

Abstract
Pregnancy in placental mammals places unique demands on the insulin-producing β-cells in the pancreatic islets of Langerhans. The pancreas anticipates the increase in insulin resistance that occurs late in pregnancy by increasing β-cell numbers and function earlier in pregnancy. In rodents, this β-cell expansion depends on secreted placental lactogens that signal through to the prolactin receptor. Then at the end of pregnancy, the β-cell population contracts back to its pre-pregnancy size. In the current review we focus on how glucose metabolism changes during pregnancy, how β-cells anticipate these changes through their response to lactogens, and what molecular mechanisms guide the adaptive compensation. In addition, we summarize current knowledge of β-cell adaptation during human pregnancy and what happens when adaptation fails and gestational diabetes ensues. A better understanding of human β-cell adaptation to pregnancy would benefit efforts to predict, prevent and treat gestational diabetes.
Keywords: β cell, gestational diabetes, Htr1d, Htr2b, Htr3a, insulin, placenta, placental lactogen, pregnancy, serotonin
Graphical Abstract
Glucose metabolism in pregnancy
During pregnancy, the proper growth and development of the fetus depends on appropriate nutrient flow from mother to fetus across the placenta. Glucose provides a substantial fraction of fetal energy needs [1,2], but its transport across the placenta is a passive process and utilizes facilitative glucose transporters [3]. Therefore, glucose delivery to the fetus depends on the concentration gradient between the fetal and maternal circulations.
Early in pregnancy, the fetal β-cells establish this gradient by maintaining low glucose in the fetal circulation through their high basal insulin secretion and relative glucose insensitivity [4,5]. In the later stages of pregnancy, however, the growing fetus diverts an increasing fraction of maternal glucose across the placenta and thereby threatens the gradient by lowering glucose in the maternal circulation. In counterbalance to the fetal glucose diversion, the placenta secretes hormones that increase maternal insulin resistance and hepatic glucose production, thus raising glucose levels in the maternal circulation and maintaining the gradient [6,7].
To prevent excessive nutrient delivery to the fetus, the increase in maternal insulin resistance must be balanced by an increase in the capacity of the maternal β-cells to respond to meals. The change in maternal β-cell capacity results from growth of both the pool of maternal β-cells and their ability to secrete insulin in response to glucose [8–17]. In rodents, expansion of maternal β-cell mass in pregnancy results predominantly, if not entirely, from proliferation of preexisting β-cells [18–21].
This balance in the mother between insulin resistance and an enlarged, hyperdynamic β-cell pool ensures a steady flow of nutrients from mother to fetus up to the end of pregnancy. Then, starting shortly before parturition and extending into the postpartum period, the β-cell mass shrinks back to its pre-pregnancy size [22,23].
The β-cell response to lactogens
Studies in rodents have shown that β-cell compensation in the mother precedes the development of insulin resistance, and thus is not simply a response to increased insulin demand [13,14]. Instead, the increased β-cell proliferation during pregnancy parallels the rise of pituitary and placental lactogens [13]. Further, treatment with prolactin and placental lactogens efficiently drives rodent β-cell proliferation and increases glucose-stimulated insulin secretion in vitro and in vivo [14,24–26]. Finally, the β-cell changes during pregnancy in mice require an intact β-cell prolactin receptor (PRLR) [27–31], which functions as the receptor for both prolactin and placental lactogen and is induced on the β-cell during pregnancy [32,33].
The prolactin receptor (PRLR) belongs to the cytokine class-1 receptor superfamily, which also includes the closely related growth hormone receptor [34]. When bound by ligand, the receptor engages and is phosphorylated by the Janus Kinase 2 (JAK2), thereby allowing the recruitment and phosphorylation of Signal Transducer and Activator of Transcription 5 (STAT5), which then moves to the nucleus where it regulates the expression of target genes [35].
Lactogen induction of serotonin and serotonin signaling in β-cells
Among the genes activated by PRLR signaling in the β-cell are the genes encoding the 2 isoforms of the enzyme that controls the rate-limiting step of serotonin synthesis, tryptophan hydroxylase 1 and 2 (TPH1 and 2); Tph1 RNA increases by as much as 3 orders of magnitude in islets during pregnancy in mice [33,36–38]. β-cells contain all of the additional machinery needed for serotonin synthesis, storage, and secretion [39], and thus fill with serotonin, and co-secrete it with insulin during pregnancy [33,37,40]. Interestingly, among the many serotonergic tissues, this pregnancy-induced activation of the TPH genes is unique to islets [33,37].
Since serotonin commonly acts locally, either as a neurotransmitter or paracrine hormone [41,42], it is a reasonable assumption that the remarkably high levels of serotonin secreted within the maternal islet during pregnancy may affect the biology of the cells within the islet. Historically, investigators have described a range of contradictory effects of serotonin on islet cell function. These differences may reflect the variety of different models (different species, different ages, different physiologic states) and experimental conditions used in these studies [43]. Further confusion may be caused by the large repertoire of serotonin receptors expressed in mammals [44], many of which are expressed on various cells within the islet. In addition, receptor expression levels change during pregnancy: expression of the Gq-coupled GPCR Htr2b goes up during pregnancy, while the Gi-coupled GPCR Htr1d goes down during pregnancy but rebounds above pre-pregnancy levels at the end of pregnancy and postpartum [33].
Evidence in mouse models suggests that the increased serotonin in islets during pregnancy drives β-cell expansion [33]. Reduction in dietary tryptophan; pharmacologic inhibition of TPH, serotonin signaling broadly and Htr2b signaling specifically; and targeted disruption of the Htr2b gene all reduce β-cell expansion and impair glucose tolerance during pregnancy in mice. Furthermore, treatment of mouse islets in vitro with serotonin induces β-cell proliferation [33] (Figure 1A).
