

Abstract
We identified two unrelated consanguineous families with three children affected by the rare association of congenital nephrotic syndrome diagnosed in the first days of life, of hypogonadism, and of prenatally detected adrenal calcifications, associated with congenital adrenal insufficiency in one case. Using exome sequencing and targeted Sanger sequencing two homozygous truncating mutations, c.1513C>T (p.Arg505*) and c.934delC (p.Leu312Phefs*30), were identified in SGPL1 encoding sphingosine-1-phosphate lyase 1. SGPL1 catalyzes the irreversible degradation of endogenous and dietary sphingosine-1-phosphate (S1P), the final step of sphingolipid catabolism, and of other phosphorylated long-chain bases. S1P is an intra- and extracellular signaling molecule involved in angiogenesis, vascular maturation, and immunity. The levels of SGPL1 substrates, S1P and sphingosine were markedly increased in the patients’ blood and fibroblasts, as determined by liquid chromatography-tandem mass spectrometry. Vascular alterations were present in a patient’s renal biopsy, in line with changes seen in Sgpl1 knockout mice that are compatible with a developmental defect in vascular maturation. In conclusion, loss of SGPL1 function is associated with congenital nephrotic syndrome, adrenal calcifications, and hypogonadism.
Keywords: Congenital nephrotic syndrome, congenital adrenal insufficiency, adrenal calcification, sphingolipids, sphingosine-1-phosphate, developmental, hypogonadism, hypergonadotropic hypogonadism, vascular
Introduction
Congenital nephrotic syndrome (CNS, MIM# 256300) is characterized by the presence of massive proteinuria, hypoalbuminemia, and generalized edema developing within the first 3 months of life. This rare disease generally results from a disturbed function of the glomerular filtration barrier. Mutations affecting the integrity of its crucial components like the podocytes or the basement membrane can be identified in the majority of patients. Therapeutic efforts aim at the stabilization of the plasma protein concentration and eventually require nephrectomy, dialysis and kidney transplantation. CNS most often presents as isolated renal disease but several syndromic forms are also recognized. Of more than 30 genes that cause CNS when mutated, NPHS1 (nephrin, MIM# 602716), NPHS2 (podocin, MIM# 604766), LAMB2 (laminin β2, MIM# 150325), WT1 (Wilms tumour suppressor 1, MIM# 607102), and PLCE1 (phospholipase C ε, MIM# 608414) account for >85% of cases (Sadowski et al., 2015; Trautmann et al., 2015).
Pre- and perinatally detected adrenal calcifications are also rare and might have resulted from certain intrauterine infections, or from adrenal hemorrhage in the context of resuscitation. It can also result from a number of monogenic steroid biosynthetic defects in the form of lipoid adrenal hyperplasia (LCAH, MIM# 201710) (Miller, 2016) and occurs in cholesteryl ester storage disease due to lysosomal acid lipase deficiency (LAL, MIM# 178000) due to LIPA (lipase A, lysosomal acid type, MIM# 613497) mutations (Anderson et al., 1994). LCAH-causing steroid biosynthetic defects and LAL deficiency do not present with CNS.
The association of congenital proteinuria and adrenal calcifications is very rare and was first reported in three patients of American Indian origin (Powers et al., 1990), and later in an Indian patient with congenital nephrotic syndrome, adrenal calcifications, and a cardiac malformation (Indumathi et al., 2005), and in two patients who were first cousins (Schreyer-Shafir et al., 2014). One male patient had non-palpable testes and micropenis, and another patient had pericardial and pleural effusions, generalized hydrops, and a cleft palate. Reported patients mostly died within the first months of life, of undocumented cause or from infections. Here we report the identification of truncating mutations in SGPL1 (sphingosine-1-phosphate lyase 1, MIM# 603729) in patients from two unrelated families with congenital proteinuria and adrenal calcifications, which represents a rare syndromic form of CNS and a novel disorder in the catabolism of sphingolipids.
Materials and Methods
Patients
The association of CNS and adrenal calcifications was reported in two male cousins (patient 1 and 2) from a consanguineous Arab family (family 1, Fig. 1A) (Schreyer-Shafir et al., 2014). Both mothers had an elevated risk for trisomy 21 on maternal serum biochemical screening and a borderline uE3 serum level. This with abnormal nuchal translucency prompted us to rule out chromosomal abnormalities and Smith-Lemli-Opiz syndrome (MIM #270400). Fetal karyotype and chromosomal microarray analysis (CMA) performed on chorionic villous cells were normal in patient 1. Maternal serological testing for Toxoplasma, Rubella, CMV and parvovirus did not suggest maternal infection during the pregnancy and sequence analysis of LIPA and WT1 was normal in patient 2. Renal autopsy findings in patient 2 who deceased at 3 months of age demonstrated a normal architecture with areas of focal segmental or global sclerosis, with foci of calcification within the interstitial and glomerular compartments (Supp. Figure S1). There was cystic dilatation of the proximal tubuli with hyaline droplets or cytoplasmic lipid droplets in a portion of tubular cells. There was expansion of the interstitial space with inter-tubular connective tissue. Of note, blood vessels showed hypertrophic thickened vessel walls and perivascular sclerosis (Fig. 1B).
