

Abstract
The diagnosis and management of children with autoimmune cytopenias can be challenging, as this diagnostic category encompasses a number of heterogeneous but closely related conditions. Children can present with immune-mediated destruction of a single cell lineage or multiple cell lineages, including platelets (immune thrombocytopenia, ITP), erythrocytes (autoimmune hemolytic anemia), and neutrophils (autoimmune neutropenia). Immune-mediated destruction can be primary or secondary to a co-morbid immunodeficiency, malignancy, rheumatologic condition, or lymphoproliferative disorder. Despite the heterogeneity, treatment options are similar and generally consist of non-specific immune suppression or modulation. Fortunately, this non-specific approach is changing as recent insights into disease biology have led to targeted therapies for certain patients, including the use of thrombopoietin mimetics in ITP and sirolimus for cytopenias associated with autoimmune lymphoproliferative syndrome (ALPS).
Keywords: Autoimmune hemolytic anemia, Immune thrombocytopenia, Autoimmune neutropenia, Evans syndrome, Autoimmune Lymphoproliferative Syndrome
Introduction
Autoimmune cytopenias are a group of heterogeneous but closely related conditions defined by immune-mediated destruction of hematologic cell lineages, including white blood cells (neutrophils), red blood cells, and platelets. This destruction can be primary or secondary to other illnesses. Primary autoimmune cytopenias, formerly classified as idiopathic, consist of single lineage destruction, including immune thrombocytopenia (ITP), autoimmune hemolytic anemia (AIHA), and autoimmune neutropenia (AIN), as well as multi-lineage destruction, known as Evans syndrome. Secondary autoimmune cytopenias result from another cause, including medications, rheumatologic disorders, immunodeficiencies, lymphoproliferative disorders, malignancies, or as a complication of organ or hematopoietic stem cell transplant (HSCT). Despite their complex and heterogeneous nature, treatment is relatively straightforward, primarily using drugs that suppress or modulate the immune system. Many patients require no treatment; however, others need multi-agent therapy and occasionally autologous or allogeneic HSCT.
This review will focus on challenges encountered in the diagnosis and management of single- and multi-lineage autoimmune cytopenias in children. New treatments for ITP and autoimmune lymphoproliferative syndrome (ALPS) will be discussed as paradigms for the translation of basic science into clinic progress, as thrombopoieitin (TPO) mimetics (TPO receptor agonists, TPO-RAs) and mTOR inhibitors have revolutionized therapy for these two conditions, respectively. With modern genomics, this paradigm will likely be used in the near future for other autoimmune cytopenia syndromes.
Primary Autoimmune Cytopenia Syndromes
Most children with autoimmune cytopenias have idiopathic destruction of a single cell lineage, most commonly idiopathic destruction of platelets, ITP. Primary autoimmune cytopenia syndromes are diagnoses of exclusion. However, in children, single lineage primary autoimmune cytopenias are far more common than secondary autoimmune cytopenias. Thus, the default is often to presume a child who presents acutely with single lineage destruction has a primary autoimmune cytopenia. Patients with chronic disease or multi-lineage cytopenias, in contrast, more commonly have a predisposing cause, necessitating a more extensive diagnostic evaluation. Importantly, patients with primary ITP frequently have a preceding viral syndrome or other immune trigger, such as vaccination. The key difference is that the triggering event is not a chronic illness or a medication. Of note, the distinction between primary and secondary autoimmune cytopenias is becoming blurred as genetic alterations and polymorphisms predisposing patients to autoimmune cytopenias are being identified.
Immune Thrombocytopenia (ITP)
Primary immune thrombocytopenia (ITP) is a fairly rare, generally benign autoimmune bleeding disorder characterized by isolated thrombocytopenia, defined as a platelet count <100 × 109/L in the absence of other causes or diseases that may cause thrombocytopenia. The thrombocytopenia is often quite severe (with platelet counts <10 × 109/L), but life-threatening bleeding is rare.1 Antibodies are often directed against the two most prevalent receptors on the platelet surface, GPIb/IX complex (vonWillebrand factor receptor) and the GPIIb/IIIa receptor (collagen/fibrinogen receptor) and are detectable in 60% of patients.2 ITP is caused by a complex interplay of immune dysregulation involving a shift toward Th1 cells, a decrease in regulatory T cell (Treg) function/number, and cytotoxic T cell mediated direct lysis of platelets and megakaryocytes.3 Finally, dysmegakaryopoiesis plays a critical role in the development of thrombocytopenia.4
In 2011, an international ITP working group recommended changing the terminology to “immune thrombocytopenia” (keeping the abbreviation ITP) to reflect that many patients may not exhibit bleeding or purpura.5 The phases of illness in ITP have also been redefined to allow for better uniformity (Table 1).5
Table 1. Revised Classification of ITP5.
| Primary ITP | Isolated thrombocytopenia (platelet count <100 × 109/L) occurring in the absence of other disease known to be associated with thrombocytopenia |
|---|---|
| Secondary ITP | All forms of immune-mediated thrombocytopenia other than primary ITP. |
| Phases of the Disease |
|
Data From: Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113:2386-2393.
Clinically, patients with ITP present most commonly with variable degrees of mucocutaneous bleeding. In a report from the International Consortium of ITP Study Group (ICIS), the majority of pediatric patients presenting with ITP (77%) had no or mild bleeding at diagnosis – despite the fact that 74% of children had platelet counts <20 × 109/L at the time of diagnosis.1 Severe bleeding is rare in pediatric ITP and in the same report, only one patient (of 863 evaluable patients) had central nervous system bleeding while 3% of patients had severe bleeding (epistaxis, mouth, GI, urinary tract, menorrhagia, or multiple sites) and 20% of patients had moderate bleeding. Given the relatively rare occurrence of significant bleeding in children with ITP, the current recommendation is careful observation for children with newly diagnosed ITP and mild to no bleeding symptoms.6
In children requiring therapy to increase the platelet count, a single dose of IVIgG or a short course of corticosteroids is recommended as first line therapy, with a single dose of anti-Rho(D) immunoglobulin as an alternative first line therapy in Rh(D) positive patients. Second line therapies addressed in the evidence-based practice guidelines include rituximab, high dose dexamethasone, and splenectomy. Splenectomy is effective in 70-80% of children with ITP, and most of these responses are durable.7 Unfortunately, 20-30% of children do not respond, and the permanent effects of splenectomy include an increased risk of infection with encapsulated organisms and the theoretical risks of venous thromboembolism and pulmonary hypertension. As most children with ITP have spontaneous remissions, splenectomy should be reserved for children who have persistent disease (>12 months) or have serious treatment-refractory disease.
Rituximab has been studied extensively in both children and adults with ITP, though there have been no pediatric randomized placebo-controlled trials. Response rates to rituximab vary from 25-80% in different series.6 Many of these responses are durable, but a significant percent of patients with chronic ITP relapse ∼12-18 months after treatment.8 A randomized trial comparing rituximab and high-dose dexamethasone versus dexamethasone alone in newly diagnosed childhood ITP demonstrated improved responses with the addition of rituximab (63% vs 36% sustained response, p = 0.004).9 More often, rituximab is used in patients with chronic ITP. Other immunosuppressive therapies, including mercaptopurine, danazol, vincristine, mycophenolate mofetil, and sirolimus, have all been used in small series of patients with reported efficacy in ∼60% of patients with ITP refractory to first and second line therapies (see treatment section).
Recently, TPO-RAs have emerged as novel therapies for ITP. TPO was described in the 1950s but was not cloned and sequenced until 1994.10 Shortly after that, recombinant TPO was developed as an adjunct for chemotherapy-induced thrombocytopenia and to stimulate increased platelet counts in platelet donors.11 Subsequently, however, several healthy volunteers developed neutralizing antibodies that cross-reacted with endogenous TPO causing severe thrombocytopenia, and development of recombinant TPO was halted due to this safety concern.12 More recently, TPO-RAs were developed that bind within the receptor pocket but do NOT share any sequence homology with the endogenous receptor. Since the development of these drugs, a few patients have developed antibodies against the TPO-RAs, but these have not been clinically significant. A number of randomized clinical trials have demonstrated durable response rates of 60-85% in adults with ITP.13 Some of these patients are able to stop taking the drugs and remain in remission; however, others have “rebound thrombocytopenia.” Less is known about the safety and efficacy of TPO-RAs in children. Several case series and one randomized clinical trial have been published suggesting that TPO-RAs may be efficacious in pediatric patients with ITP.14 Given the current lack of significant data in pediatric patients, use of these medications should be limited to ongoing clinical trials to evaluate safety and efficacy of these agents.
Autoimmune hemolytic anemia
Immune-mediated hemolytic anemia can be autoimmune (AIHA) or isoimmune/alloimmune. Isoimmune hemolytic anemia arises from alloimmunization against red blood cell-specific antigens via placental exposure (hemolytic disease of the newborn) or RBC transfusion. In contrast, AIHA is caused by autoantibodies against RBC antigens. Isoimmune hemolytic anemia and hemolytic disease of the newborn are reviewed elsewhere.15,16 Primary AIHA affects 1 in 80,000 adults and children. AIHA is classified as warm-reactive (W-AIHA), cold-agglutinin (C-AIHA), or paroxysmal cold hemoglobinuria (PCH).