Figure 1.


A proposed model is shown for the role of serotonin in the adaptation of β-cells to pregnancy. Panel (A) outlines the effects of increased expression of the serotonin synthetic enzyme TPH and the serotonin receptor Htr2b at mid gestation. Panel (B) shows the effects of decreased expression of Htr2b and increased expression of Htr1d at the end of pregnancy and during the postpartum period. Trp, tryptophan; 5HT, 5 hydroxytrytomine, serotonin; TPH, tryptophan hydroxylase; PRLR, prolactin receptor.
A role for serotonin in driving β-cell compensation during pregnancy makes some teleological sense. Much as glucokinase acts as a glucose sensor in the β-cell, TPH can act as a dietary protein sensor. As the essential amino acid with the lowest level in most diets, tryptophan acts as an indicator of dietary protein intake. Because TPH has a Km for tryptophan close to its normal tissue concentration, and controls the rate-limiting step in serotonin synthesis, production of serotonin in β-cells during pregnancy reflects dietary protein intake. Furthermore, co-secretion of serotonin with insulin [33,37,45,46] provides an additional check on the system by modulating serotonin release and signaling in the islet in parallel with insulin demand.
Other pathways involved in β-cell expansion in pregnancy
Loss of serotonin signaling in the islet does not completely block the proliferative response to pregnancy in the mouse β-cell [33], and other signals almost certainly contribute as well. Given the critical importance of nutrient balance during pregnancy, multiple pathways that provide redundancy, constraint and refinement should be expected. For example, as the pregnancy progresses, and insulin resistance begins to develop, changing glucose levels and insulin demand may modulate the serotonin-driven β-cell expansion [47]. Similarly, autonomic neural inputs controlled by central regulators of feeding and metabolism could also provide input [48,49].
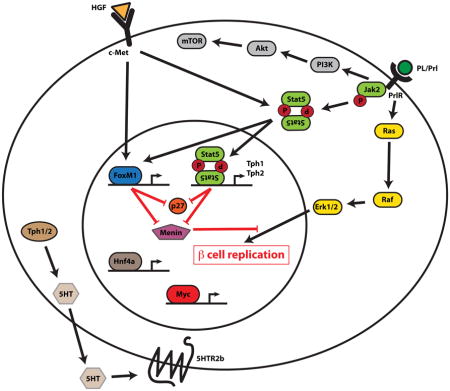
Several other genes and signaling pathways have been implicated in the β-cell expansion during pregnancy. In addition to the role of the JAK2/STAT5 cascade, PRLR signaling also activates other signaling pathways, including the Ras/Rap/MAPK and PI3K/AKT/mTOR signaling cascades, although their relative roles remain unclear [35]. Several studies have shown that pregnancy and lactogen signaling activate IRS1, IRS2, PI3K, AKT, p70S6K, and mTOR in rodent β-cells [28,50–52]. Treatment with Rapamycin, an inhibitor of mTOR signaling, limited β-cell expansion and replication in pregnant mice, but did not alter glucose tolerance [51]. Pregnancy and PRLR signaling also activate the Raf/MEK/ERK signaling cascade in rodent β-cells. However, the necessity of this pathway in maternal β-cell proliferation has not been tested, and phosphorylation of ERK does not impact β-cell proliferation in non-pregnant mice [53].
Expression of a dominant negative epidermal growth factor receptor (EGFR) blocks β-cell expansion during pregnancy in mice [54], without impacting lactogen-induced TPH expression [55]. However, it should be noted that the dominant-negative EGFR impaired β-cells in non-pregnant mice as well [56]. These studies also identified survivin/BIRC5, a cell-cycle protein and anti-apoptotic factor, as a lactogen-induced factor that depends on EGFR, mTOR and MEK/ERK signaling [55].
In pregnant rats, investigators showed that proliferation of islet endothelial cells precedes β-cell mass expansion, and when these endothelial cells were grown in culture, their conditioned media could induce β-cell proliferation [57]. During pregnancy, the maternal islet endothelial cells secrete abundant hepatocyte growth factor (HGF), and anti-serum against HGF blocked the ability of the conditioned media to dive β-cell proliferation [57]. Furthermore, deletion of the gene encoding the HNF receptor (c-met) in the pancreas reduced β-cell proliferation and β-cell mass expansion. In addition, loss of c-met signaling in the pancreas reduced β-cell proliferation in pregnant mice [58]. Interestingly, these mice also failed to increase PRLR expression on the β-cell during pregnancy and had other signs of decreased PRLR signaling, suggesting that proper PRLR signaling in the β-cell requires intact c-met signalling [58].
Studies in pregnant mice have demonstrated that levels of endocrine tumor suppressor Menin (gene name MEN1) decrease during pregnancy [15] downstream of PRLR signaling [52], and overexpression of menin induces gestational diabetes [15]. It should be noted, however, that whole transcriptome screens of gene expression in islets from pregnant rodents have not confirmed the reduction in Men1 gene expression during pregnancy [33,36–38].
Increasing evidence shows that microRNAs have important roles in modulating β-cell gene expression and thereby modify β-cell differentiation, proliferation, function, and death [59]. The expression of a number of β-cell miroRNAs change during pregnancy. Investigators showed that one of the miroRNAs downregulated during pregnancy, miR-338-3p, is inhibited by estrogen signaling through the Gpr130 estrogen receptor. Inhibiting miR-338-3p with an antisense oligonucleotide in β-cells led to an increase in proliferation [60].
