

The synchronized motions of inner core residues allosterically modulate the activity of the protein kinase A catalytic subunit.
Keywords: Biochemistry, protein kinases, signaling, NMR relaxation dispersion, Conformational Dynamics, Conformational Entropy
Abstract
Eukaryotic protein kinases (EPKs) constitute a class of allosteric switches that mediate a myriad of signaling events. It has been postulated that EPKs’ active and inactive states depend on the structural architecture of their hydrophobic cores, organized around two highly conserved spines: C-spine and R-spine. How the spines orchestrate the transition of the enzyme between catalytically uncommitted and committed states remains elusive. Using relaxation dispersion nuclear magnetic resonance spectroscopy, we found that the hydrophobic core of the catalytic subunit of protein kinase A, a prototypical and ubiquitous EPK, moves synchronously to poise the C subunit for catalysis in response to binding adenosine 5′-triphosphate. In addition to completing the C-spine, the adenine ring fuses the β structures of the N-lobe and the C-lobe. Additional residues that bridge the two spines (I150 and V104) are revealed as part of the correlated hydrophobic network; their importance was validated by mutagenesis, which led to inactivation. Because the hydrophobic architecture of the catalytic core is conserved throughout the EPK superfamily, the present study suggests a universal mechanism for dynamically driven allosteric activation of kinases mediated by coordinated signal transmission through ordered motifs in their hydrophobic cores.
INTRODUCTION
Protein phosphorylation is the most common signaling mechanism in cell biology, regulating growth, differentiation, and metabolism. Aberrant phosphorylation causes many lethal pathologies, making kinases prime targets for drug therapy (1, 2). Cyclic adenosine 5′-monophosphate (cAMP)–dependent protein kinase A (PKA) is a benchmark for the entire eukaryotic protein kinase (EPK) superfamily. The bilobal core of the catalytic subunit (PKA-C), conserved among all Ser/Thr- and Tyr-specific kinases (3), consists of a small N-lobe and a large C-lobe that harbors most of the catalytic residues as well as the substrate docking surface; adenosine 5′-triphosphate (ATP) binds in the cleft between the two lobes (4). The spatial architecture of the kinase core is also highly conserved and consists of two hydrophobic spines (regulatory R-spine and catalytic C-spine) (5), which serve as noncovalent links between the N-lobe and the C-lobe (Fig. 1). The structural assembly of the R-spine is a hallmark for activated kinases (5). Formation of the R-spine is highly regulated and typically requires phosphorylation of the activation loop (Thr197 in PKA) (6), whereas the C-spine is transiently assembled by ATP binding, which is an absolute requirement for orienting the nucleotide toward the substrate for catalysis (5). Once the spines are assembled, the enzyme binds substrates cooperatively (7, 8) and transfers the γ-phosphate of ATP to Ser/Thr acceptors. The functional relevance of the spines has been validated by the discovery of oncogenic mutations, such as the T334I mutation found in chronic myeloid leukemia that stabilizes the R-spine of the c-Abl kinase, rendering it constitutively active and resistant to imatinib (9). The constitutively activating L485F and L505F mutations found in BRAF (10, 11) further support an active role for the hydrophobic spine architecture in kinase activation and regulation.
Fig. 1. The architecture of the hydrophobic core.
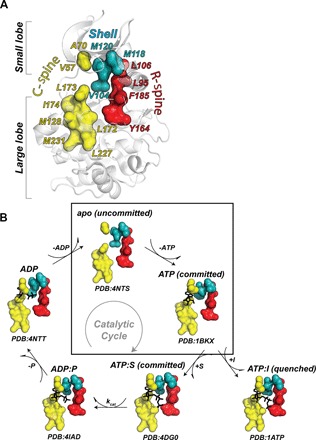
(A) Hydrophobic motifs defining active and inactive states of the kinase: C-spine (yellow), R-spine (red), and shell (teal). (B) X-ray structures of PKA-C showing the architecture of the hydrophobic core of the enzyme along the major conformational states in the catalytic cycle and the inhibited state with the pseudosubstrate. Enclosed in the box is the transition from the apo (open, uncommitted) to the ATP-bound (intermediate, committed) conformation. The synchronous motions associated with this transition are captured here with methyl-TROSY relaxation dispersion NMR spectroscopy.
RESULTS
Here, we investigated the role of conformational dynamics in the hydrophobic core of PKA-C using methyl-TROSY (transverse-relaxation optimized spectroscopy) nuclear magnetic resonance (NMR) spectroscopy, isothermal titration calorimetry, and site-directed mutagenesis. Methyl groups are ideal probes for monitoring the conformational dynamics of hydrophobic cores because they densely populate the interior of folded proteins (12). A total of 122 methyl groups [~98% of the methyl groups from isoleucine, leucine, and valine (ILV) residues] were assigned, enabling us to comprehensively map the enzyme’s hydrophobic core in the apo, binary (nucleotide-bound), and ternary (substrate or pseudosubstrate) forms (figs. S1 to S3). As a substrate, we chose a peptide encompassing the PKA-C recognition sequence of phospholamban (PLN1–19), a central regulator of heart muscle contractility (7, 13). As a pseudosubstrate, we used a peptide corresponding to the inhibitory sequence of the endogenous protein kinase inhibitor (PKI5–24). Upon nucleotide binding, the chemical shift perturbations (CSPs) follow linear trajectories along the coordinates of the open to closed states (figs. S4 and S5). When mapped onto the three-dimensional (3D) structure (Fig. 2A), we observe several spatially contiguous paths of residues crossing the inner core of the enzyme. Although the CSPs of the amide groups reported on the effects of ligand binding on the catalytic loop and the Mg-positioning loop (Mg loop), they were essentially silent for residues in the hydrophobic core (fig. S6). In contrast, methyl groups are sensitive reporters of changes occurring throughout the entire enzyme, especially across the hydrophobic core (table S1). By fusing the β sheet of the N-lobe with β7-β8 of the C-lobe, nucleotide binding exerts global effects that not only radiate to adjacent residues, as seen in Fig. 2B (for example, L173 in β7, V57 in β2, and V104 in the αC-β4 loop), but also extend to distal sites in the hydrophobic core, including L172 and I174 in β7, L180 and L182 in β8, L95 in αC, I150 in αE, and L167 in the catalytic loop, as well as the αH and αI helices, the substrate-binding cleft, and the hub for the regulatory subunit recognition (table S1). Substrate binding (PLN1–19) shifts the enzyme’s conformational equilibrium toward a closed state, which is fully reached when the pseudosubstrate inhibitor (PKI5–24) is bound (fig. S4).