W-AIHA is the most common form of AIHA in children (∼90% of cases) and is usually caused by IgG antibodies against RBC antigens; however, rarely, antibodies can be IgM or IgA.17 It is classified as a warm antibody because the maximal activity of the antibody is at approximately 37°C. Children with W-AIHA can present with mild anemia or severe, sudden onset life-threatening disease. W-AIHA is typically self-limiting in childhood. Clinical manifestations include pallor, fatigue, jaundice, dark-colored urine, and occasionally splenomegaly. The best diagnostic test is the direct antiglobulin test (DAT, Coombs test), which is usually IgG+ (with or without C3 positivity) at 37°C. Other laboratory abnormalities include anemia, reticulocytosis, elevated lactate dehydrogenase (LDH), hyperbilirubinemia, and decreased haptoglobin. Peripheral blood smear most commonly reveals polychromasia, nucleated red blood cells, spherocytes, and red cell agglutination.
Treatment of primary W-AIHA is usually immune suppression or modulation, most commonly with corticosteroids, IVIgG, or rituximab (see treatment section). Unlike patients with ITP, IVIgG is often not effective as a single agent, and usually requires treatment with very high doses for benefit, though it may be a useful adjunctive therapy in severe cases.18 Corticosteroids are the first line, especially in acutely-presenting, very ill patients.19 The majority of children with W-AIHA are corticosteroid-responsive with published response rates from 50-80% in larger series.20 Splenectomy was previously the treatment of choice for patients who failed or become steroid-refractory or intolerant, as well as those who developed chronic disease with response rates as high as 60%.21 Splenectomy is used less commonly today as better therapies have been developed and the short and long-term risks of splenectomy are increasingly apparent, especially in children. Plasmapheresis can be used for acutely ill patients; however, benefits are usually short-lived, and it is not very effective for IgG antibodies.20
Most published guidelines recommend rituximab as the agent of choice for corticosteroid-refractory patients or for children unable to wean off corticosteroids.17 Many small series and case reports have described the benefit of rituximab for children and adults with refractory W-AIHA with overall response rates ranging from 40-100% (median ∼50-60%).17,20 Some patients require multiple courses. Other agents with activity against W-AIHA include antimetabolites (mercaptopurine, azathiopurine, mycophenolate mofetil, methotrexate), calcineurin inhibitors (cyclosporine), alkylating agents (cyclosphosphamide), mTOR inhibitors (sirolimus), mitotic inhibitors (vincristine or vinblastine), or other antibody-based therapy (alemtuzumab).20 None of these have been formally studied in randomized controlled trials. Also, the published case series often group patients with primary and secondary W-AIHA or patients treated with combinations of medications. Danazol, a commonly used agent in adults, is generally not used in children with AIHA because of its masculinizing effects.22
Transfusions should be avoided if possible in W-AIHA. Identification of compatible units for transfusion is challenging, as warm antibodies are typically panreactive. Also, the panreactive autoantiboies may destroy the transfused cells in vivo, leading to increased hemolysis, hemoglobinuria, and renal failure. For patients who are very ill and/or not responding to immune suppression, safety of transfusions may be increased by using special absorption techniques to help distinguish allo- vs autoantibodies in vitro and by transfusing the patient slowly to observe for clinical reactions as well as for free hemoglobin in the plasma and urine during transfusion.
Cold agglutinin AIHA (C-AIHA) is considerably less common in children than W-AIHA and is typically caused by anti-IgM antibodies that destroy erythrocytes when exposed to cold temperatures. A DAT is typically positive for complement only. In children, C-AIHA is usually triggered by infection, mostly commonly with Mycoplasma pneumonia. Patients tend to present with less severe anemia than in W-AIHA. Peripheral smear commonly shows agglutination, but spherocytes are rare. Treatment for C-AIHA differs significantly from W-AIHA, as corticosteroids are often ineffective. A primary goal is to treat the underlying infection, if possible. The patient should remain as warm as possible until the condition improves, and if transfused, the packed red blood cells (pRBC) should be warmed. Plasmapheresis is often very effective in C-AIHA and may be combined with rituximab. Recent reports, mostly in adults, have described the efficacy of rituximab in C-AIHA with response rates over 50%.23
Rarely, children can develop paroxysmal cold hemoglobinuria (PCH), which is caused by a biphase hemolysin IgG antibody against the P-antigen on RBCs (Donath-Landsteiner antibody).24 It can bind and fix complement at cold temperatures or warm temperatures, but only after cooling and rewarming the sample, leading to complement amplification and hemolysis. Similar to C-AIHA, pRBC for transfusion should be kept warm. PCH patients often respond to plasmapheresis and several reports describe responses to rituximab.24 Unlike C-AIHA, PCH is often corticosteroid-responsive, as it is caused by an IgG-mediated antibody.
Autoimmune Neutropenia (AIN)
Similar to immune-mediated destruction of platelets and erythrocytes, immune neutropenia can be allo- or autoimmune. Neonatal alloimmune neutropenia, not discussed in detail here, is caused by alloimmunization against fetal neutrophil-specific antigens that are not present on maternal WBCs.25 Neonates can also develop autoimmune neutropenia, secondary to placental transfer of maternal anti-neutrophil autoantibodies when the mother has autoimmune neutropenia.
Primary single lineage autoimmune neutropenia (AIN) is relatively rare, most commonly presents in infancy or early childhood after viral infection, and is often self-limiting with >95% of children having a complete resolution of AIN.26 Neonatal AIN secondary to passive transfer of maternal antibodies typically resolves by 2-3 months of age.27 Other forms of childhood AIN typically recover a few months after diagnosis, with only a small percentage of children developing chronic disease.26 Similar to children with ITP or AIHA, chronic disease is more likely to be secondary to another disease process than is the acute self-resolving form.28
Patients with AIN rarely present with significant infections.28,29 Many patients develop benign infections of the skin and upper respiratory tract. Some patients develop mouth sores. Unlike children with other forms of neutropenia, patients with AIN often can mount a neutrophil response to bacteria and fungal infections. Patients who develop more severe infections should be screened for other co-morbid immune deficiencies and other causes of neutropenia. Infectious risk can correlate with degree of neutropenia; however, most patients with AIN and ANCs <200/mm3 do not develop serious infections. Patients with AIN often have a monocytosis, and bone marrow evaluation usually demonstrates normal to increased cellularity with myeloid hyperplasia. Mature neutrophils may be normal to decreased in bone marrow, and sometimes there is a maturational arrest.
Diagnosis of AIN can be difficult, because of the poor sensitivity and specificity of anti-neutrophil antibody testing.26,29 Accordingly, it is recommended that two methods of testing be used to establish the diagnosis.30 The most commonly used are the granulocyte indirect immunofluorescence test (indirect-GIFT) and the granulocyte agglutination test (GAT).
Most patients with AIN need no treatment. Preventive measures, including good oral hygiene and mouth rinses, are often used. Some centers use prophylactic antibacterial agents for these patients; however, there are no data to support this practice. For patients with serious or recurrent infections, G-CSF (granulocyte colony-stimulating factor, filgrastim) is the recommended first line treatment.28 Patients with AIN who are undergoing surgical procedures may also benefit from G-CSF. Some patients do not respond to G-CSF and second line therapies include GM-CSF (granulocyte macrophage colony-stimulating factor), a combination of G-CSF and GM-CSF, or other medicines used for immune mediated cytopenias. Patients with single lineage primary AIN rarely need these other therapies, while patients with multi-lineage cytopenias that include AIN or secondary AIN more commonly need immune modulation (see below).
Evans syndrome
The diagnosis and management of children with multi-lineage autoimmune cytopenias are more complex. Evans syndrome (ES) is a diagnosis of exclusion defined by idiopathic autoimmune destruction of multiple cell lineages. Patients with ES tend to have chronic disease and present at a young age (median 7-8 years) with symptoms based on the affected cell lineages.31,32 Different definitions for ES exist in the literature, making study comparison challenging.31-34 Some investigators and clinicians consider any patient with autoimmune disease affecting two or more cell lineages as having ES. Others restrict the definition to patients with autoimmune destruction of red cells and platelets with or without neutrophil destruction. Some studies restrict the diagnosis to patients with clinically relevant destruction of multiple lineages, whereas others will include patients with ITP and a positive DAT without obvious hemolysis. Patients often present with splenomegaly with or without lymphadenopathy and may have an increased risk of secondary lymphomas.31,32,34,35 The true risk of subsequent cancer development is difficult to determine, as studies are small, the disease is rare, definitions vary, and patients with autoimmune cytopenias due to other, potentially more cancer-prone diseases are often erroneously labeled ES. The term ES should be restricted to patients who have no identifiable underlying cause for multi-lineage autoimmune cell destruction. For example, autoimmune destruction of blood cells can occur as a consequence of SLE, but SLE patients with multi-lineage autoimmune cytopenias do not have ES. The distinction is important as the treatment and prognosis for idiopathic ES and secondary multi-lineage autoimmune cytopenias syndromes can be different.