Several transcription factors have been implicated in the β-cell expansion of pregnancy. Targeted deletion in the pancreas of the gene encoding Foxd3, a transcription factor implicated in proliferation in embryonic development and stem cells, also reduced β-cell expansion during pregnancy, although these animals also have decreased β-cell mass prior to pregnancy. Interestingly, expression of Foxd3 actually decreases during pregnancy [61]. In contrast, expression of the cell-cycle-associated transcription factor FoxM1 increases in the mouse β-cells in parallel with cell-cycle induction during pregnancy, and in mouse islets in vitro in response to lactogens [62]. Targeted deletion in the pancreas of the gene encoding of FoxM1 reduced β-cell expansion both during normal postnatal growth [63] and in pregnant female mice [62].
Finally, targeted deletion in β-cells of the gene encoding nuclear receptor HNF4α also resulted in decreased β-cell proliferation and mass in pregnant mice [64]. Heterozygous mutations in the human ortholog of this gene cause MODY1 (maturity onset diabetes of the young type 1); and prior studies of the mutant HNF4 mice demonstrated a number of β-cell defects in non-pregnant mice [65,66]. The effects of HNF4a on β-cell proliferation in pregnancy were attributed, at least in part, to its activation of Ras/MEK/ERK signaling through downregulation of Suppressor of Tumorigenicity 5 (ST5), a direct transcriptional target of HNF4a in β-cells [64].
Postpartum β-cell mass contraction
At the end of pregnancy in mice, the expression levels of the key metabotropic serotonin receptors on the maternal β-cell switch, with levels of Gq/11-coupled Htr2b decreasing and Gi/o-coupled Htr1d increasing [33] (Figure 1b). Because circulating prolactin and β-cell serotonin levels in the mother persist in the early postpartum period, this loss of Htr2b and switch to a predominantly Gi/o-coupled signaling pathway for serotonin could be expected to inhibit β-cell proliferation [33,48] and possibly even increase β-cell apoptosis, and contribute to the contraction of maternal β-cell mass postpartum [22,23]. However, again, other mechanisms contribute as well, since the high levels of circulating progesterone from the placenta at the end of pregnancy can block the pro-proliferative effects of lactogens on the β-cell and thus initiate the decline in β-cell proliferation in the latter half of pregnancy [67].
Enhanced insulin secretion
Serotonin also plays a role in the increase in glucose stimulated insulin secretion induced by pregnancy. In addition to its effects on the Htr2b and Htr1d receptors, serotonin secreted by the maternal islets binds to the Htr3a receptors on the β-cells during pregnancy [68]. The ionotropic Htr3a receptor functions as a serotonin-gated cationic ion channel. When bound by serotonin, Htr3a allows a leak of extracellular Na+ ions down the concentration gradient into the β-cell, thus mildly depolarizing the membrane and lowering the threshold for glucose-stimulated insulin secretion. Blocking Htr3a signaling reduces β-cell insulin secretion and impairs glucose tolerance in pregnant mice, but not non-pregnant mice [68].
In addition to the role of serotonin signaling, pregnancy also induce other changes that enhance insulin secretion in islets. Levels and activity of glucokinase, the β-cell glucose sensor, increase in β-cells in response to pregnancy and lactogen signaling [69], as do cAMP levels [70], intercellular junctional coupling [71], and the proteins involved in the fusion of insulin granules to the cell membrane in the final steps of insulin secretion [72].
All of these maternal β-cell changes may not occur independently, but instead act as different components of the same signaling pathways coordinated by PRLR signaling. For example, both glucokinase activity and serotonin signaling can regulate cAMP levels, which in turn could impact cell coupling and levels of the SNARE proteins. In sum, all of these enhancements in β-cell glucose sensing and insulin secretion, together with an expanded number of β-cells, provide a counterbalance to the decrease in maternal insulin sensitivity late in pregnancy, and control the steady flow of nutrients from mother to child.
β-Cell compensation in mice and women
Most of our understanding of pregnancy-mediated β-cell changes derives from studies in rodent models; our knowledge of the human β-cell in pregnancy is much more limited. What we do know, suggests both similarities and important differences between rodents and humans.
The rapid evolution of the placenta complicates the use of non-human animals to model human pregnancy [73]. The placental lactogens provide a special example of this problem [74]. Among placental mammals, placental lactogens have evolved independently in at least three different lineages, – anthropoid primates (monkeys, apes, and humans), muroid rodents (including mice and rats) and ruminants (including cows, sheep, and goats) – either through multiple duplications of the pituitary growth hormone gene (in primates) or the pituitary prolactin gene (rodents and ruminants), while many mammals (pigs for example) do not express any growth hormone or prolactin genes in the placenta, and thus must depend on pituitary prolactin alone during pregnancy. In conjunction with these gene duplications, the ancestral and duplicate genes and the genes encoding the growth hormone and prolactin receptors have undergone accelerated evolution.
Therefore, conclusions regarding the activities of mammosomatotropic hormones and their receptors developed from non-human models may not apply to human pregnancy. Furthermore, the rapid evolution of these hormones and their receptors has altered their specificity in heterologous systems. Human growth hormones (both ancestral and duplicates), for example, bind and signal efficiently through all mammalian prolactin receptors. Therefore studies of these hormones and their receptors should use both the correct ortholog and correct paralog.
Islets and β-cells also show distinct differences between humans and rodents. These differences include the distribution of islets, β-cell clusters and isolated β-cells in the pancreas, the composition and organization of the individual islets, and the patterns of the islet vasculature and neural innervation [75]. Of particular importance for our discussion here, adult human β-cells are more resistant to cell cycle entry than rodent β-cells, and have earned a reputation for rarely, if ever, dividing [76].