Fig. 2. Methyl CSPs define the conformational transition between open and closed states.
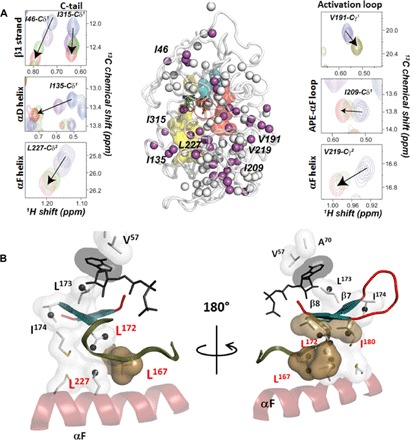
(A) Portions of the methyl-TROSY spectra showing the linear chemical shift trajectories upon ligand binding along the major states of the catalytic cycle. The residues with highly correlated chemical shift changes (R2 > 0.97) are shown with purple spheres, whereas residues with lower correlation coefficients (R2 < 0.97) are shown in white. A comprehensive map of the cross-correlation is shown in fig. S5. (B) Zoom-in of the core of the enzyme showing that the chemical shift changes with the highest correlation cluster in the proximity of the C-spine reporting on the open-to-closed conformational transition.
When analyzed using statistical methods (Materials and Methods), the CSPs display a correlated behavior along the open-to-closed path (fig. S5). Fifty-eight methyl resonances interspersed throughout the enzyme’s structure display correlated linear trajectories, indicating a concerted conformational exchange between the different states (table S1, figs. S4 and S5, and Fig. 2). The correlation map indicates the presence of several clusters of residues in physical contact throughout the kinase core; however, other clusters that are apparently isolated show the same correlated conformational equilibrium. However, when both the CSPs from amide and methyl probes are combined (fig. S6), it is apparent that the conformational changes involve the enzyme globally as predicted by the molecular dynamics simulations and community map analysis (14, 15).
To determine the contribution of the conformational entropy to ligand binding, we calculated the methyl group order parameters (S2) from 2H spin relaxation times (T2) (16). The average S2 (〈S2〉) systematically increases going from the apo to the intermediate and closed states (Fig. 3, fig. S7, and tables S2 to S5), suggesting that the enzyme’s conformational dynamics in the fast time scale (picosecond to nanosecond) plays a significant role in both nucleotide and pseudosubstrate recognition. Nucleotide binding makes the hydrophobic core of the enzyme more densely packed, with a concomitant increase of the local degree of ordering for the methyl-group bearing side chains. The C-spine and neighboring side-chain residues become less mobile when the adenine ring intercalates between A70 and V57 in the N-lobe and L173 in the C-lobe (Fig. 3B), and this locks the core into a committed conformation that is poised for catalysis. The trend and magnitude of the methyl group 〈S2〉 do not correlate with the total entropy of binding, as seen for other systems (fig. S8) (17). This is not surprising because the backbone amide groups show a net increase in the conformational dynamics upon ATP binding (18) and solvation-desolvation effects are not accounted for. Upon pseudosubstrate binding, both backbone amide and side-chain methyl group dynamics show similar trends, with a global reduction of the internal motions (that is, dynamically quenched state).
Fig. 3. Changes of the fast conformational dynamics of the hydrophobic core upon nucleotide and pseudosubstrate binding.
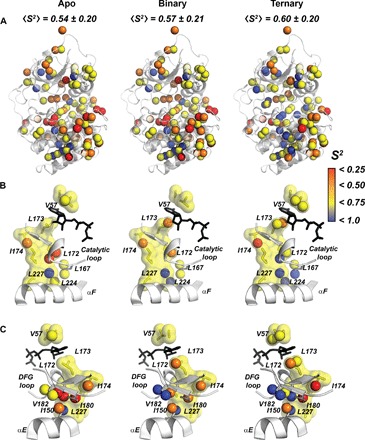
(A) Order parameters of the methyl groups mapped onto the apo, binary (ATPγC-bound), and ternary (ATPγN/PKI5–24) forms of the enzyme, showing an increasing rigidification of the hydrophobic core. (B) Ordering of the C-spine, αF, and the catalytic loop upon nucleotide and PKI binding. (C). Structural details of PKA-C showing the C-spine and the αE helix, highlighting the rigidification of I150, I180, and V182 upon ligand binding and bridging β1-β2 in the N-lobe with β7-β8 in the C-lobe.
Slow conformational dynamics (microsecond-to-millisecond time scale) of the catalytic loops are directly linked to the catalytic cycle of PKA-C (18). A dynamic knockout mutation (that is, Y204A, far from the active site) shifts the motional regime of the catalytic loops outside the microsecond-to-millisecond time scale, reducing the enzyme’s catalytic efficiency (15, 19). To assess the contribution of the hydrophobic core’s slow time scale motions to the catalytic process, we analyzed the structural fluctuations of the methyl-bearing side chains using Carr-Purcell-Meiboom-Gill (CPMG) experiments (20, 21). CPMG relaxation dispersion curves enable one to determine the exchange contribution to the transverse relaxation (Rex) and quantify both the kinetics (kex) and thermodynamics (that is, populations, pA and pB) of the conformational equilibria (tables S6 to S10 and figs. S9 and S10). The apo enzyme shows significant Rex values for several residues interspersed throughout the hydrophobic core, the C-spine (V57 and L172), the R-spine (L95), the R-spine shell (V104), and additional residues neighboring and bridging the spines (L167, I180, and I150) (Fig. 4A). Although it is possible to fit dispersion curves of isolated clusters of residues, they exchange with different rate constants, suggesting that motions in the core of the apo enzyme are essentially asynchronous. Although the nucleotide binding attenuates the magnitude of the Rex values throughout the entire enzyme, significant conformational dynamics are present for residues at the interface between the two lobes. The shapes of the CPMG dispersion curves indicate that the motions become faster and (Fig. 4A) most of the residues can be clustered into a unique group with a global exchange rate constant kex = 2500 ± 300 s−1, indicating that the enzyme’s motions in the nucleotide-bound state are faster and synchronous. The residues include the C-spine (L172, L173, and I174), the R-spine (L95), and neighboring hydrophobic residues extending toward the αC helix (V104, V182, I150, I180, L162, L103, and I94) (fig. S11). Upon PLN1–19 binding, the conformational dynamics remain persistent for these residues and become prominent for other residues away from the substrate-binding site (table S11 and fig. S9). Residues in the C-spine and R-spine, the hydrophobic shell, and those bridging the spines moving synchronously in the nucleotide-bound state retain significant motions (fig. S11). Significant Rex values were also observed for residues in and neighboring the C-spine (L227 and L49), the αC helix (L89), and the activation loop (V191), and throughout the two lobes. These motions exemplify the dynamically committed state of the enzyme in the presence of both nucleotide and substrate. In contrast, PKI5–24 binding quenches most of the conformational exchange in the microsecond-to-millisecond time scale in agreement with amide measurements (fig. S9) (22).
