

ABSTRACT
Nuclear factor of activated T cells (NFAT) was first identified as a transcription factor about 3 decades ago and was not well studied until the development of immunosuppressant. Numerous studies confirm that calcineurin/NFAT signaling is very important in the development of vasculature and cardiovascular system during embryogenesis and is involved in the development of vascular diseases such as hypertension, atherosclerosis and restenosis. Recent studies demonstrated that NFAT proteins also regulate immune response and vascular cells in the pulmonary microenvironment. In this review, we will discuss how different NFAT isoforms contribute to pulmonary vascular remodeling and potential new therapeutic targets for treating pulmonary arterial hypertension.
KEYWORDS: calcineurin, hypoxia, NFAT, proliferation, pulmonary arterial hypertension, vascular remodeling
Introduction
Members of the NFAT family regulate diverse cellular process including growth and survival and are involved in the development cancer and cardiovascular diseases. The NFAT family of proteins consist of 5 isoforms: NFAT1(NFATc2/NFATp), NFAT2(NFATc1/NFATc), NFAT3(NFATc4), NFAT4(NFATc3/NFATx) and NFAT5.1 These proteins are activated and dephosphorylated in response to various types of stimulation.
Pulmonary arterial hypertension (PAH) comprises a heterogeneous group of diseases with different etiology but similar pathology and has been subcategorized as idiopathic pulmonary arterial hypertension (IPAH), familial PAH, pulmonary hypertension (PH) associated with connective tissue diseases, portopulmonary hypertension, and PH related to human viral infection, chemicals, and toxins.2 PAH is characterized by progressive proliferation of vascular cells and can be life-threatening at end stage. Growth factors, metabolic reprogramming inflammation and NFAT have been implicated in the development of PAH.3-5 The fact that NFAT is activated in both inflammatory cells and pulmonary arterial smooth muscle cells (PASMCs) within pulmonary circulation in both animal and human PAH suggests that NFAT plays a critical role in the pathogenesis of this disease. In this paper, we will discuss how NFAT proteins regulate pulmonary vascular remodeling.
Structure ad regulation of NFAT
NFAT was first found in nuclear extracts of activated T cells6 and was subsequently reported in many different tissues such as muscle, bone, neurons, viscera and skin. NFAT has 5 members which shares an amino-terminal transactivation N-domain (TAD) and a C-terminal domain. NFAT1–4 also contains Rel-homology region (RHR) and NFAT homology region (NHR), which function as DNA binding and regulatory domain, respectively.7 NFAT5 does not contain NHR and is regulated by osmotic tension.8
The regulation of the NFAT family has been described in several excellent reviews.9,10 NFAT proteins remain phosphorylated in the cytoplasm at quiescent state. Upon stimulation, NFATs are dephosphorylated by calcineurin, a serine/threonine phosphatase dependent on calcium. Dephosphorylation of NFAT results in the exposure of its nuclear localization sequence, which leads to nuclear translocation of NFAT and initiation of transcription of target genes. The transcription also requires coactivators such as activator protein 1, myocyte enhance factor 2 and members of the GATA family.11 While calcineurin is required to keep NFAT in the nucleus, several serine/threonine kinases such as casein kinase1, glycogen synthase kinase 3β (GSK3β), p38 and C-Jun N-terminal kinase export NFAT from the nucleus by phosphorylating NFAT. Beside phosphorylation, NFAT is also modulated by ubiquitination and sumoylation.12
NFAT in pulmonary vascular remodeling
NFATc1, NFATc2 and NFATc3 are the main isotypes of NFAT involved in the pathogenic process of PAH, which is characterized by inflammation, hyperproliferation of PASMC and metabolic shift.
NFATc2
Increased expression and activation of NFATc2 in PASMC are found in both human PAH as well as in monocrotaline (MCT) induced PAH animal models. NFATc2 contribute to the development of PAH by regulating the transcription of multiple inflammatory chemokines in immune cells,13 which then recruits inflammatory cells in remodeled vessels. Along this line, cyclosporine A (CsA), a inhibitor of NFATc2, was shown to reverse right ventricular hypertrophy and pulmonary vascular remodeling in MCT rat model of PAH.14 However, CsA treatment alone failed to reverse PAH in SM22–5-HTT+ mice although NFATc2 was dephosphorylated in lungs of these mice.15 Possible explanations for this interesting phenomenon include: 1) NFATc2 is not the only target of 5-HTT in SM22–5-HTT+ mice; 2) inflammation is not a critical contributor for the development of PAH in SM22–5-HTT+ mice. In vitro, several studies reported increased activation of NFATc2 in cultured PASMC in response to 5-HT, endothelin-1(ET-1) and PDGF, which has been implicated in pulmonary vascular remodeling.16,17
NFATc2 induces PASMC proliferation and resistance to apoptosis within the remodeling PA wall by reducing the expression of Kv1.5 and upregulation of anti-apoptotic protein Bcl-2.14,18 Kv1.5 has been suggested to regulate the resting plasma membrane potential in PASMC.19 Thus dysfunction of Kv1.5 in PASMC enhances NFATc2 activation by elevating cytosolic [Ca2+] through depolarization of the plasma membrane. Increased expression of Bcl2 leads to a mitochondrial membrane potential (ΔΨm) hyperpolarization and mitochondrial-dependent resistance to apoptosis.20 Additionally, as the downstream of calcineurin, NFATc2 upregulates cyclin A expression and CDK2 activation to promote primary cultured PASMC cell cycle and proliferation.16 VIVIT or CsA induces apoptosis by inhibiting NFATc2 nuclear translocation and subsequent up regulation of Kv1.5 expression and down regulation of Bcl-2 and decrease of [Ca2+], [K+].14
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that regulates the expression of NFATc2 and subsequent proliferation of PASMC.21 STAT3 positively regulates NFATc2 by 2 mechanisms: 1) regulation of NFATc2 transcription by binding to its promoter region; 2) positive regulation of the NFATc2 activator, Pim-1, which is a oncoprotein specifically overexpressed in PAH. Pim-1 promotes cell proliferation/survival by increasing Bad phosphorylation. Apart from cytokines, the mutation of some proteins, including deficiency of uncoupling protein2 or overexpression of NogoB also lead to activation of NFATc2.22,23 On the other hand, the orphan nucleus receptor Nur77 negatively regulates PASMC proliferation through inhibition of the STAT3/Pim-1/NFATc2 pathway.24
NFATc2 is also implicated in metabolic shift to aerobic glycolysis which plays an important role in the development of PAH.25 Increased activation of NFATc2 was observed in PASMC stimulated by TNFα, which correlated with the inhibition of PASMC pyruvate dehydrogenase (PDH) and mitochondrial dysfunction.26 On the other hand, as the consequence of mitochondrial metabolic abnormality, the inhibition of metabolic signaling protein GSK3β also contributes to NFATc2 nuclear accumulation.27 The effects and regulation of NFATc2 on PAH are summarized in Fig. 1.
