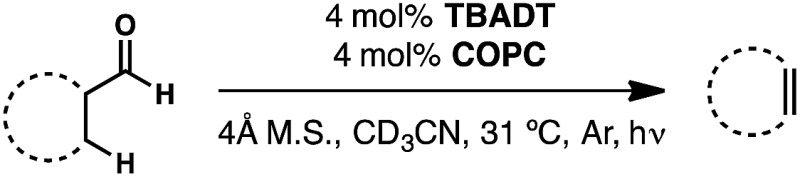
We have recently developed a cooperative catalyst system that can dehydrogenate unfunctionalized organic compounds through successive hydrogen atom transfer (HAT) events to different catalysts; this method utilizes UV light to activate one of the HAT catalysts and is capable of generating alkenes from alkanes with dihydrogen as the sole by-product (Fig. 5).19 This system features tetrabutylammonium decatungstate (TBADT), a high-valent base metal photocatalyst, to perform an initial, high-energy HAT to generate a putative, reactive organic radical intermediate. This open shell species can then transfer its highly labilized20,21 second hydrogen atom to the second catalyst, cobaloxime pyridine chloride (COPC), a vitamin B12 model compound, generating the desired alkene and two reduced catalysts. These two reduced, hydrido-catalysts can then interact to liberate dihydrogen, closing the catalytic cycle. Combined with the knowledge that both catalysts can function in the presence of CO,22–24 we considered whether such a strategy might be applicable to dehydroformylation.
Fig. 5. Cooperative hydrogen atom transfer, or the sequential removal of hydrogen atoms from a substrate by two different catalysts, has been successfully applied to the acceptorless dehydrogenation of alkanes. We reasoned that this same approach could be applied to the dehydroformylation of aldehydes.

The envisioned cycle hinges on the fact that aldehyde C–H bonds are particularly prone to homolytic activation (BDE ∼ 87 kcal mol–1) by TBADT photocatalysis.25 Following this HAT, the non-coordinated acyl radical will be free to decarbonylate, obviating the complicating formation of catalyst/carbonyl complexes through leveraging the inherent reactivity of freely diffusing acyl radicals. The ensuing alkyl radical can then transfer its labilized hydrogen atom to COPC, generating the alkene and the reduced catalysts for hydrogen evolution (Fig. 5). Entry of the Co(iii) precatalyst into the cooperative cycle is enabled by reduction by the photoreduced TBADT during the early stages of the reaction.19 Taken together, this design, a bio-inspired, “outer sphere” approach to dehydroformylation that proceeds under mild conditions using earth abundant metal catalysts, meets all the requirements stipulated at the beginning of the exercise. On the foundation of this reasoning, we set out to test the feasibility of this reaction design.
Results and discussion
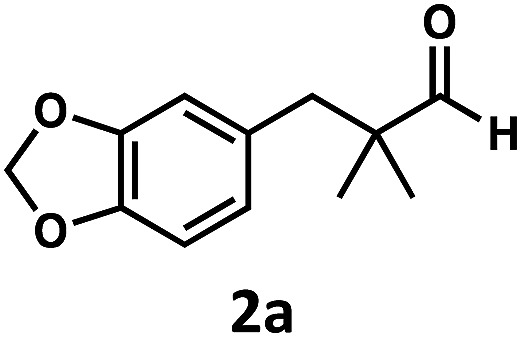
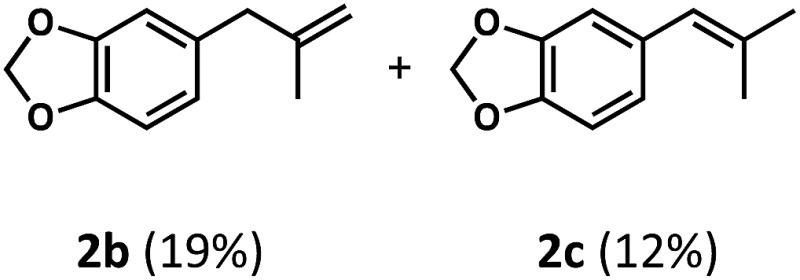
We began our studies by identifying non-enolizable aldehydes, or aldehydes with a quaternary α-carbon, as an ideal initial entry into this chemistry as their resulting acyl radicals readily decarbonylate.26 We also restricted our test substrate to one that is incapable of competitive C–H dehydrogenation under the reaction conditions, leading us to target aryl dimethyl propanal 2a. Subjecting the substrate to either catalyst with UV irradiation (TBADT maximum absorbance at 323 nm, see ESI†) yielded no or trace olefin, while subjecting it to both catalysts without irradiation similarly gave no detectable olefin (Table 1, entries 1–3). However, upon irradiation of the substrate with both catalysts at room temperature for 96 hours, two new olefins were detected in upwards of 31% yield with a terminal to internal olefin ratio of 1.6 : 1 (Table 1, entries 4–6). Hydrogen and carbon monoxide evolution were confirmed by GC-TCD analysis.