Fig. 4. Synchronization of motions within the hydrophobic core upon nucleotide binding.
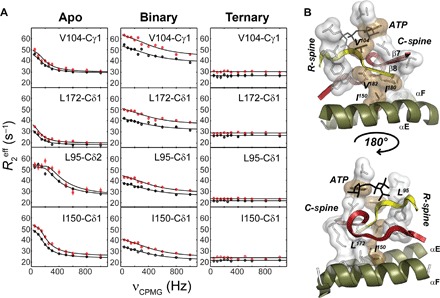
(A) 13C CPMG relaxation dispersion curves carried out at two different magnetic fields (700 MHz, black symbols; 850 MHz, red symbols) for selected residues in the hydrophobic core for the apo, binary (ATPγC-bound), and ternary (ATPγN/PKI5–24) complexes. (B) Mapping of the methyl groups showing conformational dynamics in the C-spine, R-spine, and bridging residues of the hydrophobic core. V104 bridges the R-spine and C-spine together with the adenine ring of ATP, whereas I150 bridges the C-spine to the αE helix.
To verify the importance of these motions in the allosteric network, we individually mutated two residues, V104 and I150, which display significant Rex values. These residues are highly conserved throughout the EPK family (23) and form a bridge between the two spines (Fig. 4B). Community map analysis of molecular dynamics trajectories predicted that these residues are essential to the integrity of the hydrophobic core and actively participate in the dynamically driven allosteric communication (fig. S12) (14). Upon mutating these positions (V104G and I150A), the kinase does not undergo autophosphorylation at Thr197 and S338 (figs. S13 and S14) (24), confirming that the enzyme is completely inactive. Coexpressing the mutants with PDK1, a kinase that phosphorylates Thr197, PKA-C adopts an active conformation of the activation loop, where the R-spine is assembled and the αC helix is positioned for catalysis. Remarkably, this is not sufficient to activate the kinase. Because the mutated kinases retain the overall fold, these results suggest that I150 and V104 in the inner hydrophobic core of the kinases must contribute to the intramolecular allosteric signaling for substrate recognition and catalysis.
DISCUSSION
The participation of protein hydrophobic cores in the transmission of intermolecular and intramolecular allosteric signaling was hypothesized decades ago based on free-energy considerations (25), and theoretical studies have hypothesized that correlated internal dynamics of hydrophobic cores may be required for biological function (26). In support of this, several proteins have been found to adopt loose packing of the inner cores, yet can still carry out biological functions with high specificity (27). This is reminiscent of the 3D conservation map of PKA-C with a firmly packed core (4) and site for phosphoryl transfer buried in the center. However, our NMR analysis reveals an unprecedented map of the dynamic network of communication within the hydrophobic core of PKA-C that orchestrates signal transmission between distal sites and encapsulates the active site. The apo enzyme with a dismantled C-spine is malleable and relatively accessible to water, displaying uncorrelated motions that are uncommitted for catalysis. On the other hand, the nucleotide-bound state, with the C-spine assembled, features synchronous motions, where the fluidity of the well-packed side chains is due to an extended network of correlated motions (27). We speculate that both the structure and dynamics of the core and residues surrounding the active site may have coevolved to optimize the intramolecular allosteric communication (fig. S12) (28, 29).
Computer simulations provide us with an unprecedented computational microscope and generate new mechanistic concepts whereas crystallography provides us with static snapshots of the various conformational states that reflect the catalytic cycle. We used these tools to create a community map for PKA-C, which allows us to see how elements of secondary structure are woven together in new ways (14). Using methyl relaxation dispersion NMR experiments, we can now experimentally capture the correlated internal dynamics of the hydrophobic core. The β structure of the kinase core, which embraces and surrounds the active-site cleft, provides a framework to think about how the adenine ring engages and commits the entire molecule to catalysis. The five-stranded β sheet in the N-lobe has been described extensively. It moves as a rigid body as the cleft opens and closes (30). In the apo conformation, it is not closely engaged with the C-lobe, as confirmed by earlier NMR studies (22). However, when ATP binds, the adenine ring bridges the β sheet of the N-lobe with the small β7-β8 sheets in the C-lobe (Fig. 5A). The β structure of the C-lobe received less attention, but is equally important for function. The β7 and β8 strands are directly linked to the catalytic loop and the Mg loop, respectively. The catalytic loop is anchored to the αF helix through Leu167, and this is one of the correlated residues that is quite prominent. In the assembled and active kinase, the catalytic loop is preceded by β6, whereas the Mg loop is followed by β9. The shell residues (Val104, Met11, and Met120) that bridge the R-spine and C-spine in the N-lobe have been described earlier, but there are two other residues (Val104 and Ile150) that scored high on the local spatial pattern (LSP) alignment that was used to identify the spines (5). At the time, however, their functional significance was not appreciated. Both of these residues are highly correlated in the methyl-TROSY NMR analysis and both serve as additional bridging residues that link the R-spine and C-spine (fig. S13).
Fig. 5. β structure of the kinase core is anchored to the hydrophobic core and recruited for catalysis by ATP binding.