Figure 1.
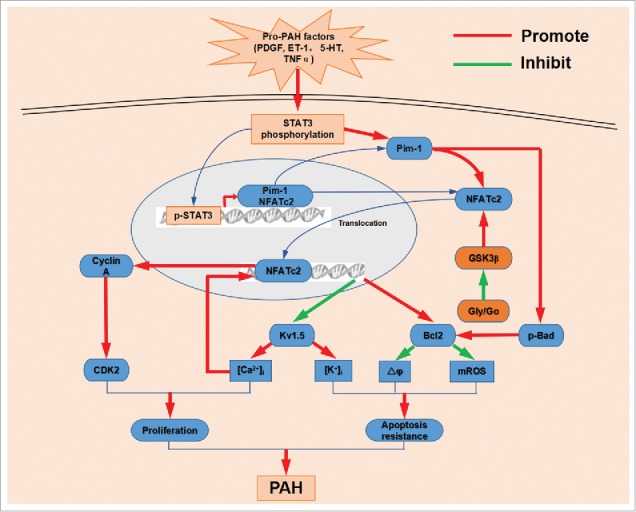
The mechanisms of NFATc2 promote PAH. Pro-PAH factors increase STAT3 phosphorylation. Then phosphorylated STAT3 translocates into nucleus and increases NFATc2 and Pim-1 expression. Pim-1 triggers NFATc2 dephosphorylation and nuclear translocation and Bad phosphorylation, which inhibits kv1.5 expression and promotes Bcl2 expression. Aerobic glycolysis inhibits GSK3β activation, which increases the nuclear localization of NFATc2. downregulation of kv1.5 results in increase of [Ca2+]i and [K+]i. Upregulated Bcl2 hyperpolarizes mitochondrial membrane potential(ΔΨm) and lowers mitochondrial ROS. Meantime, NFATc2 binding with DNA also enhances cyclin A expression and in turn promotes CDK2 activation. CDK2 activation and [Ca2+]i increase results in PASMC proliferation. Elevated [K+]i together with decreased ΔΨm and mROS inhibits PASMC apoptosis. Finally, the hyperproliferative and anti-apoptotic diathesis within the resistance pulmonary arterial wall lead to vascular remodeling and a progressive increase in pulmonary vascular resistance.
Up regulation and activation of NFATc2 has been found in circulating leukoytes in patients with PAH, including IPAH and scleroderma associated PAH, but not in healthy patients. CD3-positive T lymphocytes were found in the resistance pulmonary artery from PAH patients but not in normal controls and the majority of CD3-positive cells also showed NFATc2 activation. The generalized activation of NFAT might serve as a biomarker for PAH.14 Similarly, Pim-1 which can activate NFAT can also serve as a new biomarker in PAH.28 Large studies will be needed to determine the potential of using circulating NFATc2 as a biomarker for early diagnosis and prognosis for PAH patients who do not have additional systemic inflammatory diseases.
NFATc3
NFATc3 is activated by hypoxia in both adult and neonatal mice. In adult mice, NFATc3 activation contributes to pulmonary arterial wall thickness; conversely, NFATc3 deficient adult mice displayed normal right ventricular systemic pressure after exposure to hypoxia.29 Interestingly, nucleus accumulation of NFATc3 was observed in both endothelial cells (ECs) and PASMC in the lung of neonatal mice in response to chronic hypoxia. Compared to wild-type neonatal mice, heterozygous NFATc3 neonates showed less severity of PAH under hypoxic condition. Surprisingly, activation of other NFAT isotypes was not observed in hypoxic PAH model, which was contradictory to reports from other studies. In vitro, the inhibition of NFATc3 by VIVIT or CsA suppresses hypoxia-induced proliferation and induces apoptosis of PASMC. The results suggest that NFATc3 is involve in the hypoxic pulmonary vascular remodeling in a dose dependent manner. Interestingly, NFATc3 has been implicated in ventricular remodeling.30 Activation of myocardial NFATc3 is also involved in the pathological process of chronic hypoxia induced RV hypertrophy.