Table 1. Initial exploration of dehydroformylation a .
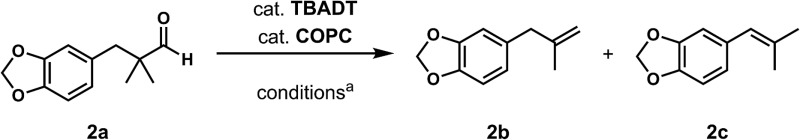
| ||||
| Entry | TBADT b (%) | COPC b (%) | Ratio (2b/2c) | Yield c (%) |
| 1 | 0 | 2 | n/a | Trace |
| 2 | 2 | 0 | n/a | 0 |
| 3 d | 2 | 2 | n/a | 0 |
| 4 e | 2 | 2 | 1.2 : 1 | 14 |
| 5 | 2 | 2 | 1.9 : 1 | 17 |
| 6 f | 4 | 4 | 1.6 : 1 | 31 |
aReaction conditions: 0.66 mmol scale, substrate concentration 0.66 M in CD3CN, 31 °C, argon, UV irradiation, 96 h.
bMol% with respect to substrate.
cYields determined via 1H NMR relative to a methyl acetate internal standard, see ESI for more detail.
dNo light.
eNo 4 Å M.S.
f0.33 mmol scale, substrate concentration 0.33 M.
This preference for the terminal olefin product reflects the known, kinetic preference of cobaloxime HAT reactions.27 Furthermore, it was found that increasing the effective catalyst loadings through reducing substrate concentration improved the outcome of the reaction; however, the limited solubility of TBADT restricted the maximum loadings achievable (See ESI for further exploration of conditions†). 4 Å molecular sieves were found to be highly beneficial to the reaction as evidenced by the reduced efficiency on their removal (Table 1, entry 4).
Closer examination of the cases without molecular sieves identified aryl aldehyde 2d as a significant side product in addition to the reduced formation of desired olefins 2b and 2c (Fig. 6). While the route by which this aldehyde product is formed is non-obvious, it is known that cobalt28 and rhodium29–33 macrocycles can promote facile C–C bond cleavage reactions in the presence of water. Both the identity of this aryl aldehyde and fact that it is not formed under anhydrous conditions is consistent with this reactivity, and further exploration of this process is a current topic of study.
Fig. 6. Aryl aldehyde 2d is a significant side product in the dehydroformylation of substrate 2a run without molecular sieves. This aldehyde can undergo the first HAT reaction but not the second, stalling the reaction through formation of a dead-end acyl radical intermediate.

Regardless of the route of formation, the inhibitory effect of aldehyde 2d can be easily understood from considering its behaviour under the reaction conditions (Fig. 6). Indeed, while the aldehyde C–H bond should still be susceptible to the first HAT step perpetrated by TBADT, the completion of the dehydroformylation is hamstrung by the reluctance of aryl acyl radicals to undergo decarbonylation. Even if this event occurs, the poor orbital overlap of the incipient singly-occupied orbital and the adjacent phenyl C–H bonds will limit the effectiveness of the second, Co-mediated HAT, rendering this reaction a dead-end process for all intents and purposes. We observed a similar dead-end, inhibitory effect on the cooperative catalyst system when attempting to dehydrogenate n-butanol,19 a case where trace n-butyraldehyde, another substrate whose acyl radical is not expected to decarbonylate readily, was formed with the subsequent stalling of the reaction. This same inhibition played out again in our study of non-α-quaternary aldehydes for dehydroformylation, vide infra (Table 3).