Similar to mouse β-cells, the number of maternal β-cells increases during human pregnancy by a factor of 1.4 to 2.4 fold, based on two autopsy series [12,16]. The mechanisms for this β-cell expansion in human pregnancy remains uncertain. A. Butler and colleagues detected no difference in β-cell proliferation as assessed by Ki67 staining when samples from pregnant women were compared to age-matched non-pregnant women, and concluded that the increase in β-cells must be due to neogenesis of β-cells from some other cell type [16]. However, neogenesis cannot be not assessed directly on autopsy samples. Furthermore, because the gestational dates of the samples spanned the full length of pregnancy, and rodent β-cell proliferation rates vary sharply with time during the pregnancy (with rates dropping below those of non-pregnant females late in pregnancy [13,15]), assessing the mean proliferation rates of these samples may have missed a proliferation peak. Also, since human pregnancy lasts for 9 month, humans require a much lower β-cell proliferation rate than rodents, which have a gestation period of 3 weeks. To achieve a 1.4 fold increase in β-cells over 9 months, the proliferation rate only has to be 0.125% per day (assuming no cell loss).
Tests of lactogen treatment of human β-cells have given varying results. Brelje and colleagues [26] treated cultured human islets with human prolactin, human placental lactogen and human growth hormone. All three hormones increased glucose stimulated insulin secretion from the cultured human islets. Significantly increased proliferation in islet cells (islets were not co-stained for insulin) as measured by BrdU incorporation was detected with hPL and hPRL treatment of islets from one donor, and with all three hormones in islets from a second donor. In contrast, Parnaud and colleagues [77] reported that hGH did not induce proliferation of human β-cells sorted and cultured in monolayers, or in intact islets, but the data was not shown, and insulin secretion was not tested. More recently, Chen and colleagues [78] tested hPRL on human islets cultured as dispersed cells, and detected no increase in β-cell proliferation. They did not test for effects on insulin secretion. They attributed this lack of response in part to low expression of the hPRLR on human β-cells. However, adenoviral overexpression of mouse STAT5a in human β-cells activated downstream targets of PRLR signaling and β-cell proliferation, but similarly expressed hPRLR (plus hPRL treatment) or human STAT5a failed to increase proliferation. They concluded that human β-cells have at least two defects in PRLR signaling, but did not explain why lactogens increase insulin secretion from human β-cells.
These studies demonstrate the dangers of trying to translate to humans the results of studies of pregnancy and the β-cell adaptation to pregnancy performed in rodent models. Although difficult, future studies in this field need to increasingly focus on the biology of human placenta and human β-cells.
Consequences of inadequate β-cell compensation: Gestational diabetes
Currently, the American Diabetes Association gestational diabetes as “diabetes diagnosed in the second or third trimester of pregnancy that is not clearly overt diabetes” [79]. However, this definition is confusing, and encompasses a heterogeneous group of women with a wide spectrum of glycemia and pathophysiology, and fails to distinguish hyperglycemia that is unique to pregnancy from permanent diabetes – diabetes that predates the pregnancy but was previously unrecognized, or diabetes that persists after pregnancy [80]. Perhaps a simpler definition might be “diabetes unique to pregnancy”.
Furthermore, the field continues to struggle to set broadly accepted criteria for diagnosing gestational diabetes [81]. Clearly hyperglycemia in pregnancy is associated with adverse pregnancy outcomes for both the mother and child, including fetal macrosomia, preeclampsia, shoulder dystocia, and cesarean section [82]. The risks of these outcomes increase as maternal fasting plasma glucose levels increase and as the one-hour and two-hour oral glucose tolerance test (OGTT) values increase. This effect is continuous, and there is no clear threshold that defines patients at increased risk of adverse outcome [82]. In addition, glucose-lowering interventions, even in pregnancies with mild hyperglycemia, improve outcomes [83].
Long-term outcomes of gestational diabetes
Women with gestational diabetes have an increased risk of developing type 2 diabetes in the years following their pregnancy. The data regarding incidence of diabetes after gestational diabetes come mostly from small studies and/or racially/ethnically homogeneous populations [84]. A 2009 systematic review and meta-analysis of 20 cohort studies demonstrated that women with gestational diabetes have at least a seven-fold increased future risk of developing type 2 diabetes relative to those who had a normoglycemic pregnancy [85]. Individual studies vary widely in their estimates of risk, ranging from 2.6% to 70%. Aside from genetic differences among populations, this large variation in the subsequent development of type 2 diabetes likely reflects the use of diverse tests for glucose tolerance in pregnancy, selection bias and, in particular, duration of follow-up. Taken together, however, the high risk of type 2 diabetes associated with gestational diabetes combined with observations that the two disorders share common risk factors and prevalence rates, suggests that gestational diabetes and type 2 diabetes might have overlapping etiologies. This line of thought has led to the prevailing view of gestational diabetes as a portent of type 2 diabetes, revealed by the increased insulin resistance during pregnancy.
However, while a significant percentage of women with GDM go on to develop type 2 diabetes, many do not. When stratified by duration of follow-up, the evidence shows that most of this risk of type 2 diabetes presents within 5–10 years postpartum in pregnancies complicated by gestational diabetes. After 10 years, the risk of type 2 diabetes decreases to the background rates in the population. In O’Sullivan’s original cohort of patients with gestational diabetes, approximately 50% of subjects did not develop diabetes during follow-up for as long as 28 years after their index pregnancies. Thus, while the diagnosis of gestational diabetes can be considered a risk factor for type 2 diabetes after pregnancy, for many women this is not true. For women with gestational diabetes who do not develop type 2 diabetes, particularly those without risk factors traditionally associated with type 2 diabetes, we contend that the development of gestational diabetes represents a problem that is pregnancy-specific, and most likely involves pathways driving gestational changes in the β-cell.
Summary
In conclusion, we have made recent progress in understanding the basic biology of the β-cell’s adaptations to pregnancy, especially non-primate models. However, much of this new understanding may not apply to human pregnancy and the human β-cell.