Hypoxia induces PASMC phenotype switching from contractile to the synthetic phenotype, increases cell proliferation, migration and thickening of the PA wall. Hypoxia is able to upregulate the expression of transient receptor potential canonical 1 (TRPC1, the store-operated Ca2+ channel) and stromal cell-interaction protein 1 (STIM1, the endoplasmic reticulum Ca2+ sensor) in PASMC, which in turn induces SOC-mediated [Ca2+] influx and subsequent activation of calcineurin phosphatase activity and accumulation of NFATc3 in the nucleus.31,32 Once activated, NFATc3 could induce the transcription of TRPC1 in a positive feedback manner. Interestingly, Kv2.1 is an oxygen sensitive potassium channel and its dysfunction contributes to the pathogenesis of PAH.33 Although activation of NFATc3 controls the excitability of cerebral arterial smooth muscle cell by downregulation of Kv2.1,34 little is known about the relationship between NFATc3 and Kv2.1 in PAH. Additionally, the overexpression of ET-1 induced by hypoxia leads to [Ca2+] mobilization and stimulates RhoA/ROK activity. ROK activiity is responsible for actin polymerization which supports nucleus translocation of NFATc3.35 Constitutively activated NFATc3 promotes soluble guonylyl cyclase-α1 and upregulation of the SM hypertrophic marker SM-α-actin in PASMC.36,37 By contrast, Kang et al reported that lentiviral overexpression NFATc3 alone in human PASMC decreased SM -α-actin expression. Co-transfecting an α-SMA promoter and NFATc1, NFATc2, or NFATc3 overexpression vector, increased the α-SMA promoter activity by about 30%. So the suppressive effect of NFAT on α-SMA might come from phenotypic change in PASMC. Therefore, the underlying molecular mechanisms by which NFAT proteins regulate the PASMC phenotype require further investigation.38 NFATc3 may be involved in the entire hypoxic pulmonary vascular remodeling process, including initial proliferation and subsequent hypertrophy of PASMC. More evidence for NFATc3 involvement in PAH was provided by studies from other animal models. Increased activation of NFATc3 has also been demonstrated in VIP−/− mice and has been proposed to partially mediate the pulmonary vascular remodeling and inflammatory response.39 In line with these findings, nuclear localization of NFATc3 but not NFATc2 was elevated by the stimulation of elevated superoxide/hydrogen peroxide ratio in superoxide dismutase-1 deficient mice. Together, these findings strongly suggest that NFATc3 isoform is involved in the development of PAH in these mice.40 But NFATc2 nuclear localization elevation has been confirmed in a rat model of MCT-induced PAH.14 These studies infer that different NFAT isoforms were activated in different models. The role of NFATc3 in hypoxia-induced PAH is summerized in Fig. 2.
Figure 2.
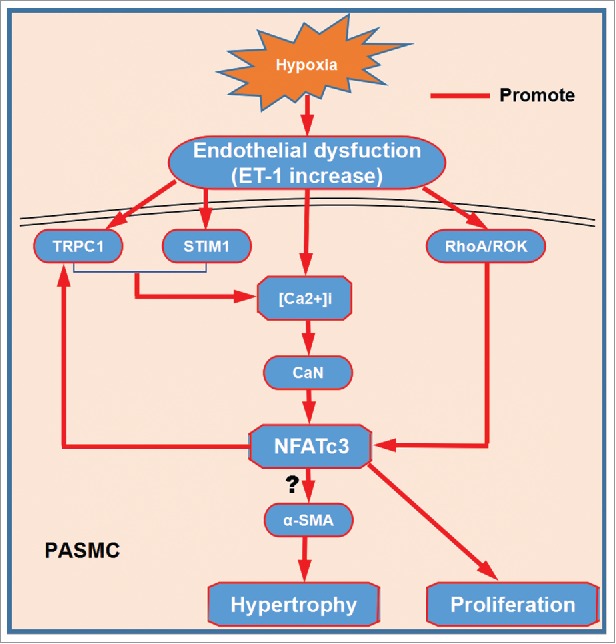
NFATc3 involves in PASMC hypertrophy and proliferation. Endothelial dysfunction induced by hypoxia upregulates the expression of TRPC1 and STIM1 in PASMC, which in turn induces SOC-mediated [Ca2+] influx and subsequent activation of calcineurin phosphatase activity and accumulation of NFATc3 in the nucleus. Once activated, NFATc3 could induce the transcription of TRPC1 in a positive feedback manner. ET-1 induced by hypoxia stimulates RhoA/ROK activity, which promotes nucleus translocation of NFATc3. Once NFATc3 translocates into nucleus, it may upregulate α-SMA in PASMC and in turn promotes PASMC hypertrophy. Moreover, NFATc3 also involves in proliferation of PASMC. PASMC hypertrophy and proliferation results in thickening of the pulmonary artery wall and remodeling.
NFATc1 and NFATc4
Compared with NFATc2 and NFATc3, the studies of other NFAT members in pulmonary vascular remodeling are relatively scarce though NFATc1 has been confirmed to play a role in neointima formation in carotid artery model, inward aortic wall remodeling induced by balloon injury.41,42
In VSMCs, the expression of NFATc1 is much higher than that of NFATc3 whereas NFATc2 and NFATc4 are not detectable.43 Subcellular localization of NFATc1 in SMC (U8A4 cell line) varies under different differentiation state: 1) widely stained in the cytoplasm in dedifferentiated SMC; 2) perinuclear in differentiated SMC; and 3) nuclear in very differentiated cells. These observations confirmed the role of calcineurin/NFATc1 pathway in SMC differentiation. The level of NFATc1 phosphorylation could influence its accumulation in the nucleus.44 Thrombin promotes SMC proliferation and inflammatory responses after vessel injury, which can be inhibited by NFATc1 inhibitors by modulating protease-activated receptor-3 expression.45 So this study indicates that NFATc1 may be implicated the vessel remodel in PAH.