Figure 1. Pedigrees, clinical findings and SGPL1 mutations.

A. Pedigree of family 1. Individuals II-1, II-2, III-1, III-2, and III-3 were available for mutation segregation analyses. B. Kidney biopsy of patient 2 at the age of 3 months (Masson’s stain) demonstrated focal segmental or global sclerosis, and hypertrophic thickened vessel walls and perivascular sclerosis of blood vessels. C. Pedigree of family 2. Individuals V-1, V-2, and VI-3 were available for mutation segregation analyses. D. Plain radiograph of patient 3 taken at 3 months of age shows bilateral adrenal calcifications (arrows). E. Sanger sequencing confirms the presence of a SGPL1 stop mutation in family 1 and a SGPL1 frameshift mutation in family 2 (F). The mutations are present in homozygous state in each patient and in heterozygous state in both parents of each patient (one carrier parent is shown in each case). Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1.
Both children were born at 38 weeks of gestation with birth weights of 3270 grams (50th percentile) and 3500 grams (50–90th percentile). Massive proteinuria was diagnosed at the ages of 6 weeks and 2 days, respectively. Both patients had a small penis and no palpable testes. Low baseline testosterone levels, lack of testosterone response to HCG test and exaggerated gonadotrophins response to early life luteinizing hormone releasing hormone test were suggestive of testicular dysfunction which was confirmed by a very low level of Mullerian inhibitory factor. Bilateral adrenal calcifications were found at 6 weeks of age and at 21weeks of gestation, respectively. The children died suddenly at the age of 7 weeks and 3 months, respectively, and autopsies were not performed.
Here, we report another patient (patient 3, Fig. 1C) with the association of CNS, congenital adrenal insufficiency and adrenal calcifications, the third child of healthy European parents who are 3rd cousins (family 2). Two older siblings were healthy.
Routine ultrasound revealed a unilateral hydrothorax and a generalized skin edema (2 to 3.7 mm) in the 12th week of gestation. However, hydrothorax and skin edema were not recognizable any more at 16 weeks of gestation.
Fetal karyotyping revealed a normal male karyotype. Maternal serological testing excluded toxoplasma, rubella, CMV and parvovirus infection during pregnancy. The patient was born at 37 weeks of gestation by Cesarean section weighing 3075 grams. A micropenis and cryptorchidism with a small inguinal testis at the right side and no testis at the left side were noted. Subsequent investigations revealed adrenal calcifications (Fig. 1D) and primary adrenal insufficiency that required corticoid supplementation. Hormones in the first week of life were altered reflecting hypergonadotropic hypogonadism with elevated LH/FSH and decreased testosterone levels. The boy developed generalized edema in the first week of life and was found to have severe hypoalbuminemia and proteinuria and was diagnosed with CNS. The patient developed end stage renal failure at one month of age and renal replacement therapy was started. In the first 5 months of life, the patient did not tolerate oral feeding and required parenteral nutrition. A gastroduodenoscopy was performed at the age of 4.5 months and revealed neither macroscopic abnormalities nor structural defects or signs of inflammation on light microscopy. Electron microscopic examination did not reveal any ultrastructural abnormalities of the duodenal epithelium; all cell types appeared normal, with moderate amounts of endosomes and lysosomes present, likely reflecting total parenteral nutrition of this patient (data not shown). Targeted sequence analyses of LIPA and WT1 were normal in this patient.
The resemblance of the perinatal presentation of these three patients born to consanguineous families of Arab and European origin were suggestive that they had the same disease due to a common cause. Bilateral adrenal calcifications and CNS with hypogonadism suggested a new clinical entity of autosomal recessive inheritance.
Whole-exome sequencing
Written informed consent for molecular research investigations was obtained from the patient’s parents, and the studies were approved by the local ethics committees. DNA was extracted from peripheral leukocytes following standard protocols. We performed whole-exome sequencing (WES) in probands from the Arab and the European family. Exonic and adjacent intronic regions were enriched from genomic DNA derived from peripheral blood via the SureSelect All Exon V4 and V6 target enrichment kits from Agilent Technologies, and paired-end sequencing was performed on Illumina HiSeq2000 and HiSeq4000 platforms, respectively. Paired-end reads of 100 bp (patient 2) and 125 bp (patient 3) were aligned to the human reference genome with Burrows-Wheeler transformation (BWA, v.0.7.12).(Li and Durbin, 2009) PCR-duplicates were removed with PICARD (v.2.0.1, http://picard.sourceforge.net) and single nucleotide substitutions, and small indels were called with SAMtools software (v.1.3). The variants were annotated using SNPeff (patient 2) (Cingolani et al., 2012) and SeattleSeq (http://snp.gs.washington.edu/SeattleSeqAnnotation/) (patient 3). PICARD’s CalculateHsMetrics function was used to search for homozygous deletions of entire exons in the patients and not present in in-house exome datasets.