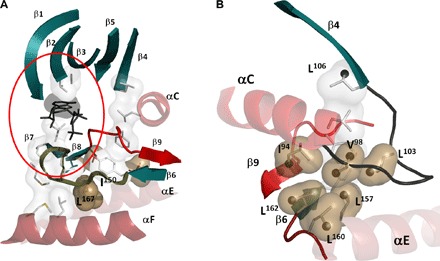
(A) The β sheet of the N-lobe (β1 to β5) is anchored to the adenine ring of ATP through V57 (β2) and A70 (β3). In the C-lobe, β7 and β8 are anchored to the adenine ring through L173 (β7). The catalytic loop that spans β6 and β7 is anchored to the F helix by L167; the Mg loop (red) bridges β8 and β9. (B) Another conserved structural element in the N-lobe is the αC-β4 loop that is characterized by a stable β turn. This loop also contains many key residues with highly correlated chemical shift changes (black spheres).
One additional element that was not fully appreciated in the initial analyses of the kinase structure is the αC-β4 loop, which is a classic β turn and also moves as a rigid body (Fig. 5B). The tip of this loop is the only piece of the N-lobe that remains anchored to the C-lobe in the open conformation (30). Val104 and Leu103 are part of this loop, and these residues are an important part of the correlated network of residues that engage one another following the binding of ATP. From earlier work, we know that Asn99, when replaced with cysteine and modified by fluorescein, is a sensor of ATP binding, and myristylation of the N terminus and the dynamic feature of this residue were also detected computationally (31, 32).
With methyl-TROSY relaxation dispersion experiments, we now have sensors for the entire β network and beyond. What is also striking about this network is that most of it is firmly in place and always anchored to the hydrophobic core even when the R-spine is not assembled. The one exception is β9 and the Mg loop that precedes it. This small segment shown in red in Fig. 6A is only assembled as a consequence of the formation of the R-spine. One can also appreciate how phosphorylation of the activation loop contributes to stabilizing both the active site and the R-spine (Fig. 6B). This small, highly dynamic segment encapsulates the essence of the switch mechanism that is an inherent feature of every protein kinase. Here, we show that once the R-spine is assembled, the molecule is poised to be committed to catalysis in a highly correlated way in response to ATP binding.
Fig. 6. Dynamic ordering of the hydrophobic core.
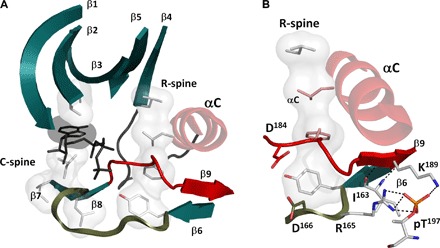
(A) Most of the elements of the hydrophobic core (teal and gray) are stable in both active and inactive conformational states. This includes β1 through β8 and the catalytic loop. These elements, as well as the C-spine, are anchored to the F helix by hydrophobic interactions. The assembly of the R-spine completes the hydrophobic core and positions the Mg loop and β9 (red). (B) This active conformation, particularly the β8-β9 sheet, is further stabilized by phosphorylation at Thr197 in the activation loop.
Because the hydrophobic core of PKA-C is highly conserved across the kinome (5), it emerges as a critical element for kinase regulation and may constitute a novel target to modulate the allosteric functions of each kinase (33). The drug imatinib, for instance, breaks the R-spine and inactivates both Src and Abl kinases (9), whereas other drugs, such as PLX4720, deform the conformation of the R-spine in BRAF and activate its noncatalytic function (fig. S15) (34). Similar strategies aimed at nonconserved allosteric pockets in kinases can be exploited to target the dynamic assembly of the spines with higher selectivity among the different kinases.
MATERIALS AND METHODS
Sample preparation
All reagents used were of analytical grade or better. Synthesis and purification of PLN1–19 and PKI5–24 have been previously described (7, 22). Details on the expression and purification procedures for PKA-C have been previously reported (35, 36). For the samples used in the NMR relaxation experiments, the murine Cα subunit of cAMP-dependent protein kinase (PKA-C) cDNA was cloned into a pRSETb vector. The kinase was expressed in Escherichia coli BL21(DE3) cells in M9 medium with 15NH4Cl as the only source of nitrogen and 100% 2H-glucose. The M9 salts were solubilized into 80% 2H2O. For selective 13CHD2 labeling, the M9 medium was supplemented with 2-ketobutyric acid-4-13C,3,3,4,4-d4 (70 mg/liter) and 2-keto-3-(methyl-13C,d2)-butyric acid-4-13C,d2 (90 mg/liter) 1 hour before induction. For selective 13CH3 labeling, the growth medium was supplemented with 2-ketobutyric acid-4-13C,3,3-d2 (70 mg/liter) and 2-keto-3-(methyl-d3)-butyric acid-4-13C,3-d1 (90 mg/liter) 1 hour before induction. Protein overexpression was induced by 0.4 mM isopropyl β-d-thiogalactopyranoside (IPTG) and carried out overnight at 24°C.
PKA-C purification was carried out by affinity chromatography using recombinant RIIα subunit (35, 36). The RIIα(R213K) was cloned into a pRSETb vector and expressed in 4 liters of LB media. Protein expression was induced by the addition of 0.4 mM IPTG and carried out for 5 hours at 24°C before harvesting the cells. Cell pellets of the catalytic subunit and Rllα(R213K) were combined and lysed in 30 mM 3-(N-morpholino)propanesulfonic acid (Mops), 200 μM ATP, 15 mM MgCl2, and 5 mM 2-mercaptoethanol (pH 8.0). After removing cell debris by centrifugation, the supernatant was incubated with Ni+2 nitrilotriacetic acid resin (Thermo Scientific, 1 ml of resin per liter of culture) at 4°C for approximately 3 hours. The resin was then washed with 30 mM Mops, 25 mM KCl, 15 mM MgCl2, and 5 mM 2-mercaptoethanol (pH 8.0) and eluted with 30 mM Mops, 25 mM KCl, 15 mM MgCl2, 5 mM 2-mercaptoethanol, and 1 mM cAMP (pH 8.0). The fractions containing PKA-C were collected and dialyzed overnight in 20 mM KH2PO4, 25 mM KCl, and 5 mM 2-mercaptoethanol (pH 6.5). The three isoforms of PKA-C (corresponding to the three different phosphorylation states with identical catalytic parameters) were separated by HiTrap SP column (GE Healthcare Life Sciences) chromatography, using a linear gradient from buffer A [20 mM KH2PO4 (pH 6.5)] to 30% buffer B [20 mM KH2PO4 and 1.0 M KCl (pH 6.5)] with a flow rate of 2.0 ml/min. Isoform II with pThr197, pSer338, and pSer10 was used for all NMR experiments (37).