At the same time, current controversies in the clinical approach to gestational diabetes complicate our approach to this important disorder. Establishing a uniform approach to diagnosing and managing GDM would benefit patients, caregivers, and policymakers. A better understanding of the underlying pathophysiology of GDM in humans would help guide these efforts to detect, prevent and treat GDM. Unfortunately our current limited knowledge of the interactions between the human placenta and human β-cells hinder these efforts.
Acknowledgments
This work was supported by the UCSF Diabetes Research Center funded by NIH grant P30 DK063720 and the Larry L. Hillblom Islet Genesis Network funded by Larry L. Hillblom Foundation grant 2014-D-004_NET.
CALLOUTS
Glucose metabolism in pregnancy
β-cells anticipate the increased insulin demand that occurs late in pregnancy by increasing their number and insulin secretory capacity early in pregnancy.
Lactogen induction of serotonin and serotonin signaling in β-cells
Placental lactogens drive the synthesis of serotonin in β-cells, which in turn drives beta-cell proliferation and glucose-stimulated insulin secretion in rodents.
β-cell compensation in mice and women
Although we know that the net effects of pregnancy on rodent and human β-cell mass and secretion are similar, the mechanisms driving these effects may be very different.
Long term outcomes of gestational diabetes
Epidemiologic studies suggest that many women with gestational diabetes may have a pregnancy-specific defect in the placental-β-cell axis.
Footnotes
Conflict of interest
The authors declare no conflict of interest
References
- 1.Hay WW., Jr Energy and substrate requirements of the placenta and fetus. Proc Nutr Soc. 1991;50:321–336. doi: 10.1079/pns19910042. [DOI] [PubMed] [Google Scholar]
- 2.Battaglia FC. Principal substrates of fetal metabolism: fuel and growth requirements of the ovine fetus. Ciba Found Symp. 1978:57–74. doi: 10.1002/9780470720462.ch4. [DOI] [PubMed] [Google Scholar]
- 3.Baumann MU, Deborde S, Illsley NP. Placental glucose transfer and fetal growth. Endocrine. 2002;19:13–22. doi: 10.1385/ENDO:19:1:13. [DOI] [PubMed] [Google Scholar]
- 4.Asplund K, Westman S, Hellerstrom C. Glucose stimulation of insulin secretion from the isolated pancreas of foetal and newborn rats. Diabetologia. 1969;5:260–262. doi: 10.1007/BF01212095. [DOI] [PubMed] [Google Scholar]
- 5.Espinosa de los M, Driscoll SG, Steinke J. Insulin release from isolated human fetal pancreatic islets. Science. 1970;168:1111–1112. doi: 10.1126/science.168.3935.1111. [DOI] [PubMed] [Google Scholar]
- 6.Nolan CJ, Proietto J. The feto-placental glucose steal phenomenon is a major cause of maternal metabolic adaptation during late pregnancy in the rat. Diabetologia. 1994;37:976–984. doi: 10.1007/BF00400460. [DOI] [PubMed] [Google Scholar]
- 7.Lain KY, Catalano PM. Metabolic changes in pregnancy. Clin Obstet Gynecol. 2007;50:938–948. doi: 10.1097/GRF.0b013e31815a5494. [DOI] [PubMed] [Google Scholar]
- 8.Hellman B. The islets of Langerhans in the rat during pregnancy and lactation, with special reference to the changes in the B/A cell ratio. Acta Obstet Gynecol Scand. 1960;39:331–342. doi: 10.3109/00016346009159930. [DOI] [PubMed] [Google Scholar]
- 9.Green IC, Taylor KW. Effects of pregnancy in the rat on the size and insulin secretory response of the islets of Langerhans. J Endocrinol. 1972;54:317–325. doi: 10.1677/joe.0.0540317. [DOI] [PubMed] [Google Scholar]
- 10.Van Assche FA. Quantitative morphologic and histoenzymatic study of the endocrine pancreas in nonpregnant and pregnant rats. Am J Obstet Gynecol. 1974;118:39–41. doi: 10.1016/s0002-9378(16)33642-0. [DOI] [PubMed] [Google Scholar]
- 11.Bone AJ, Taylor KW. Metabolic adaptation to pregnancy shown by increased biosynthesis of insulin in islets of Langerhans isolated from pregnant rat. Nature. 1976;262:501–502. doi: 10.1038/262501a0. [DOI] [PubMed] [Google Scholar]
- 12.Van Assche FA, Aerts L, De Prins F. A morphological study of the endocrine pancreas in human pregnancy. Br J Obstet Gynaecol. 1978;85:818–820. doi: 10.1111/j.1471-0528.1978.tb15835.x. [DOI] [PubMed] [Google Scholar]
- 13.Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology. 1992;130:1459–1466. doi: 10.1210/endo.130.3.1537300. [DOI] [PubMed] [Google Scholar]
- 14.Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. 1997;29:301–307. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- 15.Karnik SK, Chen H, McLean GW, et al. Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science. 2007;318:806–809. doi: 10.1126/science.1146812. [DOI] [PubMed] [Google Scholar]