NFATc1 but not NFATc2 or NFATc3 was translocated from the cytoplasm to the nucleus when VSMCs were treated with PDGF-BB or thrombin.46 In light of the findings that different isoforms of NFAT were activated in hypoxia- and MCT-induced PAH, it is possible that different NFAT isoforms may be involved in the development of different types of PAH. Kang et al observed that miRNA-124 exerted its antiproliferative and prodifferentiation effects in human PASMC by inhibiting NFATc1 dephosphorylation and nuclear translocation in chronic hypoxia-induced mouse PAH model.38 By contrast, Chan et al demonstrated that activation of NFATc1 by phenamil may exhibit protective effects in hypoxia-induced SD rat PAH model.47 The reason of the opposite effect of NFATc1 in PAH between 2 studies is unclear, but may be due to different animal models. Extracellular HMGB1 is a promoting factor for MCT-induced PAH. The blockade of HMGB1 activity improved survival of MCT-induced PAH rats, and thus might be a promising therapy for the treatment of PAH.48 Another study found that NFATc1 can inhibit HMGB1 release from THP-1 cells in vitro.49 So NFATc1 seems to protect the MCT-induced PAH, which is similar to Chan's results.47
The NFATc4 nuclear/cytoplasmic ratio increased by 39 % when PASMC was exposed to hypoxia (1% O2 for 48 h),50 which suggests that NFATc4 activation may be implicated the hypoxia-induced PAH.
Collectively, recent studies have confirmed that NFAT activation plays an important role in pulmonary artery remodeling though contradictory results have also been reported. So dissecting the role of all NFAT family members in different PAH models will help us to clarify the complicated effects of NFAT in PAH.
Targeting the NFAT pathway for PAH therapy
As mentioned above, the classical calcineurin inhibitor CsA has potential therapeutic effects in both animal models and in patients with primary PAH.51,52 This drug inactivates NFAT by inhibiting the phosphatase activity of calcineurin. The main concern is the risk of off-target effects due to the fact that broad inhibition of other substrates.53 One way to reduce side effects is to deliver the drug by local administration (inhaled therapy) into pulmonary circulation. The novel peptide VIVIT that selectively interferes the interaction between NFAT and calcineurin potently prevents NFAT nucleus accumulation and dephospherylation. The use of VIVIT for therapy is limited due to delivery issues and stability.54 Proper delivery carriers may improve the efficacy of VIVIT-related therapeutic agents. Specific NFAT inhibitors such as modulatory calcineurin-interacting proteins which restrains calcineurin activity has been used to treat cardiac hypertrophy and failure.55 NFAT inhibitor (A-285222) inhibits NFAT without affecting calcineurin activity and has no off-target side effects in either primates56,57 or rodents.58 Therefore, in PAH, NFAT inhibitors might reverse pulmonary vascular remodeling through their effects on PA-SMCs, and inflammatory cells.55 Although the RNAs (small interfering RNA and microRNA) have been demonstrated to be effective in NFAT inhibition in PASMC, the lack of suitable vectors, technical methods of extraction and dosage limit their use in vivo. The small molecular, hydrogen exerts its therapeutic effects on MCT-induced PAH in rats by modulating the STAT3/NFATc2 axis and it exhibits few side effects.59 Some compounds and drugs, including plumbagin, dichloroaletate and sildenafil have good effects in various models of PAH by inhibiting NFAT gene family.15,16,60 In clinic, efficacy and safety of endothelin-receptor antagonist has been confirmed in PAH patients.61-64 Because ET-1 (upregulated in PAH) activates NFATc1, which in turn increases bcl−2 expression, contributing to the prosurvival and antiapoptotic effects of ET-1,65 endothelin-receptor antagonist might also inhibit NFAT activation. At present, most of studies found that interventions targeting NFAT pathway may be effective for PAH therapy. However, phenamil attenuates the development of PAH and vascular remodeling via activating calcium-calcineurin-NFAT pathway and subsequent transcription of Tribbles homolog 3 gene which promotes the differentiated, contractile VSMC phenotype, which is characterized by elevated expression of contractile genes and reduced cell growth and migration.47 Mesenchymal stem cell improves right ventricular systolic pressure and pulmonary vascular remodeling through suppressing calcineurin /NFAT pathway.66,67 Nonetheless, the NFAT remains to be a good target in developing new drugs for PAH therapy.
Conclusion and perspectives
Accumulating evidence suggests that NFAT proteins are activated in PAH by modulating the process of cardiopulmonary remodeling although contradictory reports also exist. Results from preclinical and clinical studies indicate that NFATs play a key role in pathophysiological process of PAH, including inflammation, aerobic glycolysis, PASMC proliferation and even in ventricular myocyte hypertrophy. The proposed combination therapies are likely to increase the therapeutic efficacy of PAH. Therefore, therapies targeting NFATs or their up and downstream molecules will provide more clinical benefits for PAH patients. Although inflammation is the essential component of all models of PAH, little is known about the role of NFATs in innate immune response in hypoxic PAH. It remains unclear why the same NFAT isoform can have opposite functions in PAH. Further investigation is required to clarify the isoform-specific NFAT function in different types of PAH. Answers to these pressing questions will certainly enhance the development of drugs that could cure PAH with little side effects.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Natural Science Foundation of China (81670405, 81370409, 81400208, 81400269); the Natural Science Foundation of Jiangsu Province (BK20161355); Social Development Foundation of Zhenjiang under Grants SH2014025, SH2014083, SH2015041) and Key Laboratory of Cardiovascular Disease of Zhenjiang (SS2012002).