NMR experimental conditions
All NMR samples were prepared in a buffer solution containing 20 mM KH2PO4, 90 mM KCl, 10 mM MgCl2, 10 mM dithiothreitol (DTT), and 1 mM NaN3 (pH 6.5) at 27°C. The concentration of the methyl-labeled PKA-C sample was 400 μM in a 250-μl volume. The sample was first titrated with adenylyl-imidodiphosphate (ATPγN) (0.1, 0.2, 0.6, 1, 2, 3, and 12 mM) and then with PKI5–24 (100, 200, 300, and 400 μM). The titration experiments were monitored by 1H-13C–heteronuclear multiple-quantum coherence experiments. Note that we used a nonhydrolyzable nucleotide analog, ATPγC (Sigma-Aldrich), for our relaxation measurements, because ATPγN (Roche LifeScience) undergoes slow hydrolysis in a period of several days. Residue-specific resonance assignments of PKA-C were previously described (35).
Analysis of CSPs
The chemical shift trajectories were monitored using the CONCISE (COordinated ChemIcal Shifts bEhavior) method (38) to measure the change in structural equilibrium associated with each PKA-C complex (apo, ATPγN, ATPγN/PLN1–19, and ATPγN/PKI5–24). Using principal components analysis (PCA), the method identifies a set of residues whose chemical shifts respond linearly to the open-to-closed conformational transition of the kinase. Each one of these residues provides a measure of the equilibrium position for all the PKA-C complexes, scoring them along the first principal component. The equilibrium position for a given PKA-C variant is given by the average of the PC scores over all linear residues. To identify the residues whose chemical shifts follow a linear pattern, a threshold of 1.0 to 3.0 for the ratio of the SDs of PC1 over PC2 was used and residues affected by CSPs below 0.05 parts per million were discarded (38). To identify the largest group of residues that responded to ligand binding in a correlated fashion, an adapted version of the chemical shift covariance analysis (CHESCA) (39) was applied to the PCA projection of the chemical shifts. First, the correlation matrix between all linear residues’ PC1 projections was constructed and used to build a dendrogram through hierarchical clustering. After a threshold of 0.97 level of correlation coefficient was applied, a total of 58 residues formed the subset to trace the equilibrium position for each state.
Nuclear spin relaxation experiments
Side-chain dynamics in the fast time scale (picosecond to nanosecond) were determined by measuring 2H spin relaxation rates of the 13CHD2 isotopomers (40) using an 850-MHz spectrometer equipped with a Bruker AVANCE III console. For 13CHD2 methyl–labeled PKA-C, we used a 250 μM sample for the apo form, a 230 μM sample for the binary form (saturated with 12 mM ATPγC), and a 200 μM sample for the closed form (saturated with 12 mM ATPγN and 400 μM PKI5–24) in a 300-μl volume. All the samples were analyzed by dynamic light scattering to confirm sample homogeneity and determine the global rotational correlation time (τc). All spectra were collected in an interleaved manner with relaxation delays of 0.8, 1.2, 1.6, 2.0, 2.4, 3.2, 4.0, and 4.8 ms for the apo enzyme; 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, and 4.0 ms for the nucleotide-bound PKA-C; and 0.4, 0.8, 1.2, 1.6, 2.0, 2.4, 2.8, and 3.2 ms for the ternary complex. The intensities were fit to a single exponential using nonlinear fit, and the order parameter was extracted from the relaxation rate using the following relationship (40)
where QCC = e2qQ/h is the quadrupolar coupling constant (167 ± 1 kHz) and S2 is the order parameter. The order parameters were calculated directly using transverse deuterium relaxation rates and the global rotational correlation time based on the dynamic light scattering data.
Methyl relaxation dispersion experiments
Single-quantum 13C CPMG (41) experiments were carried out on 700- and 850-MHz spectrometers equipped with Bruker AVANCE and AVANCE III consoles, respectively. All the spectra were collected in an interleaved manner with CPMG field strengths of 50, 100, 150, 200, 250, 300, 400, 500, 600, 800, and 1000 Hz. Replicate experiments were performed at field strengths of 50, 300, and 1000 Hz with a constant time delay of 40 ms. Each 2D spectrum was recorded with 2048 × 160 complex points and 48 transients with a recycle delay of 2.0 s. All data were processed using NMRPipe (42), and peak intensities were picked using Sparky (43). The peak intensities were converted to transverse decay rates, R2eff, as previously described (44). The software GUARDD (45) was used to fit the relaxation data to the Carver-Richards equation (46), which describes the dependence of the relaxation contribution of chemical exchange, Rex, to transverse relaxation. A two-state fit provides the exchange rate, the population of the second state, and chemical shift difference for a two-state exchange process
where pA and pB are the populations of the two states, Δω is the chemical shift difference of the two states, τcp is the time between the π pulses, kex is the sum of the rates of the exchange process, and R20 is the intrinsic transverse relaxation rate. The fitting was performed by minimizing the function χ2 as previously described (45). The group fit of selected residues was performed if the χ2Group/χ2Local was less than 2.0 (44).
An estimate of the Rex can also be approximated by measuring the R2 for a high and low τcp value. The approximate Rex was found with the same CPMG setup but with two values (with τcp values of 1 and 20 ms). The approximate Rex for the ATPγN/PLN1–19 bound form of PKA-C was estimated by
where I is the intensity of the residue with the corresponding spin-echo period and T is the constant time period (40 ms).
Western blot analysis
QuikChange I and II site-directed mutagenesis kits (Agilent Technologies) were used to introduce mutations. Wild-type and mutants of PKA-C were tested for their autophosphorylation ability using the Western blot–based assay as previously described (6). Briefly, recombinant forms of murine PKA-C were transformed in BL21(DE3) cells and grown in 5-ml batch cultures at 37°C until an optical density at 600 nm (OD600) of 0.6 was reached. At this time, protein overexpression was induced with 0.2 mM IPTG and allowed to grow overnight. Induced cultures were lysed in a buffer containing 50 mM phosphate buffer (pH 7.0), 250 mM NaCl, and 2 mM DTT. Lysates were run on an SDS–polyacrylamide gel electrophoresis gel and tested for phosphorylation using site-specific antibodies. trans-Phosphorylation at the activation loop was probed by pThr197-specific polyclonal antibody from Invitrogen. cis-Phosphorylation at the C-tail was probed by pSer338 antibodies (BD Biosciences), and expression levels of the recombinant PKA-C were probed by monoclonal antibodies from BD Biosciences.