Children with ES must have a thorough diagnostic evaluation for underlying causes of autoimmune cytopenias, especially for those that would lead to different therapy. Often the history and physical may suggest a patient has an underlying immune deficiency, lymphoproliferative syndrome, malignancy, or rheumatologic disorder. At minimum, children with Evans syndrome should be screened for ALPS, CVID, and SLE (Table 2). Patients with significant lymphadenopathy or splenomegaly may require imaging (PET or CT) and/or biopsy to eliminate the possibility of an underlying malignancy. HIV testing should be considered, especially in teenagers.35 It is also important to re-examine for secondary causes of autoimmune disease periodically, as multi-lineage autoimmune cytopenias can be an initial presentation of SLE, and other disease manifestations can appear with time.36
Table 2. Secondary Autoimmune Cytopenias in Children.
| Diseases associated with autoimmune cytopenias in children |
|---|
| Lymphoproliferative Disorders |
| Autoimmune Lymphoproliferative Syndrome* |
| Rosai-Dorfman disease |
| Castlemans disease |
| Ras-associated leukoproliferative disorder |
|
|
| Immune deficiencies |
| Common variable immune deficiency* |
| Selective IgA deficiency |
| Chromosome 22q11.2 deletion (DiGeorge or velocardiofacial syndrome) |
| Severe combined immunodeficiency |
|
|
| Rheumatologic Conditions |
| Systemic Lupus Erythematosus* |
| Antiphospholipid antibody syndrome* |
| Juvenile Idiopathic Arthritis/Juvenile Rheumatoid Arthritis |
| Sjogren's syndrome |
| Sarcoid |
|
|
| Malignancies |
| Non-Hodgkin Lymphoma |
| Acute lymphoblastic leukemia |
| Myelodysplastic syndrome |
| Hodgkin Lymphoma |
|
|
| Chronic Infections |
| EBV |
| HIV** |
| Helicobacter pylori |
| CMV |
| HCV |
|
|
| Other |
| Celiac Disease |
| Inflammatory Bowel Disease |
|
|
| Recommended evaluation for children with chronic single lineage autoimmune cytopenias or multi-lineage autoimmune cytopenias* |
| Flow cytometry for double negative T cells (ALPS) |
| ANA (SLE) |
| Anti-phospholipid antibodies |
| Quantitative immunoglobulins (CVID) |
| Specific antibody titers (CVID) |
| T cells subsets (CD3/CD4, CD3/CD8) |
| HIV** |
Consider screening for these conditions in children with chronic single lineage autoimmune cytopenias or multi-lineage autoimmune cytopenias.
Consider also screening for HIV in adolescents with chronic single lineage or multi-lineage autoimmune cytopenias. Other diseases should be considered if the history or physical are suggestive of the underlying condition. It is extremely rare for the other conditions to present with autoimmune cytopenias and no other signs or symptoms suggestive of the underlying disease. Thus, the utility of routine screening is low.
Our group demonstrated in single- and multi-institutional trials that 30-40% of children diagnosed with ES have forme fruste ALPS (described below).33,37 We evaluated 45 children with ES and found that elevated immunoglobulins, elevated vitamin B12, and isolated lymphadenopathy without splenomegaly were predictive of ALPS, though there was likely a selection bias, as not all children with ES were captured at each institution. A recent large study from France of childhood AIHA, which included 99 patients with ES, did not find a high incidence of ALPS among children with ES.35 Of note, this study identified children with undiagnosed ALPS, and many patients were not tested for ALPS. Also, the definition of ES was restricted to patients diagnosed with AIHA. As ALPS is a genetic disease there is likely heterogeneity in the frequency in different populations. A number of studies have screened ES patients for CVID, making the diagnosis in a relatively high percentage of patients.38,39 Most of these studies were single institution evaluations with a small number of patients. In contrast, the frequency of CVID among patients with de novo single lineage autoimmune cytopenias, including newly diagnosed ITP, is very low.40
Childhood ES is often a chronic disease with a waxing and waning course. Some patients require therapy only with disease flares and others need chronic therapy. Corticosteroids are the first choice for acute flares and newly diagnosed patients. Based on the chronic nature of the illness and significant side effects of prolonged corticosteroid use (see below), we recommend alternative therapies early in the therapeutic course. Unlike single lineage autoimmune cytopenias, splenectomy is often ineffective in ES. A number of studies have shown remarkable efficacy using rituximab in ES. Unfortunately, as ES is a chronic disease, many patients relapse, typically 1-2 years after treatment.41,42 Accordingly, we have a low threshold to transition patients to single agent oral immune suppression, using mycophenolate mofetil or sirolimus and have seen marked response in a number of patients (unpublished data). The risks with a single agent are very low (see below). Anecdotal series and case reports have described success with a variety of immune suppressants, using both single agent and combination therapy (see below).
Secondary Autoimmune Cytopenia Syndromes
The diagnosis and management of secondary autoimmune cytopenias can be complex. A careful history and physical exam may identify a secondary cause in the acutely presenting patient; however, autoimmune cytopenias can be the only disease manifestation in some children with underlying immunodeficiency, rheumatologic, or lymphoproliferative disease. Splenomegaly and lymphadenopathy can often be found in children with idiopathic autoimmune cytopenias, making it difficult to use these findings to determine which patients should undergo more extensive evaluation. Nevertheless, we recommend that patients with chronic single lineage disease and lymphadenopathy or splenomegaly undergo a bone marrow aspirate and biopsy. Also, imaging for mediastinal mass and lymph node biopsy may be indicated.
Many conditions (Table 2) and medications can lead to comorbid autoimmune cytopenias. If possible, the primary goal is to treat the underlying cause of the autoimmunity. SLE patients with autoimmune cytopenias should be treated with medications active against other SLE disease manifestations. CVID patients often respond to increasing the dose of IVIgG “replacement” dosing from 400mg/kg every 3-4 weeks to “treatment” dosing (800 – 1000mg/kg every 3-4 weeks). Often there are not disease-specific medications and patients with secondary autoimmune cytopenia syndromes are treated similarly to patients with primary disease. It is beyond the scope of this manuscript to discuss all of these conditions in detail; however, ALPS will be described in detail as an example of how understanding disease biology can lead to better therapeutic options.
Patients who undergo solid organ transplant or HSCT rarely develop autoimmune cytopenias, a serious complication since these patients are often refractory to standard treatments, and escalating immune suppression can increase the risk of opportunistic infection. As transplant patients who develop immune cytopenias can be difficult to treat, aggressive multi-agent therapy, including rituximab, is often used.43,44 In addition, the trigger in many patients is calcineurin inhibitor- mediated immune dysregulation. In this case, an effective treatment strategy can be to transition to alternative immune suppression, such as sirolimus.45,46
Autoimmune Lymphoproliferative Syndrome (ALPS)
ALPS is a rare disorder of abnormal lymphocyte survival caused by dysregulation of the FAS apoptotic pathway (Figure 1).47 Normally, as part of the downregulation of the immune response, activated B and T lymphocytes up-regulate Fas, and activated T-lymphocytes upregulate Fas ligand. Fas and Fas ligand interact to trigger the caspase cascade leading to proteolysis, DNA degradation, and apoptosis. Patients with ALPS have a defect in this apoptotic pathway, leading to abnormal lymphocyte survival and subsequent lymphoproliferation, autoimmune disease, and cancer.48 ALPS is classified by mutation type: germline mutations in FAS (ALPS-FAS) occur in 70% of patients; somatic mutations in FAS limited to the double negative T-cells (DNT) compartment (see below; ALPS-sFAS) occur in 10%; rare patients have mutated FASL or CASP10, and approximately 20% have no identifiable genetic abnormality (ALPS-U).49,50
Figure 1. Fas apoptotic pathway.