References
- [1].Shou J, Jing J, Xie J, You L, Jing Z, Yao J, Han W, Pan H. Nuclear factor of activated T cells in cancer development and treatment. Cancer Lett 2015; 361(2):174-184; PMID:25766658; http://dx.doi.org/ 10.1016/j.canlet.2015.03.005 [DOI] [PubMed] [Google Scholar]
- [2].Simonneau G, Galiè N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M, et al.. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43(12 Suppl S):5S-12S; http://dx.doi.org/ 10.1016/j.jacc.2004.02.037 [DOI] [PubMed] [Google Scholar]
- [3].Stenmark KR, Tuder RM, El Kasmi KC. Metabolic reprogramming and inflammation act in concert to control vascular remodeling in hypoxicpulmonary hypertension. J Appl Physiol (1985) 2015; 119(10):1164-1172; PMID:25930027; http://dx.doi.org/ 10.1152/japplphysiol.00283.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].El Chami H, Hassoun PM. Immune and inflammatory mechanisms in pulmonary arterial hypertension. Prog Cardiovasc Dis 2012; 55(2):218-228; http://dx.doi.org/ 10.1016/j.pcad.2012.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, et al.. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol 2009; 54(1 Suppl):S10-S19; http://dx.doi.org/ 10.1016/j.jacc.2009.04.006 [DOI] [PubMed] [Google Scholar]
- [6].Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification of a putative regulator of early T cell activation genes. Science. 1988; 241(4862):202-205; PMID:3260404; http://dx.doi.org/ 10.1126/science.3260404 [DOI] [PubMed] [Google Scholar]
- [7].Nilsson LM, Nilsson-Ohman J, Zetterqvist AV, Gomez MF. Nuclear factor of activated T-cells transcription factors in the vasculature: the good guys or the bad guys? Curr Opin Lipidol 2008; 19(5):483-490 [DOI] [PubMed] [Google Scholar]
- [8].Lorgen M, Jorgensen EH, Jordan WC, Martin SA, Hazlerigg DG. NFAT5 genes are part of the osmotic regulatory system in Atlantic salmon (Salmo salar). Mar Genomics 2016; pii: S1874-7787(16)30058-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mancini M, Toker A. NFAT proteins: emerging roles in cancer progression. Nat Rev Cancer 2009; 9(11):810-820; PMID:19851316; http://dx.doi.org/ 10.1038/nrc2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pan MG, Xiong Y, Chen F. NFAT gene family in inflammation and cancer. Curr Mol Med 2013; 13(4):543-554; PMID:22950383; http://dx.doi.org/ 10.2174/1566524011313040007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen L, Glover JN, Hogan PG, Rao A, Harrison SC. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998; 392(6671):42-48; PMID:9510247; http://dx.doi.org/ 10.1038/32100 [DOI] [PubMed] [Google Scholar]
- [12].Medyouf H, Ghysdael J. The calcineurin/NFAT signaling pathway: a novel therapeutic target in leukemia and solid tumors. Cell Cycle 2008; 7(3):297-303. [DOI] [PubMed] [Google Scholar]
- [13].Fric J, Zelante T, Wong AY, Mertes A, Yu HB, Ricciardi-Castagnoli P. NFAT control of innate immunity. Blood 2012; 120(7):1380-1389; PMID:22611159; http://dx.doi.org/ 10.1182/blood-2012-02-404475 [DOI] [PubMed] [Google Scholar]
- [14].Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN, Michelakis ED. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci USA 2007;104(27):11418-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Guignabert C, Tu L, Izikki M, Dewachter L, Zadigue P, Humbert M, Adnot S, Fadel E, Eddahibi S. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22alpha-targeted overexpression of the serotonin transporter. FASEB J 2009; 23(12):4135-4147; PMID:19679640; http://dx.doi.org/ 10.1096/fj.09-131664 [DOI] [PubMed] [Google Scholar]
- [16].Li M, Liu Y, Sun X, Li Z, Liu Y, Fang P, He P, Shi H, Xie M, Wang X, et al.. Sildenafil inhibits calcineurin/NFATc2-mediated cyclin A expression in pulmonary artery smooth muscle cells. Life Sci 2011; 89(17–18):644-649. [DOI] [PubMed] [Google Scholar]
- [17].Paulin R, Courboulin A, Meloche J, Mainguy V, Dumas de la Roque E, Saksouk N, Côté J, Provencher S, Sussman MA, Bonnet S. Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation 2011; 123(11):1205-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zimmer J, Takahashi T, Hofmann AD, Puri P. Imbalance of NFATc2 and KV1.5 Expression in Rat Pulmonary Vasculature of Nitrofen-Induced Congenital Diaphragmatic Hernia. Eur J Pediatr Surg. 2017;27(1):68–73. doi: 10.1055/s-0036-1587589. [DOI] [PubMed] [Google Scholar]
- [19].Moudgil R, Michelakis ED, Archer SL. The role of k+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 2006; 13(8):615-632; PMID:17085423; http://dx.doi.org/ 10.1080/10739680600930222 [DOI] [PubMed] [Google Scholar]
- [20].Gross A. BCL-2 family proteins as regulators of mitochondria metabolism. Biochim Biophys Acta 2016; 1857(8):1243-1246; http://dx.doi.org/ 10.1016/j.bbabio.2016.01.017 [DOI] [PubMed] [Google Scholar]