ConSurf analysis
The sequence conservation was plotted onto the protein structure using ConSurf-DB (47, 48). Initially, a CS-BLAST (49) search against the SWISS-PROT database was performed to obtain at least 50 unique hits. The list of collected homologs was subsequently filtered by coverage (minimum of 80%) and sequence identity (between 60 and 95%). The remaining homologs were filtered again using Cluster Database at High Identity with Tolerance algorithm with 90% sequence identity clustering threshold (50). The decision on whether to proceed with the search for homologs or abort and move to the next step was based on the number of sequences after filtration. For PKA-C, 301 unique homologs were collected from SWISS-PROT for this calculation. A multiple sequence alignment (MSA) of the homologs was constructed using MAFFT (51). The MSA was used to build a phylogenetic tree using the neighbor-joining algorithm (52), as implemented in the Rate4Site (53) program. Position-specific conservation scores were computed using the Bayesian algorithm and JTT evolutionary substitution model and were mapped to a single-digit scale from 0 to 9 and plotted as colors onto the Protein Data Bank (PDB) structure using PyMOL (Schrodinger Inc.).
Dynamic light scattering
The homogeneity of the PKA-C samples prepared for NMR relaxation experiments was analyzed with a Malvern Zetasizer μV system. The sample (100 μl) was placed in a polystyrene microcuvette and equilibrated for 5 min at 25°C. All of the measurements were performed at 25°C after automatic optical adjustment. Six runs were carried out, with each run consisting of an average of 11 measurements. The Z-average diameter, reporting on the hydrodynamic radius of the protein, was determined from a cumulative analysis of the measured intensity autocorrelation function using the Zetasizer software Version 6.34. Assuming isotropic molecular tumbling, using the Z-average diameter, the rotation correlation time (τc) was calculated using the Stokes-Einstein equation at 27°C where all NMR experiments were performed.
Supplementary Material
Acknowledgments
We thank A. Cembran (University of Minnesota Duluth) and G. Li for helpful discussions. Funding: This work was supported by the NIH (GM100310 and GM 72701 to G.V. and T32AR007612 to J.K.). NMR experiments were carried out at the Minnesota NMR Center. Author contributions: J.K. prepared the PKA-C samples; designed, executed, and analyzed the NMR experiments; and contributed to the writing of the manuscript. L.G.A. carried out mutagenesis and activity experiments. F.-A.C. prepared the PKA-C samples and performed NMR experiments. Y.X. optimized NMR pulse sequences and helped set up NMR experiments. C.L.M. calculated the conservation maps of PKA-C residues using ConSurf. A.P.K. performed data analysis, prepared figures, and contributed to the writing of the manuscript. S.S.T. designed the mutagenesis experiments and contributed to the writing of the manuscript. G.V. designed the NMR experiments and contributed to the writing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/4/e1600663/DC1
fig. S1. [1H-13C] methyl-TROSY spectra of apo, nucleotide-bound (ATPγC), and ternary (ATPγN/PKI5-24 and ATPγN/PLN1–19) forms of 2H,13C-isoleucine, leucine, and valine (IVL)–labeled PKA-C.
fig. S2. Plots of the IVL methyl group CSPs upon ligand binding.
fig. S3. Mapping of the CSPs of the IVL methyl side-chain groups onto the crystal structure of PKA-C (PDB: 1ATP).
fig. S4. Statistical analysis of the chemical shift changes.
fig. S5. CHESCA (39) correlation matrix showing the degree of correlated chemical shift changes for the methyl side chains of IVL residues.
fig. S6. Global conformational transitions mapped via linear CSPs probed by amide backbone resonances and methyl group resonances.
fig. S7. Fast time scale (picosecond to nanosecond) conformational dynamics of the kinase upon ligand binding.
fig. S8. Thermodynamics of ligand binding for PKA-C as measured by isothermal titration calorimetry (ITC).
fig. S9. Slow time scale (microsecond to millisecond) conformational dynamics of the kinase.
fig. S10. Slow time scale conformational dynamics of the kinase.
fig. S11. Synchronous dynamics occurring in the highly conserved hydrophobic core.
fig. S12. Location of I150 at the interface between different communities of PKA-C.
fig. S13. Bridging residues connect the R-spine and C-spine at the PKA hydrophobic core.
fig. S14. Western blot–based activity assay.
fig. S15. Assembly of the R-spine for active protein kinases.
fig. S16. Expression and purification of recombinant 2H, 15N, 13CH3-ILV, and PKA-C from E. coli bacteria.
table S1. Classification of residues undergoing correlated chemical shift changes and their respective location in a specific community as identified by community map analysis.
table S2. The dynamic light scattering data for three different forms of PKA-C.
table S3. T2 and S2 values for methyl side-chain groups of apo PKA-C.
table S4. T2 and S2 values for methyl side-chain groups of the ATPγC-bound state of PKA-C.
table S5. T2 and S2 values for methyl side-chain groups of the ATPγN/PKI5-24-bound state of PKA-C.
table S6. Group fits of CPMG dispersion curves measured at 700 and 850 MHz of the apo form of PKA-C.
table S7. Group fits of the CPMG relaxation dispersion curves measured at 700 and 850 MHz of the ATPγC form of PKA-C.
table S8. Single-quantum individual site fits of CPMG relaxation dispersion curves measured at 700 and 850 MHz of the apo form of PKA-C.
table S9. Individual fits of the CPMG dispersion curves measured at 700 and 850 MHz of the ATPγC-bound state of PKA-C.
table S10. Single-quantum individual site fits of CPMG curves at 700 and 850 MHz of the ATPγN/PKI5-24-bound state of PKA-C.
table S11. Approximate Rex values from two points of the CPMG experiment for the ATPγN/PLN1–19-bound form of PKA-C.