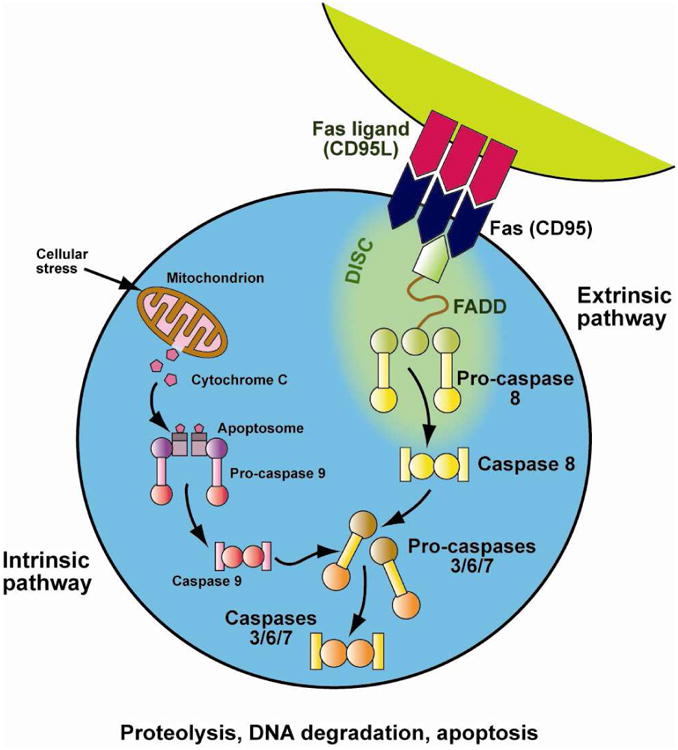
Normally, as part of the down-regulation of the immune response, activated B and T lymphocytes up-regulate FAS and activated T lymphocytes up-regulate FAS-ligand. These interact and trigger the caspase cascade leading to proteolysis, DNA degradation, and apoptosis. This FAS-mediated pathway is part of the extrinsic apoptotic pathway. In contrast, mitochondrial-induced apoptosis after cellular stress is part of the intrinsic apoptotic pathway. Patients with ALPS have a defect in the FAS-apoptotic pathway, leading to abnormal lymphocyte survival. Courtesy of © Sue Seif.
Most ALPS patients present at a young age (median age 11.5 months).51 Lymphoproliferation is ubiquitous and presents as lymphadenopathy, splenomegaly, and/or hepatomegaly, ranging from mild to massive. Lymphadenopathy rarely requires medical intervention; however, some patients have organ compromise from bulky disease, and others develop splenic sequestration. Autoimmunity is the second most common clinical manifestation, affecting over 70% of patients, these disease manifestations most commonly require medical intervention. Most commonly seen are mild to severe autoimmune cytopenias affecting one or more cell lineages. Some patients require treatment for periodic disease flares after infections, and others need chronic therapy. Similar to SLE, patients with ALPS can also develop autoimmune disease affecting virtually any organ system.52 Patients with ALPS may have an increased risk of secondary cancers, most commonly EBER+ non-Hodgkin lymphoma.53 This risk is approximately 5-10% and is most common in patients with FAS mutations.
Until recently, in order for a patient to be diagnosed with ALPS, the patient had to meet three mandatory diagnostic criteria: (1) chronic non-malignant lymphoproliferation (lymphadenopathy or splenomegaly > 6 months); (2) elevated peripheral blood DNTs; and (3) in vitro evidence of defective Fas-mediated apoptosis. These diagnostic criteria were revised in 2010 based on an international consensus conference.49 The first two criteria continue to be mandatory under the new diagnostic algorithm. All patients with ALPS have elevated peripheral blood DNTs, T cells that are CD3+/CD4-/CD8- and are positive for the TCRα/β receptor.54 The third criterion, Fas-mediated apoptosis testing, is still used for the diagnosis in some patients; however, is not needed for the majority of patients. This testing is labor-intensive and only performed in a few specialized laboratories in the world. In addition, this testing is negative in the second most common form of ALPS: ALPS-sFAS.50 Most patients can be diagnosed with genetic testing or by measuring biomarkers that are highly predictive of ALPS, including elevated levels of vitamin B12, IL-10, sFASL, and IL-18 or a combination of hypergammaglobulinemia and autoimmune cytopenias.55,56
The new diagnostic algorithm (Table 3) categorizes ALPS as definitive or probable.49 Definitive ALPS is defined as meeting the two required criteria of lymphoproliferation and elevated DNTs with either a genetic mutation in an ALPS causative gene OR defective FAS mediated apoptosis in vitro. Probable ALPS is defined as meeting the two required criteria and the presence of a biomarker, positive family history, or histopathologic finding of ALPS. Patients with probable and definitive ALPS should be counseled that they have ALPS and subsequently managed using the same approaches, with the “probable” distinction used only for consistency in the medical literature and for clinical trials.
Table 3. ALPS diagnostic criteria.
| Old diagnostic criteria (Prior to 2010) |
|---|
| Required |
| 1. Chronic (>6 months) non-malignant lymphoproliferation (lymphadenopathy or splenomegaly) |
| 2. Elevated DNTs in peripheral blood |
| 3. Laboratory evidence of defective Fas mediated apoptosis in vitro |
| New diagnostic criteria |
| Required |
| 1. Chronic non-malignant lymphoproliferation |
| 2. Elevated DNTs in peripheral blood |
| Accessory |
| Primary |
| 1. Laboratory evidence of defective Fas mediated apoptosis (on 2 separate assays) |
| 2. Mutation in ALPS causative gene (FAS, FASL, CASP10) (somatic or germline) |
| Secondary |
1. Elevated biomarkers (One or more of the following)
|
| 2. Histopathologic findings on biopsy consistent with ALPS |
| 3. Autoimmune cytopenias and polyclonal hypergammaglobulinemia |
| 4. Family history of ALPS or nonmalignant lymphoproliferation |
|
Definitive Diagnosis: Both required criteria plus one primary accessory criteria Probable Diagnosis: Both required criteria plus one secondary accessory criteria Patients with probable and definitive ALPS should be treated the same. |
Corticosteroids are the first-line agent of choice for acute flares.57 ALPS is a chronic disease, and many patients need chronic treatment. IVIgG is usually ineffective in ALPS except for patients with single lineage autoimmune thrombocytopenia.58 The second line therapies most commonly used in idiopathic ES, rituximab and splenectomy, are relatively contraindicated in ALPS.59,60 Thus, it is very important to distinguish ES from ALPS. ALPS patients who undergo splenectomy have a markedly increased lifetime risk of pneumococcal sepsis (∼30%) even with vaccination and antimicrobial prophylaxis.58,61 Rituximab can be useful in treating cytopenias in ALPS, but some patients never recover B cell function, leading to life-long IVIgG replacement.62
The two most commonly used second line therapies for ALPS are mycophenolate mofetil (MMF, Cellcept) and sirolimus (rapamycin, Rapamune). MMF has been shown to be effective in improving autoimmune cytopenias in approximately 80% of ALPS patients based on clinical trial results from the NIH.63 While many patients respond, these were frequently partial responses; some patients needed continued corticosteroid treatment; and, some patients relapsed. MMF is not effective against lymphoproliferation and does not reduce DNTs. We have studied the activity of the mTOR inhibitor sirolimus in ALPS. We hypothesized that targeting the PI3K/Akt/mTOR signaling pathway with sirolimus would be effective in ALPS. We tested this hypothesis first in a murine model and found rapid improvement in all disease manifestations and superior activity compared to other drugs, including MMF.64 Abnormal DNTs were eliminated and other lymphocyte subsets were relatively spared. Furthermore, the PI3K/Akt/mTOR signaling pathway was abnormally activated in the DNT compartment, suggesting sirolimus is a targeted therapy for the disease (unpublished data). Based on these results, we currently have an open clinical trial of sirolimus in pediatric ALPS (NCT00392951). We published the results of the first cohort of five patients.65 The majority of patients in the clinical trial (as well as 50 international patients for whom we have assisted with sirolimus management) had a rapid, complete, and durable response with resolution of autoimmune cytopenias and other autoimmune disease manifestations and elimination of lymphoproliferation and DNTs, results confirmed by other groups.66,67 The majority of children were refractory to or intolerant of other therapies, including MMF.
Treatment of single and multi-lineage autoimmune cytopenias
As mentioned above, the treatment for autoimmune cytopenias is heterogeneous and complex. Until recently, options were limited, and the initial management for patients with acute or chronic disease, single lineage or multi-lineage, and primary or secondary disease was IVIgG or corticosteroids followed by splenectomy for intolerant or refractory patients. Recently, this paradigm has added rituximab early into therapy for many patients. Next line treatments varied widely because of the lack of randomized trials and preclinical data, preventing disease-specific target identification. As described above with TPO-RAs in ITP and sirolimus in ALPS, disease biology can often lead to superior therapies. The genomic age may lead to a better understanding of the biology of autoimmune cytopenias.
Medication choice is often based on the experience of the practicing physician instead of evidence or biologic rationale. In addition, there are considerable misconceptions surrounding the safety of a number of oral immune suppressants. Drugs such as MMF, sirolimus, tacrolimus, and mercaptopurine are often used as part of a multi-agent immunosuppressant backbone after organ or marrow transplant or as a part of a chemotherapy regimen for lymphoid malignancies. The risk of infection is markedly higher with multi-agent therapy compared with single agent therapy. Some practitioners falsely assume that these drugs are more immunosuppressive and carry a higher infectious risk than corticosteroids. The post-transplant literature has clearly established that the agents most likely to predispose to opportunistic infection are corticosteroids.68,69 Chronic corticosteroid use also has significant long-term morbidity, especially on bone health, as its use leads to osteoporosis and avascular necrosis. Among oral immune suppressants, the degree of immune suppression varies as is evidenced by development of progressive multifocal leukoencephalopathy with some drugs but not others (Table 4).