Table 3. Dehydroformylation of non-α-quaternary aldehydes through cooperative catalysis a .
aReaction conditions: 0.33 mmol scale, substrate concentration 0.33 M.
bYields determined via 1H NMR relative to a methyl acetate internal standard, see ESI for more detail. n.d. = not detected.
c96 h.
d72 h.
e48 h.
f24 h.
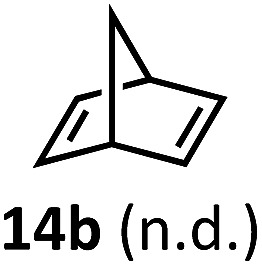
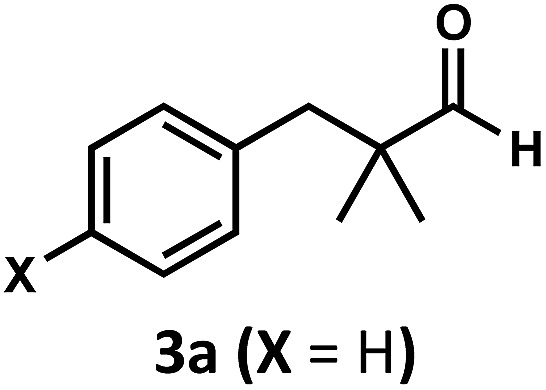
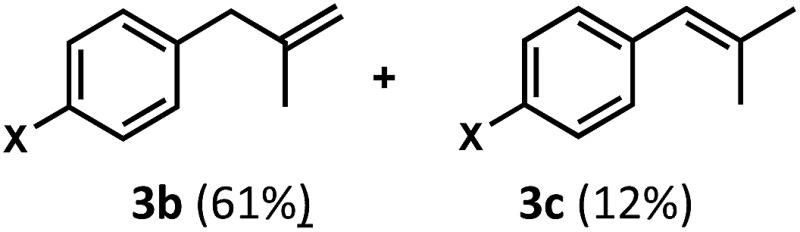
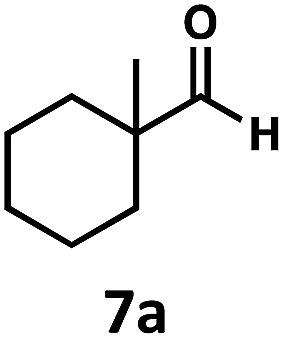
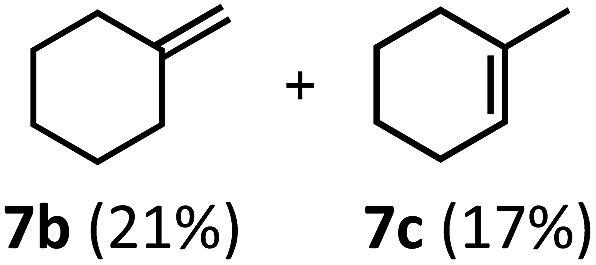
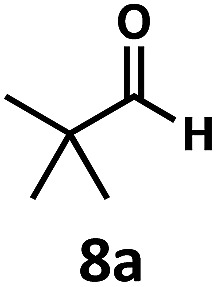
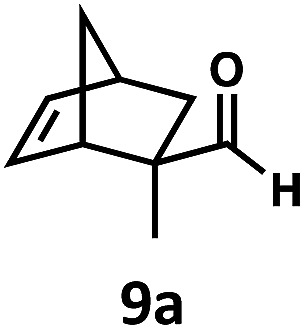
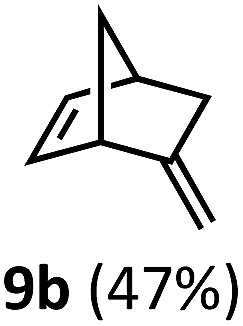
Regardless, with promising conditions in hand, we looked to other substrates to determine an initial substrate tolerance of the dehydroformylation. While appealing from a synthetic standpoint, we recognized that the dioxolane methylene of substrate 2a served as a competitive site of activation by TBADT as demonstrated by Fagnoni and coworkers,34 suggesting that the elimination of this group would increase the reaction efficiency. To our delight, subjecting phenyl dimethyl propanal 3a to the same conditions afforded two alkenes in a combined yield of 73%, and a higher selectivity of 6.3 : 1 (Table 2). The reaction was found to tolerate substitution at the para position of this framework, with the 4-cyano and 4-methyl ester compounds 4a and 5a furnishing their dehydroformylation products in high yields, including an isolated yield of 40% for products 4b and 4c and 41% for 5b and 5c. Fully substituted α-stereocenters containing a heteroatom were also cooperative, with the subjection of aryloxyaldehyde 6a to the reaction conditions providing a 1 : 1 mixture of 4-chlorophenol (6b) and acetone (6c), the hydrolysis products of the presumed enol ether product. A dehydroformylation of the purely aliphatic substrate, methylcyclohexanecarboxaldehyde 7a, also afforded the expected alkenes, although with a decreased regioselectivity of 1.2 : 1. Additionally, 2,2-dimethylpropanal 8a was rapidly transformed into isobutylene (8b) using the optimized conditions. Interestingly, 2-methyl-5-norbornene-2-carboxaldehyde 9a (4 : 1 mixture of exo : endo isomers) underwent dehydroformylation to give exclusively the exocyclic 2-methylene-5-norbornene (9b).