Reference (54)
REFERENCES AND NOTES
- 1.Stenberg K. A., Riikonen P. T., Vihinen M., KinMutBase, a database of human disease-causing protein kinase mutations. Nucleic Acids Res. 27, 362–364 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S., The protein kinase complement of the human genome. Science 298, 1912–1934 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Huse M., Kuriyan J., The conformational plasticity of protein kinases. Cell 109, 275–282 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Johnson D. A., Akamine P., Radzio-Andzelm E., Madhusudan M., Taylor S. S., Dynamics of cAMP-dependent protein kinase. Chem. Rev. 101, 2243–2270 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Kornev A. P., Taylor S. S., Ten Eyck L. F., A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. U.S.A. 105, 14377–14382 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meharena H. S., Chang P., Keshwani M. M., Oruganty K., Nene A. K., Kannan N., Taylor S. S., Kornev A. P., Deciphering the structural basis of eukaryotic protein kinase regulation. PLOS Biol. 11, e1001680 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim J., Masterson L. R., Cembran A., Verardi R., Shi L., Gao J., Taylor S. S., Veglia G., Dysfunctional conformational dynamics of protein kinase A induced by a lethal mutant of phospholamban hinder phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 112, 3716–3721 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masterson L. R., Mascioni A., Traaseth N. J., Taylor S. S., Veglia G., Allosteric cooperativity in protein kinase A. Proc. Natl. Acad. Sci. U.S.A. 105, 506–511 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azam M., Seeliger M. A., Gray N. S., Kuriyan J., Daley G. Q., Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat. Struct. Mol. Biol. 15, 1109–1118 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu J., Yu H., Kornev A. P., Zhao J., Filbert E. L., Taylor S. S., Shawa A. S., Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc. Natl. Acad. Sci. U.S.A. 108, 6067–6072 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu J., Stites E. C., Yu H., Germino E. A., Meharena H. S., Stork P. J. S., Kornev A. P., Taylor S. S., Shaw A. S., Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154, 1036–1046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sprangers R., Kay L. E., Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature 445, 618–622 (2007). [DOI] [PubMed] [Google Scholar]
- 13.MacLennan D. H., Kranias E. G., Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 4, 566–577 (2003). [DOI] [PubMed] [Google Scholar]
- 14.McClendon C. L., Kornev A. P., Gilson M. K., Taylor S. S., Dynamic architecture of a protein kinase. Proc. Natl. Acad. Sci. U.S.A. 111, E4623–E4631 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahuja L. G., Kornev A. P., McClendon C. L., Veglia G., Taylor S. S., Mutation of a kinase allosteric node uncouples dynamics linked to phosphotransfer. Proc. Natl. Acad. Sci. U.S.A. 114, E931–E940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marlow M. S., Dogan J., Frederick K. K., Valentine K. G., Wand A. J., The role of conformational entropy in molecular recognition by calmodulin. Nat. Chem. Biol. 6, 352–358 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tzeng S.-R., Kalodimos C. G., Allosteric inhibition through suppression of transient conformational states. Nat. Chem. Biol. 9, 462–465 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Masterson L. R., Cheng C., Yu T., Tonelli M., Kornev A., Taylor S. S., Veglia G., Dynamics connect substrate recognition to catalysis in protein kinase A. Nat. Chem. Biol. 6, 821–828 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srivastava A. K., McDonald L. R., Cembran A., Kim J., Masterson L. R., McClendon C. L., Taylor S. S., Veglia G., Synchronous opening and closing motions are essential for cAMP-dependent protein kinase A signaling. Structure 22, 1735–1743 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmer A. G. III, Kroenke C. D., Loria J. P., Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 339, 204–238 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Baldwin A. J., Kay L. E., NMR spectroscopy brings invisible protein states into focus. Nat. Chem. Biol. 5, 808–814 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Masterson L. R., Shi L., Metcalfe E., Gao J., Taylor S. S., Veglia G., Dynamically committed, uncommitted, and quenched states encoded in protein kinase A revealed by NMR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 108, 6969–6974 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kannan N., Neuwald A. F., Did protein kinase regulatory mechanisms evolve through elaboration of a simple structural component? J. Mol. Biol. 351, 956–972 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Taylor S. S., Zhang P., Steichen J. M., Keshwani M. M., Kornev A. P., PKA: Lessons learned after twenty years. Biochim. Biophys. Acta 1834, 1271–1278 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gregory R. B., Lumry R., Hydrogen-exchange evidence for distinct structural classes in globular proteins. Biopolymers 24, 301–326 (1985). [DOI] [PubMed] [Google Scholar]
- 26.Bowman G. R., Geissler P. L., Extensive conformational heterogeneity within protein cores. J. Phys. Chem. B 118, 6417–6423 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baldwin R. L., Rose G. D., Molten globules, entropy-driven conformational change and protein folding. Curr. Opin. Struct. Biol. 23, 4–10 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Xiao Y., Lee T., Latham M. P., Warner L. R., Tanimoto A., Pardi A., Ahn N. G., Phosphorylation releases constraints to domain motion in ERK2. Proc. Natl. Acad. Sci. U.S.A. 111, 2506–2511 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyken S. E., Chopra N., Xie Q., Joseph R. E., Wales T. E., Bruce Fulton D., Engen J. R., Jernigan R. L., Andreotti A. H., A conserved isoleucine maintains the inactive state of Bruton’s tyrosine kinase. J. Mol. Biol. 426, 3656–3669 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsigelny I., Greenberg J. P., Cox S., Nichols W. L., Taylor S. S., Ten Eyck L. F., 600 ps molecular dynamics reveals stable substructures and flexible hinge points in cAMP dependent protein kinase. Biopolymers 50, 513–524 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Bastidas A. C., Pierce L. C., Walker R. C., Johnson D. A., Taylor S. S., Influence of N-myristylation and ligand binding on the flexibility of the catalytic subunit of protein kinase A. Biochemistry 52, 6368–6379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cembran A., Masterson L. R., McClendon C. L., Taylor S. S., Gao J., Veglia G., Conformational equilibrium of N-myristoylated cAMP-dependent protein kinase A by molecular dynamics simulations. Biochemistry 51, 10186–10196 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J., Yang P. L., Gray N. S., Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dar A. C., Shokat K. M., The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 80, 769–795 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Chao F.-A., Kim J., Xia Y., Milligan M., Rowe N., Veglia G., FLAMEnGO 2.0: An enhanced fuzzy logic algorithm for structure-based assignment of methyl group resonances. J. Magn. Reson. 245, 17–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hemmer W., McGlone M., Taylor S. S., Recombinant strategies for rapid purification of catalytic subunits of cAMP-dependent protein kinase. Anal. Biochem. 245, 115–122 (1997). [DOI] [PubMed] [Google Scholar]