- [21].Paulin R, Meloche J, Bonnet S. STAT3 signaling in pulmonary arterial hypertension. JAKSTAT 2012; 1(4):223-233; PMID:24058777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, Michelakis ED. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res 2013; 113(2):126-136. [DOI] [PubMed] [Google Scholar]
- [23].Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC, et al.. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med 2011; 3:88ra55; PMID:21697531; http://dx.doi.org/ 10.1126/scitranslmed.3002194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu Y, Zhang J, Yi B, Chen M, Qi J, Yin Y, Lu X, Jasmin JF, Sun J. Nur77 suppresses pulmonary artery smooth muscle cell proliferation through inhibition of the STAT3/Pim-1/NFAT pathway. Am J Respir Cell Mol Biol 2014; 50(2):379-388; PMID:24047441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Assad TR, Hemnes AR. Metabolic Dysfunction in Pulmonary Arterial Hypertension. Curr Hypertens Rep 2015; 17(3):20; PMID:25754317; http://dx.doi.org/ 10.1007/s11906-014-0524-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sutendra G, Dromparis P, Bonnet S, Haromy A, McMurtry MS, Bleackley RC, Michelakis ED. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFα contributes to the pathogenesis of pulmonary arterial hypertension. J Mol Med (Berl) 2011; 89(8):771-783; PMID:21809123; http://dx.doi.org/ 10.1007/s00109-011-0762-2 [DOI] [PubMed] [Google Scholar]
- [27].Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, Dyck JR, Michelakis ED. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med 2010; 2(44):44ra58; PMID:20702857; http://dx.doi.org/ 10.1126/scitranslmed.3001327 [DOI] [PubMed] [Google Scholar]
- [28].Renard S, Paulin R, Breuils-Bonnet S, Simard S, Pibarot P, Bonnet S, Provencher S. Pim-1: A new biomarker in pulmonary arterial hypertension. Pulm Circ 2013; 3(1):74-81; PMID:23662177; http://dx.doi.org/ 10.4103/2045-8932.109917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bierer R, Nitta CH, Friedman J, Codianni S, de Frutos S, Dominguez-Bautista JA, Howard TA, Resta TC, Bosc LV. NFATc3 is required for chronic hypoxia-induced pulmonary hypertension in adultand neonatal mice. Am J Physiol Lung Cell Mol Physiol 2011; 301(6):L872-880; PMID:21908592; http://dx.doi.org/ 10.1152/ajplung.00405.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Walther S, Awad S, Lonchyna VA, Blatter LA. NFAT transcription factor regulation by urocortin II in cardiac myocytes and heart failure. Am J Physiol Heart Circ Physiol 2014; 306(6):H856-866; PMID:24441548; http://dx.doi.org/ 10.1152/ajpheart.00353.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang C, Li JF, Zhao L, Liu J, Wan J, Wang YX, Wang J, Wang C. Inhibition of SOC/Ca2+/NFAT pathway is involved in the antiproliferative effect of sildenafil on pulmonaryartery smooth muscle cells. Respir Res 2009; 10:123; PMID:20003325; http://dx.doi.org/ 10.1186/1465-9921-10-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hou X, Chen J, Luo Y, Liu F, Xu G, Gao Y. Silencing of STIM1 attenuates hypoxia-induced PASMCs proliferation via inhibition of the SOC/Ca2+/NFAT pathway. Respir Res 2013; 14:2; PMID:23289723; http://dx.doi.org/ 10.1186/1465-9921-14-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Guignabert C, Tu L, Le Hiress M, Ricard N, Sattler C, Seferian A, Huertas A, Humbert M, Montani D. Pathogenesis of pulmonary arterial hypertension: lessons from cancer. Eur Respir Rev 2013; 22(130):543-551; PMID:24293470; http://dx.doi.org/ 10.1183/09059180.00007513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Amberg GC, Rossow CF, Navedo MF, Santana LF. NFATc3 regulates Kv2.1 expression in arterial smooth muscle. J Biol Chem 2004; 279(45):47326-47334; PMID:15322114; http://dx.doi.org/ 10.1074/jbc.M408789200 [DOI] [PubMed] [Google Scholar]
- [35].de Frutos S, Diaz JM, Nitta CH, Sherpa ML, Bosc LV. Endothelin-1 contributes to increased NFATc3 activation by chronic hypoxia in pulmonary arteries. Am J Physiol Cell Physiol 2011; 301(2):C441-450; PMID:21525433; http://dx.doi.org/ 10.1152/ajpcell.00029.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].de Frutos S, Nitta CH, Caldwell E, Friedman J, González Bosc LV. Regulation of soluble guanylyl cyclase- alpha 1 expression in chronic hypoxia-induced pulmonary hypertension: role of NFATc3 and HuR. Am J Physiol Lung Cell Mol Physiol 2009; 297(3):L475-486; PMID:19592461; http://dx.doi.org/ 10.1152/ajplung.00060.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].de Frutos S, Spangler R, Alò D, Bosc LV. NFATc3 mediates chronic hypoxi-induced pulmonary arterial remodeling with alpha-actin up-regulation. J Biol Chem 2007; 282(20):15081-15089; PMID:17403661; http://dx.doi.org/ 10.1074/jbc.M702679200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kang K, Peng X, Zhang X, Wang Y, Zhang L, Gao L, Weng T, Zhang H, Ramchandran R, Raj JU, et al.. MicroRNA 124 suppresses the transactivation of nuclear factor of activated T cells by targeting multiple genesand inhibits the proliferation of pulmonary artery smooth muscle cells. J Biol Chem 2013; 288(35):25414-25427; PMID:23853098; http://dx.doi.org/ 10.1074/jbc.M113.460287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Said SI. The vasoactive intestinal peptide gene is a key modulator of pulmonary vascular remodeling and inflammation. Ann N Y Acad Sci 2008; 1144:148-153. [DOI] [PubMed] [Google Scholar]