Table 2. Dehydroformylation of α-quaternary aldehydes through cooperative catalysis a .
| ||
| Entry | substrate | Products b (% yield) |
| 1 c |
|
|
| 2 d |
|
|
| 3 d | 4a (X = CN) | 4b (65%, 32%), 4c (13%, 8%) |
| 4 d | 5a (X = CO2CH3) | 5b (59%, 35%), 5c (9%, 6%) |
| 5 d |
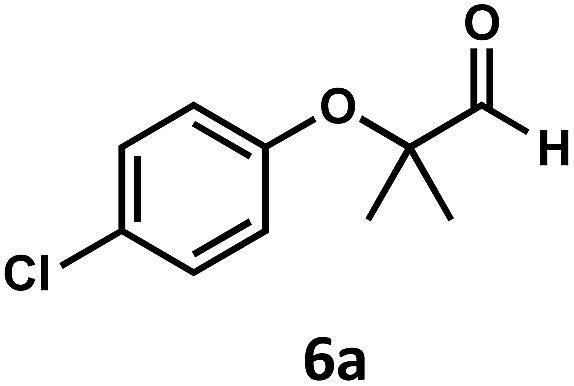
|
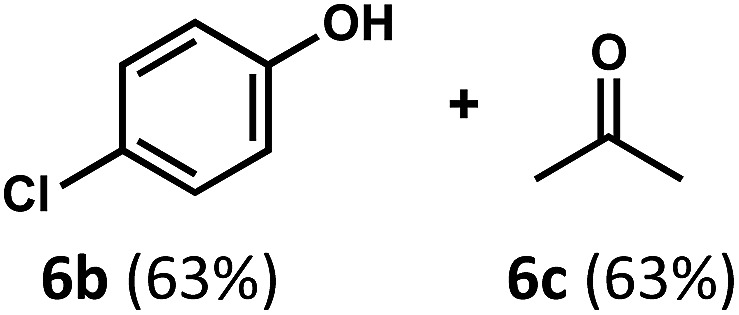
|
| 6 e |
|
|
| 7 e |
|
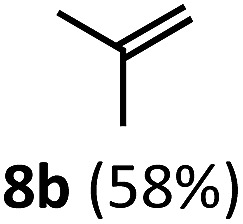
|
| 8 e |
|
|
aReaction conditions: 0.33 mmol scale, substrate concentration 0.33 M.
bYields determined via 1H NMR relative to a methyl acetate internal standard, see ESI for more detail. Underlined values correspond to isolated yields n.d. = not detected.
c96 h.
d72 h.
e48 h.