Table 4. Immunosuppressive agents used for childhood autoimmune cytopenias and association with progressive multifocal leukoencephalopathy.
| Drugs associated with PML | Drugs not associated with PML |
|---|---|
|
| |
| Azathioprine | Everolimus |
| Cyclosporine | Methotrexate |
| Cyclophosphamide | Sirolimus |
| Mycophenolate Mofetil | |
| Rituximab | |
| Tacrolimus | |
Data From: Schmedt N, Andersohn F, Garbe E. Signals of progressive multifocal leukoencephalopathy for immunosuppressants: a disproportionality analysis of spontaneous reports within the US Adverse Event Reporting System (AERS). Pharmacoepidemiology and drug safety. 2012;21:1216-1220. 70
Rituximab has been shown to be very effective against autoimmune cytopenias in a number of trials as described above. Whether it is a better drug than the myriad of available oral immune suppressants is not known and, unfortunately, based on the rarity and heterogeneity of childhood autoimmune cytopenia syndromes, it is unlikely there will be randomized trials for many of these drugs. This raises the question: how should the child who fails standard front-line therapy be treated? One approach is to tailor the therapy based on response to medication. A patient who has no response to rituximab should be approached differently than a patient who has a partial response yet is unable to wean off corticosteroids or a patient who has a complete response and relapses one year later. Rituximab exclusively targets B cells. If a patient has no response to rituximab, it is likely that B cells are not the principal driver of the autoimmune disease, and T-cell directed therapy, for example with a calcineurin inhibitor, could be considered. In contrast, the patient with a partial response to rituximab may benefit from a drug that suppresses both B and T cells, such as MMF. Mercaptopurine and azathioprine are metabolized into the same active compound. Accordingly, it will likely be ineffective to use mercaptopurine in a patient who fails azathioprine. Some agents require therapeutic drug monitoring, whereas others do not. Toxicity profiles are vastly different between agents and should be considered in the context of each patient's co-morbidities. Finally, some agents take time to be effective, occasionally up to 2-3 months. Thus, time and patience are a must and patients should be given a chance to respond to a new drug prior to switching therapies. Tables 5-10 list and describe the various immunosuppressive compounds that have been used to treat autoimmune cytopenias. A basic understanding of the drugs, their mechanism of action, the cells targeted, and the side effects of the drugs can help a clinician make rational choices when it comes to choosing the best agent to use for a given patient.
Table 5. Immune modulators for autoimmune cytopenias.
| Immune Modulators |
|---|
| Corticosteroids (Prednisone and Dexamethasone) |
| Mechanism of Action: (Multi-factorial) Down-regulates inflammatory cytokine production at transcriptional and translational level. Decreases leukocyte trafficking, including neutrophil migration. Decreases dendritic cells. Depletes T cells and B cells (T≫B). Also, effects T-lineage commitment.71 |
| Lymphocytes targeted: B, T, NK cells, monocytes, macrophages, neutrophils, and eosinophils |
| Pros: Most active drug for majority of patients with autoimmune cytopenias. Fast-acting. |
| Ability to titrate dose to response. Lots of data using it in combination with other drugs |
| Cons: Short term toxicities: Mild (hypertension, hyperglycemia, irritability, weight gain) |
| Long term toxicities: Potentially severe (avascular necrosis, cataracts, growth delay) |
| Infectious risk: Single agent - low. Combination therapy - Higher risk than most other oral agents. PCP prophylaxis should be strongly considered when steroids combined with other immune suppressants. Antifungal prophylaxis may also be needed. |
|
|
| IVIgG |
Mechanism of Action: (Multi-factorial). Poorly defined but proposed hypotheses include:72
|
| Pros: Active against ITP in majority of patients. Well-tolerated. Quick acting (<48 hours) |
| Cons: Short duration of activity. Patients with chronic disease may need frequent retreatment. Limited activity against AIHA or AIN. Aseptic meningitis & severe allergic reactions. |
| Infectious Risk: None with current products. Protective against certain infections. |
|
|
| Anti-D immunoglobulin (anti-D)73 |
| Mechanism of Action: Only works in patients who are Rh+ by saturating FcyRs in spleen |
| Pros: Active against ITP in a majority of patients. Well-tolerated. Quick acting (<48 hours). |
| Cons: May cause hemolytic anemia. Only recommended for children with ITP who are Rh+, have a negative DAT, no anemia, and have not undergone splenectomy.6 Can also cause headache, fever, and chills. Rarely, can cause life-threatening DIC. NOT for children with AIHA or AIN. |
| Infectious Risk: None reported. |
Table 10. Other therapies for autoimmune cytopenias.
| Other Therapies |
|---|
| Splenectomy77 |
| Mechanism of action: Removes main site of platelet destruction and site of anti-platelet antibody production |
| Lymphocytes targeted: N/A |
| Pros: Curative in majority of patients with ITP. Often effective in AIHA. Well-characterized risks. Does not affect fertility or harm fetus if pregnant |
| Cons: Removing a healthy organ and cannot predict who will respond. Not very effective for AIN, or multi-lineage cytopenias. Risk of infection with encapsulated organisms. |
| Increased risk of venous thromboembolism and artherosclerosis. ?Increased risk of portal hypertension and pulmonary arterial hypertension. |
| Infectious risk: Encapsulated organisms. Relatively contraindicated in patients with ALPS. |
|
|
| TPO mimetics (TPO-RAS, Romiplostim and eltrombopag)77 |
| Mechanism of action: Agonists for TPO receptor, the major regulator of megakaryocytopoiesis and platelet formation |
| Lymphocytes targeted: N/A |
| Pros: Only second line agent studied in randomized placebo controlled trials (adults) showing clear efficacy. High rates of response and most durable. |
| Cons: Likely need long-term chronic treatment. Abrupt discontinuation of drug can cause “rebound thrombocytopenia.” Theoretical risk of bone marrow fibrosis and leukemic transformation. Increased risk of venous thromboembolism. Eltrombopag is hepatotoxic. |
| Infectious risk: None |
|
|
| Hematopoietic Stem Cell Transplant78-80 |
| Mechanism of action: Allogeneic replaces all blood producing cells with donor cells. Mechanism of activity of autologous may be secondary to intensive immune suppression or due to “immune reset.” |
| Lymphocytes targeted: All |
| Pros: Can be curative |
| Cons: Very toxic. ∼10-50% mortality and significant morbidity. Often ineffective ∼50% response rates. Should be reserved for patients with severe and refractory autoimmune cytopenias and proven success. Most commonly used for secondary autoimmune cytopenias (immune deficiency, cancer, some lymphoproliferative disorders, some rheumatologic conditions). Can worsen autoimmune disease. |
| Infections: Extremely high risk. Bacteria, Fungi, Viruses, others. |
|
|
| Plasmapheresis |
| Mechanism of action: Mechanical plasma filtration to remove antibodies |
| Lymphocytes targeted: N/A |
| Pros: Very rapid onset. Effective for IgG mediated autoimmune cytopenias |
| Cons: Improvements short-lived and often see relapse after stopping. Often used in conjunction with immune suppression. Requires central venous catheters in young children. Limited activity against IgM mediated disease. Hypotension, hypocalcemia. Removes beneficial antibodies too and medications. |
| Infections: Biggest risk is from the central line |
Conclusion
In conclusion, the diagnosis, management, and treatment of childhood autoimmune cytopenias can be complex. Fortunately, the past 10 years have seen the discovery and use of new agents that have improved efficacy. The genomic era will certainly lead to novel insights into disease biology, allowing for the rational selection of targeted therapies, better determination of which patients need treatment, and improved prediction of which patients will have short-term disease.
Table 6. Monoclonal Antibodies for autoimmune cytopenias.
| Monoclonal Antibodies |
|---|
| Rituximab74 |
| Mechanism of Action: Anti-CD20 monoclonal antibody |
| Lymphocytes targeted: B cells |
| Pros: Very active drug for many patients with autoimmune cytopenias |
| Studied in randomized trials |
| Does not require a patient to take daily medication. |
| Cons: Very long half-life with B cell depletion lasting up to a year in most patients. Thus, non-responders have continued B-cell depletion yet no benefit |
| Infectious risk: Single agent low. Risk of viral reactivation |
|
|
| Alemtuzumab (Campath) |
| Mechanism of Action: Anti-CD52 monoclonal antibody |
| Lymphocytes targeted: B, T, and NK cells |
| Pros: Very active against large number of autoimmune diseases, including cytopenias |
| Cons: Limited data for childhood idiopathic autoimmune cytopenias (Most of the data in adults with autoimmune cytopenias secondary to chronic lymphocytic leukemia). Very long half-life with B, T, and NK depletion lasting many months. Very high infectious risk. |
| No consistent recommendations on best dose or schedule. Can be immunoablative at high doses, requiring stem cell rescue for immune recovery. |
| Infectious risk: Very high as a single agent. Risk of viral and fungal infections. Prophylactic antifungal and antiviral medications should be considered. Routine viral monitoring needed especially for CMV reactivation. Consider IVIgG replacement therapy. |
|
|
| Anti-thymocyte globulin (ATG and ALG) |
| Mechanism of Action: Horse (ATG) or rabbit (ALG) antibodies against human T cells |
| Lymphocytes targeted: T cells |
| Pros: Very active against large number of autoimmune diseases, including cytopenias |
| Cons: Limited data in childhood idiopathic or secondary autoimmune cytopenias. Most data anecdotal; however, numerous studies showing activity in autoimmune aplastic anemia and for prevention and treatment of GVHD in children. |
| High infectious risk. Long half-life. Severe infusional toxicities and risk for serum sickness. |
| Infectious risk: High. Largely unknown as single agent as most commonly combined with other drugs. PCP, antifungal, and anti-viral prophylaxis should be considered, especially if combined with other agents |
Table 7. Anti-metabolites for Autoimmune Cytopenias.