- 16.Butler AE, Cao-Minh L, Galasso R, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia. 2010;53:2167–2176. doi: 10.1007/s00125-010-1809-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab. 2010;21:151–158. doi: 10.1016/j.tem.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell. 2007;12:817–826. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Abouna S, Old RW, Pelengaris S, et al. Non-β-cell progenitors of β-cells in pregnant mice. Organogenesis. 2010;6:125–133. doi: 10.4161/org.6.2.10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao X, Chen Z, Shiota C, et al. No evidence for beta cell neogenesis in murine adult pancreas. J Clin Invest. 2013;123:2207–2217. doi: 10.1172/JCI66323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toselli C, Hyslop CM, Hughes M, Natale DR, Santamaria P, Huang CT. Contribution of a non-beta-cell source to beta-cell mass during pregnancy. PLoS ONE. 2014;9:e100398. doi: 10.1371/journal.pone.0100398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marynissen G, Aerts L, Van Assche FA. The endocrine pancreas during pregnancy and lactation in the rat. Journal of developmental physiology. 1983;5:373–381. [PubMed] [Google Scholar]
- 23.Scaglia L, Smith FE, Bonner-Weir S. Apoptosis contributes to the involution of beta cell mass in the post partum rat pancreas. Endocrinology. 1995;136:5461–5468. doi: 10.1210/endo.136.12.7588296. [DOI] [PubMed] [Google Scholar]
- 24.Brelje TC, Allaire P, Hegre O, Sorenson RL. Effect of prolactin versus growth hormone on islet function and the importance of using homologous mammosomatotropic hormones. Endocrinology. 1989;125:2392–2399. doi: 10.1210/endo-125-5-2392. [DOI] [PubMed] [Google Scholar]
- 25.Brelje TC, Sorenson RL. Role of prolactin versus growth hormone on islet B-cell proliferation in vitro: implications for pregnancy. Endocrinology. 1991;128:45–57. doi: 10.1210/endo-128-1-45. [DOI] [PubMed] [Google Scholar]
- 26.Brelje TC, Scharp DW, Lacy PE, et al. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology. 1993;132:879–887. doi: 10.1210/endo.132.2.8425500. [DOI] [PubMed] [Google Scholar]
- 27.Freemark M, Avril I, Fleenor D, et al. Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology. 2002;143:1378–1385. doi: 10.1210/endo.143.4.8722. [DOI] [PubMed] [Google Scholar]
- 28.Amaral ME, Cunha DA, Anhe GF, et al. Participation of prolactin receptors and phosphatidylinositol 3-kinase and MAP kinase pathways in the increase in pancreatic islet mass and sensitivity to glucose during pregnancy. J Endocrinol. 2004;183:469–476. doi: 10.1677/joe.1.05547. [DOI] [PubMed] [Google Scholar]
- 29.Huang C, Snider F, Cross JC. Prolactin receptor is required for normal glucose homeostasis and modulation of beta-cell mass during pregnancy. Endocrinology. 2009;150:1618–1626. doi: 10.1210/en.2008-1003. [DOI] [PubMed] [Google Scholar]
- 30.Rawn SM, Huang C, Hughes M, Shaykhutdinov R, Vogel HJ, Cross JC. Pregnancy hyperglycemia in prolactin receptor mutant, but not prolactin mutant, mice and feeding-responsive regulation of placental lactogen genes implies placental control of maternal glucose homeostasis. Biol Reprod. 2015;93:75. doi: 10.1095/biolreprod.115.132431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goyvaerts L, Lemaire K, Arijs I, et al. Prolactin receptors and placental lactogen drive male mouse pancreatic islets to pregnancy-related mRNA changes. PLoS ONE. 2015;10:e0121868. doi: 10.1371/journal.pone.0121868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moldrup A, Petersen ED, Nielsen JH. Effects of sex and pregnancy hormones on growth hormone and prolactin receptor gene expression in insulin-producing cells. Endocrinology. 1993;133:1165–1172. doi: 10.1210/endo.133.3.8365359. [DOI] [PubMed] [Google Scholar]
- 33.Kim H, Toyofuku Y, Lynn FC, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med. 2010;16:804–808. doi: 10.1038/nm.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brooks CL. Molecular mechanisms of prolactin and its receptor. Endocr Rev. 2012;33:504–525. doi: 10.1210/er.2011-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr Rev. 1998;19:225–268. doi: 10.1210/edrv.19.3.0334. [DOI] [PubMed] [Google Scholar]
- 36.Rieck S, White P, Schug J, et al. The transcriptional response of the islet to pregnancy in mice. Mol Endocrinol. 2009;23:1702–1712. doi: 10.1210/me.2009-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schraenen A, Lemaire K, de Faudeur G, et al. Placental lactogens induce serotonin biosynthesis in a subset of mouse beta cells during pregnancy. Diabetologia. 2010;53:2589–2599. doi: 10.1007/s00125-010-1913-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Layden BT, Durai V, Newman MV, et al. Regulation of pancreatic islet gene expression in mouse islets by pregnancy. J Endocrinol. 2010;207:265–279. doi: 10.1677/JOE-10-0298. [DOI] [PubMed] [Google Scholar]
- 39.Ohta Y, Kosaka Y, Kishimoto N, et al. Convergence of the insulin and serotonin programs in the pancreatic beta-cell. Diabetes. 2011;60:3208–3216. doi: 10.2337/db10-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goyvaerts L, Schraenen A, Schuit F. Serotonin competence of mouse beta cells during pregnancy. Diabetologia. doi: 10.1007/s00125-016-3951-2. In press. [DOI] [PubMed] [Google Scholar]
- 41.Amireault P, Sibon D, Cote F. Life without peripheral serotonin: insights from tryptophan hydroxylase 1 knockout mice reveal the existence of paracrine/autocrine serotonergic networks. ACS Chem Neurosci. 2013;4:64–71. doi: 10.1021/cn300154j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Ann Rev Med. 2009;60:355–366. doi: 10.1146/annurev.med.60.042307.110802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zawalich WS, Tesz GJ, Zawalich KC. Are 5-hydroxytryptamine-preloaded beta-cells an appropriate physiologic model system for establishing that insulin stimulates insulin secretion? J Biol Chem. 2001;276:37120–37123. doi: 10.1074/jbc.M105008200. [DOI] [PubMed] [Google Scholar]