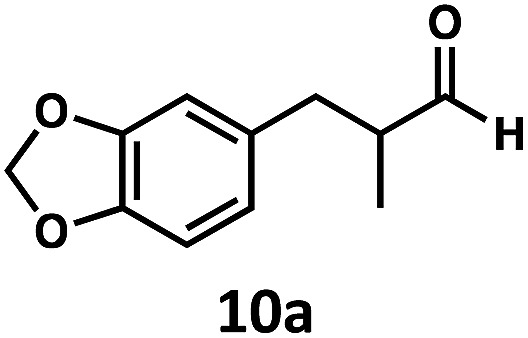
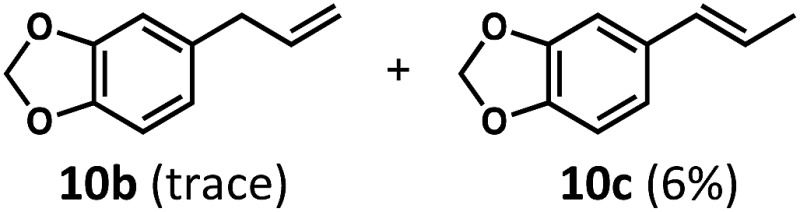
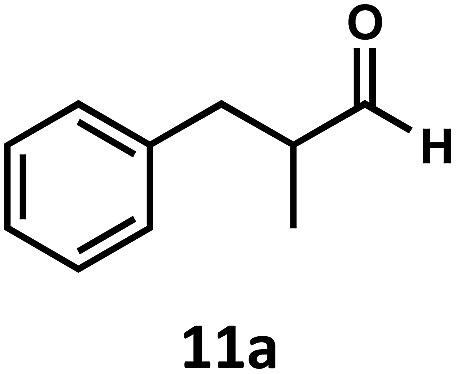
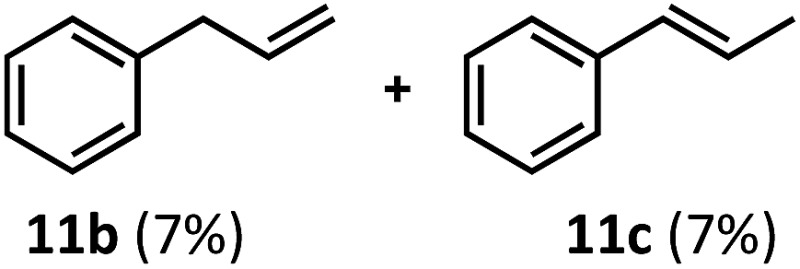
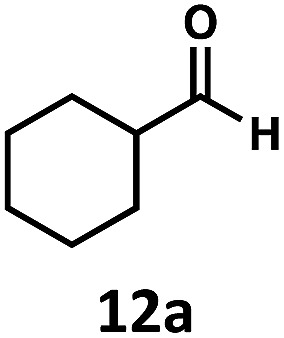
Having demonstrated the suitability of α-quaternary substrates, we wondered if α-tertiary and secondary substrates, where decarbonylation would be distinctly slower, could likewise function as suitable inputs. Aryl methyl propanal 10a yielded the terminal (safrole, 10b) and internal (isosafrole, 10c) olefins in a 1 : 2.8 ratio and combined 8% yield (Table 3). Elimination of the dioxolane functionality moderately improved the reaction outcome, with substrate 11a providing a combined 14% yield of olefins 11b and 11c. Cyclohexanecarboxaldehyde 12a produced only trace amounts of cyclohexene despite significant conversion, with the formation of extensive precipitate suggesting a polymerization-related side process. Additionally, n-octanal (13a) and norbornenecarboxaldehyde (14a) both gave no identifiable olefin products. While this limited suitability of α-tertiary and secondary aldehydes using the current conditions is disappointing, it is consistent with the greater barrier to decarbonylation of their respective acyl radicals.35 Alternatively, the lessened steric demand of non-α-quaternary acyl radicals might enable their addition to the Co(ii) metalloradical, removing the cobalt from the catalytic cycle through formation of stable36 Co–C complexes.
Although the mild dehydroformylations described herein are consistent with the cooperative HAT process outlined in Fig. 5, we sought to gain additional insight into its operative mechanism. Subjecting the reaction of 3a to a stoichiometric quantity of the radical inhibitor 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) significantly impeded the reaction (Fig. 7), a result consistent with the intermediacy of free radicals (though radical trap experiments should be interpreted with care37). This information, combined with the spectroscopic data described in our dehydrogenation study19 provide a collective body of evidence that supports a reaction mechanism featuring an irreversible loss of two gases and two catalyst-dependent hydrogen atom transfer steps.
Fig. 7. The addition of stoichiometric TEMPO significantly impedes reaction progress.