| Anti-Metabolites |
|---|
| Purine Analogues (Mercaptopurine (6MP), Azathioprine, and Thioguanine) |
| Mechanism of Action: Inhibits purine synthesis. Azathioprine is metabolized into 6MP. |
| This class of drugs also includes fludarabine, pentostatin, and cladribine; however, these are almost never used for autoimmune cytopenias in children. |
| Lymphocytes targeted: B, T, and NK cells |
| Pros: Well-tolerated agents with demonstrated activity in children. Numerous studies in childhood cancer have shown can be safely combined with other drugs. |
| Cons: Often takes 1-3 months for response.75, 76 Polymorphisms in the enzyme thiopurine methyltransferase (TPMT) can lead to decreased activity of enzyme, inability to metabolize the drugs and markedly increased toxicity. Can cause myelosuppression and hepatic toxicity. |
| Infectious risk: Single agent low except in patients with absent TPMT activity. |
|
|
| Methotrexate |
| Mechanism of Action: Inhibits folate synthesis, which is needed for thymidine synthesis. Inhibits purine metabolism |
| Lymphocytes targeted: B, T, and NK cells (T > B) |
| Pros: Well-tolerated agent with demonstrated activity in children, especially in children with rheumatologic autoimmune diseases with secondary autoimmune cytopenias. Can be given by oral, subcutaneous injection, or intravenous route. Numerous studies in childhood cancer have shown can be safely combined with other drugs. |
| Cons: Dose dependent myelosuppression, liver, neurologic, and renal toxicity. These side effects very rare if dosed for autoimmune diseases (as compared to dosing for cancer). |
| Infectious risk: Single agent low when dosed for autoimmune diseases |
|
|
| Mycophenolate Mofetil |
| Mechanism of Action: Inhibits iosine monophosphate dehydrogenase in purine synthesis |
| Lymphocytes targeted: B, T, and NK cells |
| Pros: Well-tolerated with demonstrated activity in children. |
| Cons: Often results in partial responses. Side effects include diarrhea and neutropenia. |
| Infectious risk: Single agent low. Recently, FDA released “black-box” warning because of risk of progressive multifocal leukoencephalopathy (PML). Prescribers now need REMS certification because of potential risks to fetus in pregnant women. |
Table 8. Signaling pathway inhibitors for autoimmune cytopenais.
| Calcineurin Inhibitors |
|---|
| Tacrolimus and Cyclosporine (CSA) |
| Mechanism of action: Prevents T-cell activation and cytokine release by inhibiting calcineurin. |
| Calcinuerin normally dephosphorylates nuclear factor of activated T-cells (NF-AT) Lymphocytes targeted: T cells only |
| Pros: Well-tolerated. Anecdotal reports showing activity in childhood autoimmune cytopenias |
| Cons: Can cause autoimmune cytopenias and microangiopathic hemolytic anemia (MAH). CSA has more side effect than tacrolimus and it is not more effective. Nephrotoxic. Can cause seizures from combination of hypomagnesemia and hypertension. Requires therapeutic drug monitoring. |
| Infectious risk: Single agent low. Consider PCP prophylaxis. |
|
|
| mTOR Inhibitors |
|
|
| Sirolimus (Rapamycin) |
| Mechanism of action: Targets mTORC1 complex in PI3K/Akt/mTOR signaling pathway. Other mTOR inhibitors, including everolimus are being studied in autoimmune disease. |
| Lymphocytes targeted: B and T cells. Spares Regulatory T cells (Tregs) |
| Pros: Well-tolerated. Very active in ALPS associated autoimmune cytopenias. Demonstrated activity and arguably treatment of choice for post solid organ transplant autoimmune cytopenias. Least potential for secondary cancers with chronic use based on preclinical data. One of a small handful of immune suppressants shown not to be associated with PML. |
| Cons: Requires therapeutic drug monitoring. Significant drug-drug interactions. Has few side effects; however, inhibits endothelial and epithelial healing and tends to magnify the side effects of other drugs. For example, does not cause MAH; however, when combined with drugs that cause MAH (eg calcineurin inhibitors) greatly increases the risk over the other drug alone. Can cause mucositis and hyperlipidemia. |
| Infectious risk: Single agent very low. May have activity against EBV, CMV, and fungi. |
Table 9. Cytotoxic chemotherapeutics for autoimmune cytopenias.
| Cytotoxic Chemotherapeutics |
|---|
| Cyclophosphamide |
| Mechanism of action: Alkylates DNA |
| Lymphocytes targeted: B, T and NK cells. Eliminates Tregs. |
| Pros: Long and established track record of use in rheumatologic autoimmune disease, including autoimmune cytopenias. |
| Cons: Toxicity (dose dependent). Short term: myelosuppression, GI toxicity, alopecia. Long term: Associated with increased risk of Monosomy 7 MDS/AML. Infertility in males. Duration of response often limited to a few months. |
| Infectious risk: Dose dependent. Low risk with doses used commonly for autoimmune disease. Very high risk with myeloablative dosing. |
|
|
| Vincristine and Vinblastine |
| Mechanism of action: mitotic inhibitor |
| Lymphocytes targeted: B, T, and NK cells. |
| Pros: Long and established track record of use and documented efficacy |
| Cons: Infrequently used because of development of “newer” agents. Side effects for vincristine include neuropathy and constipation. Side effects for vinblastine include myelosuppression, GI toxicity, and alopecia. Duration of response often limited to a few months. |
| Infectious risk: Low with monotherapy. Higher when combined with other drugs |
Key points.
Most children with autoimmune cytopenias have idiopathic disease with no secondary cause and will enter a spontaneous remission with time.
Chronic or multi-lineage disease should prompt testing for secondary causes of autoimmune cytopenias, including HIV, systemic lupus erythematosis, ALPS, and common variable immune deficiency (CVID)
First-line treatments typically include corticosteroids and IVIgG. Second-line treatments vary depending on the cell lineage(s) affected and whether or not there is an underlying cause
Corticosteroids can cause significant long-term morbidity; therefore, their long-term use should be avoided, as numerous safe and effective alternatives exist.
Recent advances have led to targeted therapies for select patients, including the use of thrombopoeitin receptor agonists (TPO mimetics) for ITP and sirolimus for ALPS
Acknowledgments
COI: MPL has participated in clinical trials funded by GSK and Amgen.