- 44.Hoyer D, Clarke DE, Fozard JR, et al. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin) Pharmacol Rev. 1994;46:157–203. [PubMed] [Google Scholar]
- 45.Gylfe E. Association between 5-hydroxytryptamine release and insulin secretion. J Endocrinol. 1978;78:239–248. doi: 10.1677/joe.0.0780239. [DOI] [PubMed] [Google Scholar]
- 46.Smith PA, Proks P, Ashcroft FM. Quantal analysis of 5-hydroxytryptamine release from mouse pancreatic beta-cells. J Physiol. 1999;521(Pt 3):651–664. doi: 10.1111/j.1469-7793.1999.00651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dadon D, Tornovsky-Babaey S, Furth-Lavi J, et al. Glucose metabolism: key endogenous regulator of beta-cell replication and survival. Diabetes Obes Metab. 2012;14(Suppl 3):101–108. doi: 10.1111/j.1463-1326.2012.01646.x. [DOI] [PubMed] [Google Scholar]
- 48.Berger M, Scheel DW, Macias H, et al. Galphai/o-coupled receptor signaling restricts pancreatic beta-cell expansion. Proc Natl Acad Sci U S A. 2015;112:2888–2893. doi: 10.1073/pnas.1319378112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guettier JM, Gautam D, Scarselli M, et al. A chemical-genetic approach to study G protein regulation of beta cell function in vivo. Proc Natl Acad Sci U S A. 2009;106:19197–19202. doi: 10.1073/pnas.0906593106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amaral ME, Ueno M, Carvalheira JB, et al. Prolactin-signal transduction in neonatal rat pancreatic islets and interaction with the insulin-signaling pathway. Horm Metab Res. 2003;35:282–289. doi: 10.1055/s-2003-41303. [DOI] [PubMed] [Google Scholar]
- 51.Zahr E, Molano RD, Pileggi A, et al. Rapamycin impairs in vivo proliferation of islet beta-cells. Transplantation. 2007;84:1576–1583. doi: 10.1097/01.tp.0000296035.48728.28. [DOI] [PubMed] [Google Scholar]
- 52.Hughes E, Huang C. Participation of Akt, menin, and p21 in pregnancy-induced beta-cell proliferation. Endocrinology. 2011;152:847–855. doi: 10.1210/en.2010-1250. [DOI] [PubMed] [Google Scholar]
- 53.Chamberlain CE, Scheel DW, McGlynn K, et al. Menin determines K-RAS proliferative outputs in endocrine cells. J Clin Invest. 2014;124:4093–4101. doi: 10.1172/JCI69004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hakonen E, Ustinov J, Mathijs I, et al. Epidermal growth factor (EGF)-receptor signalling is needed for murine beta cell mass expansion in response to high-fat diet and pregnancy but not after pancreatic duct ligation. Diabetologia. 2011;54:1735–1743. doi: 10.1007/s00125-011-2153-1. [DOI] [PubMed] [Google Scholar]
- 55.Hakonen E, Ustinov J, Palgi J, Miettinen PJ, Otonkoski T. EGFR signaling promotes beta-cell proliferation and survivin expression during pregnancy. PLoS ONE. 2014;9:e93651. doi: 10.1371/journal.pone.0093651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miettinen PJ, Ustinov J, Ormio P, et al. Downregulation of EGF receptor signaling in pancreatic islets causes diabetes due to impaired postnatal beta-cell growth. Diabetes. 2006;55:3299–3308. doi: 10.2337/db06-0413. [DOI] [PubMed] [Google Scholar]
- 57.Johansson M, Mattsson G, Andersson A, Jansson L, Carlsson PO. Islet endothelial cells and pancreatic beta-cell proliferation: studies in vitro and during pregnancy in adult rats. Endocrinology. 2006;147:2315–2324. doi: 10.1210/en.2005-0997. [DOI] [PubMed] [Google Scholar]
- 58.Demirci C, Ernst S, Alvarez-Perez JC, et al. Loss of HGF/c-Met signaling in pancreatic beta-cells leads to incomplete maternal beta-cell adaptation and gestational diabetes mellitus. Diabetes. 2012;61:1143–1152. doi: 10.2337/db11-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Filios SR, Shalev A. Beta-cell MicroRNAs: Small but powerful. Diabetes. 2015;64:3631–3644. doi: 10.2337/db15-0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacovetti C, Abderrahmani A, Parnaud G, et al. MicroRNAs contribute to compensatory beta cell expansion during pregnancy and obesity. J Clin Invest. 2012;122:3541–3551. doi: 10.1172/JCI64151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Plank JL, Frist AY, LeGrone AW, Magnuson MA, Labosky PA. Loss of Foxd3 results in decreased beta-cell proliferation and glucose intolerance during pregnancy. Endocrinology. 2011;152:4589–4600. doi: 10.1210/en.2010-1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang H, Zhang J, Pope CF, et al. Gestational diabetes mellitus resulting from impaired beta-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes. 2010;59:143–152. doi: 10.2337/db09-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang H, Ackermann AM, Gusarova GA, et al. The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Mol Endocrinol. 2006;20:1853–1866. doi: 10.1210/me.2006-0056. [DOI] [PubMed] [Google Scholar]