Finally, in our initial report, we also showed the capability of this dual catalytic system to perform dehydrogenations of alcohols to ketones.19 Leveraging this observation, we wondered whether the dehydroxymethylation transformation, as performed enzymatically, might be possible using this catalyst system (Fig. 4). Subjecting dimethyl phenyl propanol 15 to the optimized dehydroformylation conditions resulted in both dimethyl phenyl propanal (3a), along with the dehydroxymethylation olefin products (3b/3c), in a 1.7 : 6:1 ratio (Fig. 8), showing that such a bio-inspired, tandem dehydrogenation/dehydroformylation reaction is possible using cooperative catalysis. The presence of both the intermediate aldehyde, and an olefin ratio consistent with the relevant dehydroformylation reaction above (Table 2, entry 2), suggests to us that the alcohol is proceeding through an oxidation/dehydroformylation sequence, conceptually similar to the enzymatic strategy outlined in Fig. 4 though mechanistically distinct. Although reductive dehydroxymethylation (that is, replacing –CH2OH with –H) has been described on multiple occasions in the literature,38–40 to our knowledge this represents the first loss of a hydroxymethyl group coupled with olefin formation in a non-enzymatic transformation.
Fig. 8. The acceptorless dehydroxymethylation of primary alcohols, a hitherto-unknown chemical reaction, is possible using cooperative catalysis.

Conclusion
Herein, we report a conceptually unique approach to dehydroformylation. By considering the benefits and drawbacks of the known catalytic dehydroformylations, we developed a dual-catalytic method featuring successive HATs that, while currently limited, requires neither harsh conditions, nor sacrificial reagents. This method establishes another link between α,α′-disubstituted aldehydes and alkenes and also permits alkene formations from neopentyl alcohols by the rare process of dehydroxymethylation. Applications of these methods in organic synthesis, our efforts to achieve new bond formations between the discrete, catalyst-dependent HAT reactions, and our ongoing focus on the concept of cooperative base-metal catalysis will be described in forthcoming reports.
Acknowledgments
Financial support was provided by the National Institute of General Medical Sciences (R01 GM065483), Princeton University and a National Science Foundation Graduate Research Fellowship to J. G. W (DGE 1148900). We thank Prof. Andrew Bocarsly for the use of his GC/TCD instrument, and we thank Dr Istvaán Pelczer for his help with analyzing complex NMR spectra. Finally, we would like to thank the members of the National Science Foundation CCI Center for Selective C–H Functionalization (CHE-1205646) for inspiring our interest in C–H functionalization chemistry and useful discussions throughout our studies.
Footnotes
References
- (a) Roelen O., German Patent DE 849548, 1938/1952; US Patent 2327066, 1943; Chem. Abstr., 1944, 38, 3631. [Google Scholar]; (b) Franke R., Selent D., Borner A. Chem. Rev. 2012;112:5675–5732. doi: 10.1021/cr3001803. [DOI] [PubMed] [Google Scholar]
- Fleischer I., Wu L., Profir I., Jackstell R., Franke R., Beller M. Chem.–Eur. J. 2013;19:10589–10594. doi: 10.1002/chem.201300899. [DOI] [PubMed] [Google Scholar]
- Breit B., Seiche W. Synthesis. 2001;1:1–36. [Google Scholar]
- Nandakumar A., Sahoo M. K., Balaraman E. Org. Chem. Front. 2015;2:1422–1424. [Google Scholar]
- Prince R. H., Raspin K. A. Chem. Commun. 1966;6:156. [Google Scholar]
- United States Patent Office, US4,272,444, 1981, pp. 1–4.