This work was supported by R56A1091791, R21A1099301, and the Cures within Reach (formerly Partnership for Cures) (DTT)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Neunert CE, Buchanan GR, Imbach P, et al. Severe hemorrhage in children with newly diagnosed immune thrombocytopenic purpura. Blood. 2008;112:4003–4008. doi: 10.1182/blood-2008-03-138487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMillan R. Antiplatelet antibodies in chronic immune thrombocytopenia and their role in platelet destruction and defective platelet production. Hematology/oncology clinics of North America. 2009;23:1163–1175. doi: 10.1016/j.hoc.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Yazdanbakhsh K, Zhong H, Bao W. Immune dysregulation in immune thrombocytopenia. Seminars in hematology. 2013;50(1):S63–67. doi: 10.1053/j.seminhematol.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bussel JB, Kuter DJ. New thrombopoietic agents: introduction. Seminars in hematology. 2010;47:211. doi: 10.1053/j.seminhematol.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113:2386–2393. doi: 10.1182/blood-2008-07-162503. [DOI] [PubMed] [Google Scholar]
- 6.Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117:4190–4207. doi: 10.1182/blood-2010-08-302984. [DOI] [PubMed] [Google Scholar]
- 7.Aronis S, Platokouki H, Avgeri M, et al. Retrospective evaluation of long-term efficacy and safety of splenectomy in chronic idiopathic thrombocytopenic purpura in children. Acta paediatrica. 2004;93:638–642. [PubMed] [Google Scholar]
- 8.Patel VL, Mahevas M, Lee SY, et al. Outcomes 5 years after response to rituximab therapy in children and adults with immune thrombocytopenia. Blood. 2012;119:5989–5995. doi: 10.1182/blood-2011-11-393975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hedlund-Treutiger I, Henter JI, et al. Randomized study of IVIg and high-dose dexamethasone therapy for children with chronic idiopathic thrombocytopenic purpura. Journal of pediatric hematology/oncology. 2003;25:139–144. doi: 10.1097/00043426-200302000-00011. [DOI] [PubMed] [Google Scholar]
- 10.Kaushansky K, Lok S, Holly RD, et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature. 1994;369:568–571. doi: 10.1038/369568a0. [DOI] [PubMed] [Google Scholar]
- 11.Vadhan-Raj S, Murray LJ, Bueso-Ramos C, et al. Stimulation of megakaryocyte and platelet production by a single dose of recombinant human thrombopoietin in patients with cancer. Annals of internal medicine. 1997;126:673–681. doi: 10.7326/0003-4819-126-9-199705010-00001. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Yang C, Xia Y, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood. 2001;98:3241–3248. doi: 10.1182/blood.v98.12.3241. [DOI] [PubMed] [Google Scholar]
- 13.Arnold DM, Nazi I, Kelton JG. New treatments for idiopathic thrombocytopenic purpura: rethinking old hypotheses. Expert opinion on investigational drugs. 2009;18:805–819. doi: 10.1517/13543780902905848. [DOI] [PubMed] [Google Scholar]
- 14.Bussel JB, Buchanan GR, Nugent DJ, et al. A randomized, double-blind study of romiplostim to determine its safety and efficacy in children with immune thrombocytopenia. Blood. 2011;118:28–36. doi: 10.1182/blood-2010-10-313908. [DOI] [PubMed] [Google Scholar]
- 15.Letsky EA, Greaves M. Guidelines on the investigation and management of thrombocytopenia in pregnancy and neonatal alloimmune thrombocytopenia. Maternal and Neonatal Haemostasis Working Party of the Haemostasis and Thrombosis Task Force of the British Society for Haematology. British journal of haematology. 1996;95:21–26. [PubMed] [Google Scholar]
- 16.Roberts IA. The changing face of haemolytic disease of the newborn. Early human development. 2008;84:515–523. doi: 10.1016/j.earlhumdev.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Seve P, Philippe P, Dufour JF, et al. Autoimmune hemolytic anemia: classification and therapeutic approaches. Expert review of hematology. 2008;1:189–204. doi: 10.1586/17474086.1.2.189. [DOI] [PubMed] [Google Scholar]
- 18.Bussel JB, Cunningham-Rundles C, Abraham C. Intravenous treatment of autoimmune hemolytic anemia with very high dose gammaglobulin. Vox sanguinis. 1986;51:264–269. doi: 10.1111/j.1423-0410.1986.tb01967.x. [DOI] [PubMed] [Google Scholar]
- 19.Petz LD. Treatment of autoimmune hemolytic anemias. Current opinion in hematology. 2001;8:411–416. doi: 10.1097/00062752-200111000-00016. [DOI] [PubMed] [Google Scholar]
- 20.Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert review of hematology. 2011;4:607–618. doi: 10.1586/ehm.11.60. [DOI] [PubMed] [Google Scholar]
- 21.Chertkow G, Dacie JV. Results of splenectomy in auto-immune haemolytic anaemia. British journal of haematology. 1956;2:237–249. [PubMed] [Google Scholar]
- 22.Pignon JM, Poirson E, Rochant H. Danazol in autoimmune haemolytic anaemia. British journal of haematology. 1993;83:343–345. doi: 10.1111/j.1365-2141.1993.tb08293.x. [DOI] [PubMed] [Google Scholar]
- 23.Berentsen S. How I manage cold agglutinin disease. British journal of haematology. 2011;153:309–317. doi: 10.1111/j.1365-2141.2011.08643.x. [DOI] [PubMed] [Google Scholar]
- 24.Petz LD. Cold antibody autoimmune hemolytic anemias. Blood reviews. 2008;22:1–15. doi: 10.1016/j.blre.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 25.Maheshwari A, Christensen RD, Calhoun DA. Immune-mediated neutropenia in the neonate. Acta paediatrica. 2002;91:98–103. doi: 10.1111/j.1651-2227.2002.tb02912.x. [DOI] [PubMed] [Google Scholar]
- 26.Bussel JB, Abboud MR. Autoimmune neutropenia of childhood. Critical reviews in oncology/hematology. 1987;7:37–51. doi: 10.1016/s1040-8428(87)80013-6. [DOI] [PubMed] [Google Scholar]
- 27.Bruin MC, von dem Borne AE, Tamminga RY, et al. Neutrophil antibody specificity in different types of childhood autoimmune neutropenia. Blood. 1999;94:1797–1802. [PubMed] [Google Scholar]
- 28.Capsoni F, Sarzi-Puttini P, Zanella A. Primary and secondary autoimmune neutropenia. Arthritis research & therapy. 2005;7:208–214. doi: 10.1186/ar1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bux J, Behrens G, Jaeger G, et al. Diagnosis and clinical course of autoimmune neutropenia in infancy: analysis of 240 cases. Blood. 1998;91:181–186. [PubMed] [Google Scholar]
- 30.Bux J, Chapman J. Report on the second international granulocyte serology workshop. Transfusion. 1997;37:977–983. doi: 10.1046/j.1537-2995.1997.37997454028.x. [DOI] [PubMed] [Google Scholar]
- 31.Mathew P, Chen G, Wang W. Evans syndrome: results of a national survey. Journal of pediatric hematology/oncology. 1997;19:433–437. doi: 10.1097/00043426-199709000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Wang WC. Evans syndrome in childhood: pathophysiology, clinical course, and treatment. The American journal of pediatric hematology/oncology. 1988;10:330–338. doi: 10.1097/00043426-198824000-00013. [DOI] [PubMed] [Google Scholar]
- 33.Seif AE, Manno CS, Sheen C, et al. Identifying autoimmune lymphoproliferative syndrome in children with Evans syndrome: a multi-institutional study. Blood. 2010;115:2142–2145. doi: 10.1182/blood-2009-08-239525. [DOI] [PubMed] [Google Scholar]
- 34.Savasan S, Warrier I, Ravindranath Y. The spectrum of Evans' syndrome. Archives of disease in childhood. 1997;77:245–248. doi: 10.1136/adc.77.3.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aladjidi N, Leverger G, Leblanc T, et al. New insights into childhood autoimmune hemolytic anemia: a French national observational study of 265 children. Haematologica. 2011;96:655–663. doi: 10.3324/haematol.2010.036053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miescher PA, Tucci A, Beris P, et al. Autoimmune hemolytic anemia and/or thrombocytopenia associated with lupus parameters. Seminars in hematology. 1992;29:13–17. [PubMed] [Google Scholar]
- 37.Teachey DT, Manno CS, Axsom KM, et al. Unmasking Evans syndrome: T-cell phenotype and apoptotic response reveal autoimmune lymphoproliferative syndrome (ALPS) Blood. 2005;105:2443–2448. doi: 10.1182/blood-2004-09-3542. [DOI] [PubMed] [Google Scholar]
- 38.Savasan S, Warrier I, Buck S, et al. Increased lymphocyte Fas expression and high incidence of common variable immunodeficiency disorder in childhood Evans' syndrome. Clinical immunology. 2007;125:224–229. doi: 10.1016/j.clim.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 39.Heeney MM, Zimmerman SA, Ware RE. Childhood autoimmune cytopenia secondary to unsuspected common variable immunodeficiency. The Journal of pediatrics. 2003;143:662–665. doi: 10.1067/S0022-3476(03)00445-1. [DOI] [PubMed] [Google Scholar]
- 40.George JN, Woolf SH, Raskob GE, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood. 1996;88:3–40. [PubMed] [Google Scholar]
- 41.Bader-Meunier B, Aladjidi N, Bellmann F, et al. Rituximab therapy for childhood Evans syndrome. Haematologica. 2007;92:1691–1694. doi: 10.3324/haematol.11540. [DOI] [PubMed] [Google Scholar]
- 42.Rao A, Kelly M, Musselman M, et al. Safety, efficacy, and immune reconstitution after rituximab therapy in pediatric patients with chronic or refractory hematologic autoimmune cytopenias. Pediatric blood & cancer. 2008;50:822–825. doi: 10.1002/pbc.21264. [DOI] [PubMed] [Google Scholar]