- 64.Gupta RK, Gao N, Gorski RK, et al. Expansion of adult beta-cell mass in response to increased metabolic demand is dependent on HNF-4alpha. Genes Dev. 2007;21:756–769. doi: 10.1101/gad.1535507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gupta RK, Vatamaniuk MZ, Lee CS, et al. The MODY1 gene HNF-4α regulates selected genes involved in insulin secretion. J Clin Invest. 2005;115:1006–1015. doi: 10.1172/JCI200522365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miura A, Yamagata K, Kakei M, et al. Hepatocyte nuclear factor-4α is essential for glucose-stimulated insulin secretion by pancreatic β-cells. J Biol Chem. 2006;281:5246–5257. doi: 10.1074/jbc.M507496200. [DOI] [PubMed] [Google Scholar]
- 67.Sorenson RL, Brelje TC, Roth C. Effects of steroid and lactogenic hormones on islets of Langerhans: a new hypothesis for the role of pregnancy steroids in the adaptation of islets to pregnancy. Endocrinology. 1993;133:2227–2234. doi: 10.1210/endo.133.5.8404674. [DOI] [PubMed] [Google Scholar]
- 68.Ohara-Imaizumi M, Kim H, Yoshida M, et al. Serotonin regulates glucose-stimulated insulin secretion from pancreatic beta cells during pregnancy. Proc Natl Acad Sci U S A. 2013;110:19420–19425. doi: 10.1073/pnas.1310953110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weinhaus AJ, Stout LE, Sorenson RL. Glucokinase, hexokinase, glucose transporter 2, and glucose metabolism in islets during pregnancy and prolactin-treated islets in vitro: mechanisms for long term up-regulation of islets. Endocrinology. 1996;137:1640–1649. doi: 10.1210/endo.137.5.8612496. [DOI] [PubMed] [Google Scholar]
- 70.Weinhaus AJ, Bhagroo NV, Brelje TC, Sorenson RL. Role of cAMP in upregulation of insulin secretion during the adaptation of islets of Langerhans to pregnancy. Diabetes. 1998;47:1426–1435. doi: 10.2337/diabetes.47.9.1426. [DOI] [PubMed] [Google Scholar]
- 71.Sorenson RL, Brelje TC, Hegre OD, Marshall S, Anaya P, Sheridan JD. Prolactin (in vitro) decreases the glucose stimulation threshold, enhances insulin secretion, and increases dye coupling among islet B cells. Endocrinology. 1987;121:1447–1453. doi: 10.1210/endo-121-4-1447. [DOI] [PubMed] [Google Scholar]
- 72.Cunha DA, Amaral ME, Carvalho CP, Collares-Buzato CB, Carneiro EM, Boschero AC. Increased expression of SNARE proteins and synaptotagmin IV in islets from pregnant rats and in vitro prolactin-treated neonatal islets. Biol Res. 2006;39:555–566. doi: 10.4067/s0716-97602006000300016. [DOI] [PubMed] [Google Scholar]
- 73.Chuong EB, Hannibal RL, Green SL, Baker JC. Evolutionary perspectives into placental biology and disease. Appl Transl Genom. 2013;2:64–69. doi: 10.1016/j.atg.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haig D. Placental growth hormone-related proteins and prolactin-related proteins. Placenta. 2008;29(Suppl):36–41. doi: 10.1016/j.placenta.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 75.Dolensek J, Rupnik MS, Stozer A. Structural similarities and differences between the human and the mouse pancreas. Islets. 2015;7:e1024405. doi: 10.1080/19382014.2015.1024405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang P, Fiaschi-Taesch NM, Vasavada RC, Scott DK, Garcia-Ocana A, Stewart AF. Diabetes mellitus--advances and challenges in human beta-cell proliferation. Nat Rev Endocrinol. 2015;11:201–212. doi: 10.1038/nrendo.2015.9. [DOI] [PubMed] [Google Scholar]
- 77.Parnaud G, Bosco D, Berney T, et al. Proliferation of sorted human and rat beta cells. Diabetologia. 2008;51:91–100. doi: 10.1007/s00125-007-0855-1. [DOI] [PubMed] [Google Scholar]
- 78.Chen H, Kleinberger JW, Takane KK, et al. Augmented Stat5 signaling bypasses multiple impediments to lactogen-medi ated proliferation in human beta-cells. Diabetes. 2015;64:3784–3797. doi: 10.2337/db15-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.American Diabetes Association. 2 Classification and diagnosis of diabetes. Diabetes Care. 2016;39(Suppl 1):S13–S22. doi: 10.2337/dc16-er09. [DOI] [PubMed] [Google Scholar]
- 80.Buchanan TA, Xiang A, Kjos SL, Watanabe R. What is gestational diabetes? Diabetes Care. 2007;30(Suppl 2):S105–S111. doi: 10.2337/dc07-s201. [DOI] [PubMed] [Google Scholar]
- 81.VanDorsten JP, Dodson WC, Espeland MA, et al. National Institutes of Health consensus development conference statement: diagnosing gestational diabetes mellitus, March 4–6, 2013. Obstet Gynecol. 2013;122:358–369. doi: 10.1097/AOG.0b013e31829c3e64. [DOI] [PubMed] [Google Scholar]
- 82.Group HSCR, Metzger BE, Lowe LP, et al. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. 2008;358:1991–2002. doi: 10.1056/NEJMoa0707943. [DOI] [PubMed] [Google Scholar]
- 83.Landon MB, Spong CY, Thom E, et al. A multicenter, randomized trial of treatment for mild gestational diabetes. N Engl J Med. 2009;361:1339–1348. doi: 10.1056/NEJMoa0902430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim C, Newton KM, Knopp RH. Gestational diabetes and the incidence of type 2 diabetes: a systematic review. Diabetes Care. 2002;25:1862–1868. doi: 10.2337/diacare.25.10.1862. [DOI] [PubMed] [Google Scholar]
- 85.Bellamy L, Casas JP, Hingorani AD, Williams D. Type 2 diabetes mellitus after gestational diabetes: a systematic review and meta-analysis. Lancet. 2009;373:1773–1779. doi: 10.1016/S0140-6736(09)60731-5. [DOI] [PubMed] [Google Scholar]
- 86.O’Sullivan JB. The Boston Gestational Diabetes Studies: Review and Perspectives. In: Sutherland HW, Stowers JM, Pearson DWM, editors. Carbohydrate Metabolism in Pregnancy and the Newborn · IV. London: Springer London; 1989. pp. 287–294. [Google Scholar]