- Falbe J., Hahn H. D. Chem.–Ztg. 1972;96:164. [Google Scholar]
- Kondo T., Tsuji Y., Watanabe Y. J. Org. Chem. 1990;55:1286–1291. [Google Scholar]
- Landis C. R. Science. 2015;347:29–30. doi: 10.1126/science.aaa2329. [DOI] [PubMed] [Google Scholar]
- Labinger J. A., Bercaw J. E. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]
- Iwai T., Fujihara T., Tsuji Y. Chem. Commun. 2008:6215–6217. doi: 10.1039/b813171f. [DOI] [PubMed] [Google Scholar]
- Lenges C. P., Brookhart M. Angew. Chem., Int. Ed. 1999;38:3533–3537. doi: 10.1002/(sici)1521-3773(19991203)38:23<3533::aid-anie3533>3.3.co;2-5. [DOI] [PubMed] [Google Scholar]
- Murphy S. K., Park J.-W., Cruz F. A., Dong V. M. Science. 2015;347:56–60. doi: 10.1126/science.1261232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumoto S., Tatsuki T., Nozaki K. Angew. Chem., Int. Ed. 2015;54:8458–8461. doi: 10.1002/anie.201503620. [DOI] [PubMed] [Google Scholar]
- Sen K., Hackett J. C. J. Am. Chem. Soc. 2010;132:10293–10305. doi: 10.1021/ja906192b. [DOI] [PubMed] [Google Scholar]
- Lepesheva G. I., Waterman M. R. Biochim. Biophys. Acta, Gen. Subj. 2007;1770:467–477. doi: 10.1016/j.bbagen.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y., Wada S., Morishima I., Watanabe Y. J. Inorg. Biochem. 1998;69:241–247. [Google Scholar]
- Wertz D. L., Sisemore M. F., Selke M., Driscoll J., Valentine J. S. J. Am. Chem. Soc. 1998;120:5331–5332. [Google Scholar]
- West J. G., Huang D., Sorensen E. J. Nat. Commun. 2015;6:10093. doi: 10.1038/ncomms10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. J. Org. Chem. 1998;3263:1872–1877. [Google Scholar]
- Blanksby S. J., Ellison G. B. Acc. Chem. Res. 2003;36:255–263. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- Okada M., Yamada K., Fukuyama T., Ravelli D., Fagnoni M., Ryu I. J. Org. Chem. 2015;80:9365–9369. doi: 10.1021/acs.joc.5b01850. [DOI] [PubMed] [Google Scholar]
- Lee L.-P., Schrauzer G. J. Am. Chem. Soc. 1968;90:5274–5276. doi: 10.1021/ja01021a043. [DOI] [PubMed] [Google Scholar]
- Schrauzer G. N., Weber J. H., Beckham T. M. J. Am. Chem. Soc. 1970;92:7078–7086. [Google Scholar]
- Tzirakis M. D., Lykakis I. N., Orfanopoulos M. Chem. Soc. Rev. 2009;38:2609–2621. doi: 10.1039/b812100c. [DOI] [PubMed] [Google Scholar]
- Esposti S., Dondi D., Fagnoni M., Albini A. Angew. Chem., Int. Ed. 2007;46:2531–2534. doi: 10.1002/anie.200604820. [DOI] [PubMed] [Google Scholar]
- Gridnev A. A., Ittel S. D. Chem. Rev. 2001;101:3611–3659. doi: 10.1021/cr9901236. [DOI] [PubMed] [Google Scholar]
- Lee S. Y., Fung H. S., Feng S., Chan K. S. Organometallics. 2016;35:2480–2487. [Google Scholar]
- Zhang L., Chan K. S. J. Organomet. Chem. 2006;691:3782–3787. [Google Scholar]
- Lee S. Y., Feng S., Chan K. S. Dalton Trans. 2016;45:3522–3527. doi: 10.1039/c5dt04149j. [DOI] [PubMed] [Google Scholar]
- Lee S. Y., Lai T. H., Choi K. S., Chan K. S. Organometallics. 2011;30:3691–3693. [Google Scholar]
- Lee S. Y., Chan K. S. Organometallics. 2013;32:5391–5401. [Google Scholar]
- Fung H. S., Li B. Z., Chan K. S. Organometallics. 2010;29:4421–4423. [Google Scholar]
- Murphy J. J., Bastida D., Paria S., Fagnoni M., Melchiorre P. Nature. 2016;532:218–222. doi: 10.1038/nature17438. [DOI] [PubMed] [Google Scholar]
- Chatgilialoglu C., Crich D., Komatsu M., Ryu I. Chem. Rev. 1999;99:1991–2069. doi: 10.1021/cr9601425. [DOI] [PubMed] [Google Scholar]
- Crossley S. W. M., Barabe F., Shenvi R. A. J. Am. Chem. Soc. 2014;136:16788–16791. doi: 10.1021/ja5105602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albéniz A. C., Espinet P., López-Fernández R., Sen A. J. Am. Chem. Soc. 2002;124:11278–11279. doi: 10.1021/ja0271126. [DOI] [PubMed] [Google Scholar]
- Pines H., Shamaiengar M., Postl W. S. J. Am. Chem. Soc. 1955;77:5099–5102. [Google Scholar]
- Peng L., Ma M., Zhang X., Zhang S., Wang J. Tetrahedron Lett. 2006;47:8175–8178. [Google Scholar]
- Modak A., Naveen T., Maiti D. Chem. Commun. 2013;49:252–254. doi: 10.1039/c2cc36951f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.