- [40].Ramiro-Diaz JM, Nitta CH, Maston LD, Codianni S, Giermakowska W, Resta TC, Gonzalez Bosc LV. NFAT is required for spontaneous pulmonary hypertension in superoxide dismutase 1 knockout mice. Am J Physiol Lung Cell Mol Physiol 2013; 304(9):L613-625; PMID:23475768; http://dx.doi.org/ 10.1152/ajplung.00408.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Singh NK, Kundumani-Sridharan V, Kumar S, Verma SK, Kotla S, Mukai H, Heckle MR, Rao GN. Protein kinase N1 is a novel substrate of NFATc1-mediated cyclin D1-CDK6 activity and modulates vascular smooth muscle cell division and migration leading to inward blood vessel wall remodeling. J Biol Chem 2012; 287(43):36291-36304; PMID:22893700; http://dx.doi.org/ 10.1074/jbc.M112.361220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liu Z, Zhang C, Dronadula N, Li Q, Rao GN. Blockade of nuclear factor of activated T cells activation signaling suppresses balloon injury-induced neointima formation in a rat carotid artery model. J Biol Chem 2005; 280(15):14700-14708; PMID:15681847; http://dx.doi.org/ 10.1074/jbc.M500322200 [DOI] [PubMed] [Google Scholar]
- [43].Boss V, Abbott KL, Wang XF, Pavlath GK, Murphy TJ. The cyclosporin A-sensitive nuclear factor of activated T cells (NFAT) proteins are expressed in vascular smooth muscle cells. Differential localization of NFAT isoforms and induction of NFAT-mediated transcription by phospholipase C-coupled cell surface receptors. J Biol Chem 1998; 273(31):19664-19671. [DOI] [PubMed] [Google Scholar]
- [44].Larrieu D, Thiébaud P, Duplàa C, Sibon I, Thézé N, Lamazière JM. Activation of the Ca(2+)/calcineurin/NFAT2 pathway controls smooth muscle cell differentiation. Exp Cell Res 2005; 310(1):166-175; PMID:16129432; http://dx.doi.org/ 10.1016/j.yexcr.2005.07.021 [DOI] [PubMed] [Google Scholar]
- [45].Rosenkranz AC, Rauch BH, Doller A, Eberhardt W, Böhm A, Bretschneider E, Schrör K. Regulation of human vascular protease-activated receptor-3 through mRNA stabilization and the transcription factor nuclear factor of activated T cells (NFAT). Mol Pharmacol 2011; 80(2):337-344. [DOI] [PubMed] [Google Scholar]
- [46].Yellaturu CR, Ghosh SK, Rao RK, Jennings LK, Hassid A, Rao GN. A potential role for nuclear factor of activated T-cells in receptor tyrosine kinase and G-protein-coupledreceptor agonist-induced cell proliferation. Biochem J 2002; 368(Pt 1):183-190; PMID:12188924; http://dx.doi.org/ 10.1042/bj20020347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chan MC, Weisman AS, Kang H, Nguyen PH, Hickman T, Mecker SV, Hill NS, Lagna G, Hata A. The amiloride derivative phenamil attenuates pulmonary vascular remodeling by activating NFAT and the bone morphogenetic protein signaling pathway. Mol Cell Biol 2011; 31(3):517-530; PMID:21135135; http://dx.doi.org/ 10.1128/MCB.00884-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sadamura-Takenaka Y, Ito T, Noma S, Oyama Y, Yamada S, Kawahara K, Inoue H, Maruyama I. HMGB1 promotes the development of pulmonary arterial hypertension in rats. PLoS One 2014; 9(7):e102482; PMID:25032709; http://dx.doi.org/ 10.1371/journal.pone.0102482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhao Q, Wang L, Hu J, Liu H. Role of nuclear factor of activated T cells-2 in high mobility protein box-1 release in human monocytic THP-1 cells in vitro. Nan Fang Yi Ke Da Xue Xue Bao 2016; 36(1):8-12; PMID:26806731 [PubMed] [Google Scholar]
- [50].Parpaite T, Cardouat G, Mauroux M, Gillibert-Duplantier J, Robillard P, Quignard JF, Marthan R, Savineau JP, Ducret T. Effect of hypoxia on TRPV1 and TRPV4 channels in rat pulmonary arterial smooth muscle cells. Pflugers Arch 2016; 468(1):111-130; PMID:25799977; http://dx.doi.org/ 10.1007/s00424-015-1704-6 [DOI] [PubMed] [Google Scholar]
- [51].Morelli S, Giordano M, De Marzio P, Priori R, Sgreccia A, Valesini G. Pulmonary arterial hypertension responsive to immunosuppressive therapy in systemic lupus erythematosus. Lupus. 1993; 2(6):367-369; PMID:8136819; http://dx.doi.org/ 10.1177/096120339300200606 [DOI] [PubMed] [Google Scholar]