- 37.Yonemoto W., Garrod S. M., Bell S. M., Taylor S. S., Identification of phosphorylation sites in the recombinant catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem. 268, 18626–18632 (1993). [PubMed] [Google Scholar]
- 38.Cembran A., Kim J., Gao J., Veglia G., NMR mapping of protein conformational landscapes using coordinated behavior of chemical shifts upon ligand binding. Phys. Chem. Chem. Phys. 16, 6508–6518 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selvaratnam R., Chowdhury S., VanSchouwen B., Melacini G., Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. U.S.A. 108, 6133–6138 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tugarinov V., Ollerenshaw J. E., Kay L. E., Probing side-chain dynamics in high molecular weight proteins by deuterium NMR spin relaxation: An application to an 82-kDa enzyme. J. Am. Chem. Soc. 127, 8214–8225 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Lundström P., Vallurupalli P., Religa T. L., Dahlquist F. W., Kay L. E., A single-quantum methyl 13C-relaxation dispersion experiment with improved sensitivity. J. Biomol. NMR 38, 79–88 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 43.Lee W., Tonelli M., Markley J. L., NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDonald L. R., Boyer J. A., Lee A. L., Segmental motions, not a two-state concerted switch, underlie allostery in CheY. Structure 20, 1363–1373 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kleckner I. R., Foster M. P., GUARDD: User-friendly MATLAB software for rigorous analysis of CPMG RD NMR data. J. Biomol. NMR 52, 11–22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carver J. P., Richards R. E., A general two-site solution for the chemical exchange produced dependence of T2 upon the Carr-Purcell pulse separation. J. Magn. Reson. 6, 89–105 (1972). [Google Scholar]
- 47.Celniker G.,Nimrod G., Ashkenazy H., Glaser F., Martz E., Mayrose I., Pupko T., Ben-Tal N., ConSurf: Using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 53, 199–206 (2013). [Google Scholar]
- 48.Goldenberg O., Erez E., Nimrod G., Ben-Tal N., The ConSurf-DB: Pre-calculated evolutionary conservation profiles of protein structures. Nucleic Acids Res. 37, D323–D327 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Angermüller C., Biegert A., Söding J., Discriminative modelling of context-specific amino acid substitution probabilities. Bioinformatics 28, 3240–3247 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Huang Y., Niu B., Gao Y., Fu L., Li W., CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katoh K., Toh H., Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26, 1899–1900 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saitou N., Nei M., The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 (1987). [DOI] [PubMed] [Google Scholar]
- 53.Pupko T., Bell R. E., Mayrose I., Glaser F., Ben-Tal N., Rate4Site: An algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 18, S71–S77 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Bastidas A. C., Deal M. S., Steichen J. M., Guo Y., Wu J., Taylor S. S., Phosphoryl transfer by protein kinase A is captured in a crystal lattice. J. Am. Chem. Soc. 135, 4788–4798 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/4/e1600663/DC1
fig. S1. [1H-13C] methyl-TROSY spectra of apo, nucleotide-bound (ATPγC), and ternary (ATPγN/PKI5-24 and ATPγN/PLN1–19) forms of 2H,13C-isoleucine, leucine, and valine (IVL)–labeled PKA-C.
fig. S2. Plots of the IVL methyl group CSPs upon ligand binding.
fig. S3. Mapping of the CSPs of the IVL methyl side-chain groups onto the crystal structure of PKA-C (PDB: 1ATP).
fig. S4. Statistical analysis of the chemical shift changes.
fig. S5. CHESCA (39) correlation matrix showing the degree of correlated chemical shift changes for the methyl side chains of IVL residues.
fig. S6. Global conformational transitions mapped via linear CSPs probed by amide backbone resonances and methyl group resonances.
fig. S7. Fast time scale (picosecond to nanosecond) conformational dynamics of the kinase upon ligand binding.
fig. S8. Thermodynamics of ligand binding for PKA-C as measured by isothermal titration calorimetry (ITC).
fig. S9. Slow time scale (microsecond to millisecond) conformational dynamics of the kinase.
fig. S10. Slow time scale conformational dynamics of the kinase.
fig. S11. Synchronous dynamics occurring in the highly conserved hydrophobic core.
fig. S12. Location of I150 at the interface between different communities of PKA-C.
fig. S13. Bridging residues connect the R-spine and C-spine at the PKA hydrophobic core.
fig. S14. Western blot–based activity assay.
fig. S15. Assembly of the R-spine for active protein kinases.
fig. S16. Expression and purification of recombinant 2H, 15N, 13CH3-ILV, and PKA-C from E. coli bacteria.
table S1. Classification of residues undergoing correlated chemical shift changes and their respective location in a specific community as identified by community map analysis.
table S2. The dynamic light scattering data for three different forms of PKA-C.
table S3. T2 and S2 values for methyl side-chain groups of apo PKA-C.
table S4. T2 and S2 values for methyl side-chain groups of the ATPγC-bound state of PKA-C.
table S5. T2 and S2 values for methyl side-chain groups of the ATPγN/PKI5-24-bound state of PKA-C.
table S6. Group fits of CPMG dispersion curves measured at 700 and 850 MHz of the apo form of PKA-C.
table S7. Group fits of the CPMG relaxation dispersion curves measured at 700 and 850 MHz of the ATPγC form of PKA-C.
table S8. Single-quantum individual site fits of CPMG relaxation dispersion curves measured at 700 and 850 MHz of the apo form of PKA-C.
table S9. Individual fits of the CPMG dispersion curves measured at 700 and 850 MHz of the ATPγC-bound state of PKA-C.
table S10. Single-quantum individual site fits of CPMG curves at 700 and 850 MHz of the ATPγN/PKI5-24-bound state of PKA-C.
table S11. Approximate Rex values from two points of the CPMG experiment for the ATPγN/PLN1–19-bound form of PKA-C.
Reference (54)