- 43.Miloh T, Arnon R, Roman E, et al. Autoimmune hemolytic anemia and idiopathic thrombocytopenic purpura in pediatric solid organ transplant recipients, report of five cases and review of the literature. Pediatric transplantation. 2011;15:870–878. doi: 10.1111/j.1399-3046.2011.01596.x. [DOI] [PubMed] [Google Scholar]
- 44.Li M, Goldfinger D, Yuan S. Autoimmune hemolytic anemia in pediatric liver or combined liver and small bowel transplant patients: a case series and review of the literature. Transfusion. 2012;52:48–54. doi: 10.1111/j.1537-2995.2011.03254.x. [DOI] [PubMed] [Google Scholar]
- 45.Acquazzino MA, Fischer RT, Langnas A, et al. Refractory autoimmune hemolytic anemia after intestinal transplant responding to conversion from a calcineurin to mTOR inhibitor. Pediatric transplantation. 2013 doi: 10.1111/petr.12101. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 46.Teachey DT, Jubelirer T, Baluarte HJ, et al. Treatment with sirolimus ameliorates tacrolimus-induced autoimmune cytopenias after solid organ transplant. Pediatric blood & cancer. 2009;53:1114–1116. doi: 10.1002/pbc.22183. [DOI] [PubMed] [Google Scholar]
- 47.Rieux-Laucat F, Le Deist F, Fischer A. Autoimmune lymphoproliferative syndromes: genetic defects of apoptosis pathways. Cell Death Differ. 2003;10:124–133. doi: 10.1038/sj.cdd.4401190. [DOI] [PubMed] [Google Scholar]
- 48.Bleesing JJ. Autoimmune lymphoproliferative syndrome: A genetic disorder of abnormal lymphocyte apoptosis. Immunology and Allergy Clinics of North America. 2002;22:339–349. doi: 10.1016/s0031-3955(05)70272-8. [DOI] [PubMed] [Google Scholar]
- 49.Oliveira JB, Bleesing JJ, Dianzani U, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116:e35–40. doi: 10.1182/blood-2010-04-280347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holzelova E, Vonarbourg C, Stolzenberg MC, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351:1409–1418. doi: 10.1056/NEJMoa040036. [DOI] [PubMed] [Google Scholar]
- 51.Jackson CE, Puck JM. Autoimmune lymphoproliferative syndrome, a disorder of apoptosis. Curr Opin Pediatr. 1999;11:521–527. doi: 10.1097/00008480-199912000-00009. [DOI] [PubMed] [Google Scholar]
- 52.Sneller MC, Wang J, Dale JK, et al. Clincial, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89:1341–1348. [PubMed] [Google Scholar]
- 53.Straus SE, Jaffe ES, Puck JM, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001;98:194–200. doi: 10.1182/blood.v98.1.194. [DOI] [PubMed] [Google Scholar]
- 54.Bleesing JJ, Brown MR, Dale JK, et al. TcR-alpha/beta(+) CD4(-)CD8(-) T cells in humans with the autoimmune lymphoproliferative syndrome express a novel CD45 isoform that is analogous to murine B220 and represents a marker of altered O-glycan biosynthesis. Clin Immunol. 2001;100:314–324. doi: 10.1006/clim.2001.5069. [DOI] [PubMed] [Google Scholar]
- 55.Magerus-Chatinet A, Stolzenberg MC, Loffredo MS, et al. FAS-L, IL-10, and double-negative CD4- CD8-TCR alpha/beta+ T cells are reliable markers of autoimmune lymphoproliferative syndrome (ALPS) associated with FAS loss of function. Blood. 2009;113:3027–3030. doi: 10.1182/blood-2008-09-179630. [DOI] [PubMed] [Google Scholar]
- 56.Caminha I, Fleisher TA, Hornung RL, et al. Using biomarkers to predict the presence of FAS mutations in patients with features of the autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. 2010;125:946–949 e946. doi: 10.1016/j.jaci.2009.12.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bleesing JJ, Straus SE, Fleisher TA. Autoimmune lymphoproliferative syndrome. A human disorder of abnormal lymphocyte survival. Pediatr Clin North Am. 2000;47:1291–1310. doi: 10.1016/s0031-3955(05)70272-8. [DOI] [PubMed] [Google Scholar]
- 58.Rao VK, Oliveira JB. How I treat autoimmune lymphoproliferative syndrome. Blood. 2011 doi: 10.1182/blood-2011-07-325217. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teachey DT. Autoimmune lymphoproliferative syndrome: new approaches to diagnosis and management. Clin Adv Hematol Oncol. 2011;9:233–235. [PubMed] [Google Scholar]
- 60.Teachey DT. New advances in the diagnosis and treatment of autoimmune lymphoproliferative syndrome. Curr Opin Pediatr. 2012;24:1–8. doi: 10.1097/MOP.0b013e32834ea739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neven B, Magerus-Chatinet A, Florkin B, et al. A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood. 2011 doi: 10.1182/blood-2011-04-347641. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 62.Rao VK, Price S, Perkins K, et al. Use of rituximab for refractory cytopenias associated with autoimmune lymphoproliferative syndrome (ALPS) Pediatr Blood Cancer. 2009;52:847–852. doi: 10.1002/pbc.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rao VK, Dugan F, Dale JK, et al. Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br J Haematol. 2005;129:534–538. doi: 10.1111/j.1365-2141.2005.05496.x. [DOI] [PubMed] [Google Scholar]
- 64.Teachey DT, Obzut D, Axsom KM, et al. Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS) Blood. 2006 doi: 10.1182/blood-2006-01-010124. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Teachey DT, Greiner R, Seif A, et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009;145:101–106. doi: 10.1111/j.1365-2141.2009.07595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Janic MD, Brasanac CD, Jankovic JS, et al. Rapid regression of lymphadenopathy upon rapamycin treatment in a child with autoimmune lymphoproliferative syndrome. Pediatric blood & cancer. 2009;53:1117–1119. doi: 10.1002/pbc.22151. [DOI] [PubMed] [Google Scholar]
- 67.Tommasini A, Valencic E, Piscianz E, et al. Immunomodulatory drugs in autoimmune lymphoproliferative syndrome (ALPS) Pediatric blood & cancer. 2012;58:310. doi: 10.1002/pbc.23205. author reply 311. [DOI] [PubMed] [Google Scholar]
- 68.Mikulska M, Raiola AM, Bruno B, et al. Risk factors for invasive aspergillosis and related mortality in recipients of allogeneic SCT from alternative donors: an analysis of 306 patients. Bone marrow transplantation. 2009;44:361–370. doi: 10.1038/bmt.2009.39. [DOI] [PubMed] [Google Scholar]
- 69.Bodro M, Sabe N, Gomila A, et al. Risk factors, clinical characteristics, and outcomes of invasive fungal infections in solid organ transplant recipients. Transplantation proceedings. 2012;44:2682–2685. doi: 10.1016/j.transproceed.2012.09.059. [DOI] [PubMed] [Google Scholar]
- 70.Schmedt N, Andersohn F, Garbe E. Signals of progressive multifocal leukoencephalopathy for immunosuppressants: a disproportionality analysis of spontaneous reports within the US Adverse Event Reporting System (AERS) Pharmacoepidemiology and drug safety. 2012;21:1216–1220. doi: 10.1002/pds.3320. [DOI] [PubMed] [Google Scholar]
- 71.Van Molle W, Libert C. How glucocorticoids control their own strength and the balance between pro- and anti-inflammatory mediators. European journal of immunology. 2005;35:3396–3399. doi: 10.1002/eji.200535556. [DOI] [PubMed] [Google Scholar]
- 72.Nimmerjahn F, Ravetch JV. The antiinflammatory activity of IgG: the intravenous IgG paradox. The Journal of experimental medicine. 2007;204:11–15. doi: 10.1084/jem.20061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Despotovic JM, Lambert MP, Herman JH, et al. RhIG for the treatment of immune thrombocytopenia: consensus and controversy (CME) Transfusion. 2012;52:1126–1136. doi: 10.1111/j.1537-2995.2011.03384.x. quiz 1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grace RF, Bennett CM, Ritchey AK, et al. Response to steroids predicts response to rituximab in pediatric chronic immune thrombocytopenia. Pediatric blood & cancer. 2012;58:221–225. doi: 10.1002/pbc.23130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boruchov DM, Gururangan S, Driscoll MC, et al. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP) Blood. 2007;110:3526–3531. doi: 10.1182/blood-2007-01-065763. [DOI] [PubMed] [Google Scholar]
- 76.Sobota A, Neufeld EJ, Lapsia S, et al. Response to mercaptopurine for refractory autoimmune cytopenias in children. Pediatric blood & cancer. 2009;52:80–84. doi: 10.1002/pbc.21729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ghanima W, Godeau B, Cines DB, et al. How I treat immune thrombocytopenia: the choice between splenectomy or a medical therapy as a second-line treatment. Blood. 2012;120:960–969. doi: 10.1182/blood-2011-12-309153. [DOI] [PubMed] [Google Scholar]
- 78.Rabusin M, Snowden JA, Veys P, et al. Long-term outcomes of hematopoietic stem cell transplantation for severe treatment-resistant autoimmune cytopenia in children. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2013;19:666–669. doi: 10.1016/j.bbmt.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 79.Snowden JA, Pearce RM, Lee J, et al. Haematopoietic stem cell transplantation (HSCT) in severe autoimmune diseases: analysis of UK outcomes from the British Society of Blood and Marrow Transplantation (BSBMT) data registry 1997-2009. British journal of haematology. 2012;157:742–746. doi: 10.1111/j.1365-2141.2012.09122.x. [DOI] [PubMed] [Google Scholar]
- 80.Pession A, Zama D, Masetti R, et al. Hematopoietic stem cell transplantation for curing children with severe autoimmune diseases: is this a valid option? Pediatric transplantation. 2012;16:413–425. doi: 10.1111/j.1399-3046.2012.01691.x. [DOI] [PubMed] [Google Scholar]