- [52].Padeh S, Laxer RM, Silver MM, Silverman ED. Primary pulmonary hypertension in a patient with systemic-onset juvenile arthritis. Arthritis Rheum. 1991; 34(12):1575-1579; PMID:1747143; http://dx.doi.org/ 10.1002/art.1780341216 [DOI] [PubMed] [Google Scholar]
- [53].Kockx M, Jessup W, Kritharides L. Cyclosporin A and atherosclerosis–cellular pathways in atherogenesis. Pharmacol Ther 2010; 128(1):106-118. doi: 10.1016/j.pharmthera.2010.06.001 [DOI] [PubMed] [Google Scholar]
- [54].Yu H, van Berkel TJ, Biessen EA. Therapeutic potential of VIVIT, a selective peptide inhibitor of nuclear factor of activated T cells, incardiovascular disorders. Cardiovasc Drug Rev 2007; 25(2):175-187; PMID:17614939; http://dx.doi.org/ 10.1111/j.1527-3466.2007.00011.x [DOI] [PubMed] [Google Scholar]
- [55].McKinsey TA, Olson EN. Toward transcriptional therapies for the failing heart: chemical screens to modulate genes. J Clin Invest 2005; 115(3):538-546; PMID:15765135; http://dx.doi.org/ 10.1172/JCI24144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bîrsan T, Dambrin C, Marsh KC, Jacobsen W, Djuric SW, Mollison KW, Christians U, Carter GW, Morris RE. Preliminary in vivo pharmacokinetic and pharmacodynamic evaluation of a novel calcineurin-independent inhibitor of NFAT. Transpl Int 2004; 17(3):145-150; PMID:14735234; http://dx.doi.org/ 10.1111/j.1432-2277.2004.tb00419.x [DOI] [PubMed] [Google Scholar]
- [57].Trevillyan JM, Chiou XG, Chen YW, Ballaron SJ, Sheets MP, Smith ML, Wiedeman PE, Warrior U, Wilkins J, Gubbins EJ, et al.. Potent inhibition of NFAT activation and T cell cytokine production by novel low molecular weight pyrazole compounds. J Biol Chem 2001; 276(51):48118-48126; PMID:11592964 [DOI] [PubMed] [Google Scholar]
- [58].Nilsson-Berglund LM, Zetterqvist AV, Nilsson-Ohman J, Sigvardsson M, Gonzalez Bosc LV, Smith ML, Salehi A, Agardh E, Fredrikson GN, Agardh CD, et al.. Nuclear factor of activated T cells regulates osteopontin expression in arterial smooth muscle in response to diabetes-induced hyperglycemia. Arterioscler Thromb Vasc Biol 2010; 30(2):218-224; http://dx.doi.org/ 10.1161/ATVBAHA.109.199299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kishimoto Y, Kato T, Ito M, Azuma Y, Fukasawa Y, Ohno K, Kojima S. Hydrogen ameliorates pulmonary hypertension in rats by anti-inflammatory and antioxidant effects. J Thorac Cardiovasc Surg 2015; 150(3):645-654; http://dx.doi.org/ 10.1016/j.jtcvs.2015.05.052 [DOI] [PubMed] [Google Scholar]
- [60].Courboulin A, Barrier M, Perreault T, Bonnet P, Tremblay VL, Paulin R, Tremblay E, Lambert C, Jacob MH, Bonnet SN, et al.. Plumbagin reverses proliferation and resistance to apoptosis in experimental PAH. Eur Respir J 2012; 40(3):618-629; PMID:22496325; http://dx.doi.org/ 10.1183/09031936.00084211 [DOI] [PubMed] [Google Scholar]
- [61].Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, et al.. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002; 346(12):896-903; PMID:11907289; http://dx.doi.org/ 10.1056/NEJMoa012212 [DOI] [PubMed] [Google Scholar]
- [62].Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, Badesch DB, Roux S, Rainisio M, Bodin F, et al.. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet 2001; 358(9288):1119-1123; PMID:11597664; http://dx.doi.org/ 10.1016/S0140-6736(01)06250-X [DOI] [PubMed] [Google Scholar]
- [63].Williamson DJ, Wallman LL, Jones R, Keogh AM, Scroope F, Penny R, Weber C, Macdonald PS. Hemodynamic effects of Bosentan, an endothelin receptor antagonist, in patients with pulmonary hypertension. Circulation 2000; 102(4):411-418; http://dx.doi.org/ 10.1161/01.CIR.102.4.411 [DOI] [PubMed] [Google Scholar]
- [64].Barst RJ, Langleben D, Badesch D, Frost A, Lawrence EC, Shapiro S, Naeije R, Galie N, STRIDE-2 Study Group . Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol 2006; 47(10):2049-2056; PMID:16697324; http://dx.doi.org/ 10.1016/j.jacc.2006.01.057 [DOI] [PubMed] [Google Scholar]
- [65].Kawamura T, Ono K, Morimoto T, Akao M, Iwai-Kanai E, Wada H, Sowa N, Kita T, Hasegawa K. Endothelin-1-dependent nuclear factor of activated T lymphocyte signaling associates with transcriptional coactivator p300 in the activation of the B cell leukemia-2 promoter in cardiac myocytes. Circ Res 2004; 94(11):1492-1499. [DOI] [PubMed] [Google Scholar]
- [66].Liu J, Han Z, Han Z, He Z. Mesenchymal stem cell-conditioned media suppresses inflammation-associated overproliferation of pulmonary artery smooth muscle cells in a rat model of pulmonary hypertension. Exp Ther Med 2016; 11(2):467-475; PMID:26893632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Liu J, Han Z, Han Z, He Z. Mesenchymal stem cells suppress CaN/NFAT expression in the pulmonary arteries of rats with pulmonary hypertension. Exp Ther Med 2015; 10(5):1657-166; PMID:26640533 [DOI] [PMC free article] [PubMed] [Google Scholar]