Exons 14 and 11 of SGPL1 were PCR-amplified and directly sequenced in available samples from families 1 and 2, respectively, with primers available from the authors. The SGPL1 variant designation is based on the NCBI reference sequence for transcript NM_003901.3 (corresponding to Ensembl reference sequence ENST00000373202.7) and the genomic reference sequence ENSG00000166224. Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1.
Sphingolipid analysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS)
Blood samples were taken from patient 3 with a homozygous truncating mutation in the SGPL1 gene at age 5 months and from 2 healthy adult controls (36-year old male, 32-year old female) within the same hour and were transported and processed simultaneously. Fibroblast cultures from patient 3 and from two healthy adult controls (44-year old male, 39-year old female) were derived from skin punch biopsies, taken at different times. They had similar passage times when they were grown to 90% confluence in Innsbruck and shipped and processed together at the biochemistry lab in New York. Two ml of RPMI were added to 100 ul of each blood sample. To extract lipids, 2 ml extraction buffer, ethyl acetate/isopropanol/water (60/30/10, v/v) were added. After the extraction, formic acid was added to the organic phase. The lipids were extracted from the blood by the extraction buffer once more. After 2 extractions, they were combined and dried down under N2 gas stream. Sphingolipids were determined by LC-MS/MS performed on a TSQ 7000 triple quadruple mass spectrometer (Thermo Finnigan; Ringoes, NJ) as described (Bielawski et al., 2006). Skin fibroblasts were cultured to 90% confluence in DMEM medium containing 20% FBS before being harvested for sphingolipid profiling by LC-MS/MS.
Results
Detection of homozygous truncating SGPL1 variants
The mean on-target coverage and the percentage of bases covered at least 20 times of their exomes were 50× and 56% for patient 2, and 114× and 95.7% for patient 3. Filtering the WES data for variants predicted to affect protein sequence or exon splicing with a MAF < 0.001 in dbSNP, in the Exome Variant Server, the 1000 Genomes Project, or in in-house databases under an autosomal-recessive disease model immediately provided one plausible candidate gene, SGPL1 (RefSeq accession number NM_003901.3): Both patients had homozygous mutations, (NM_003901.3) c.1513C>T (p.Arg505*) (patient 2) and (NM_003901.3) c.934delC (p.Leu312Phefs*30) (patient 3), in SGPL1 that predicted severe truncation of the encoded 568-amino acid protein, or rather nonsense-mediated mRNA decay of the transcripts. Both variants were not listed in dbSNP, ESP, and EXAC databases. Both parents of patient 2 were heterozygous carriers of the c.1513C>T variant (Fig. 1E) and both parents of patient 3 were heterozygous carriers of the c.934delC variant (Fig. 1F). The phenotype and variant information was submitted to the LOVD database (http://databases.lovd.nl/shared/individuals/00095243 (patient 3) and /00095244 (patient 2)). Under an autosomal recessive model and segregation analyses in families (Fig. 1) by Sanger sequencing there were no other rare or private, potentially disease causing variants, in homozygous or compound-heterozygous state seen in patient 3 and there were two missense variants of unknown significance present in IL17RC (MIM #610925) and ANLN (MIM #616027) genes in patient 2, respectively. Unfortunately, we could not study patient 1 and his parents.
The stop mutation in SGPL1 alters sphingolipid levels in blood and in cultured fibroblasts
SGPL1 (also named SPL; EC 4.1.2.27) catalyzes the irreversible degradation of endogenous and dietary sphingosine-1-phosphate (S1P) to hexadecenal and ethanolamine phosphate by cleaving the C2–3 carbon bond in S1P (van Veldhoven and Mannaerts, 1993). It is an endoplasmic reticulum-resident, integral membrane protein with the active, cofactor pyridoxal 5′-phosphate binding domain exposed to the cytosol (Zhou and Saba, 1998; Van Veldhoven et al., 2000; Ikeda et al., 2004). The sphingolipid metabolism relating to SGPL1 is outlined in Figure 2. To investigate how the stop mutation in the SGPL1 gene affected sphingolipid metabolism in the patients, we performed the profiling of sphingolipids in blood samples from the patient and two healthy individuals. As expected, we found that the patient had higher blood levels of S1P (Fig. 3A) compared to the healthy individuals because S1P is the major endogenous substrate of SGPL1. The patient also had much higher levels of sphingosine (SPH), the immediate precursor of S1P (Fig. 3B). Interestingly, the patient had lower levels of dihydrosphingosine (DHS) (Fig. 3C), dihydrosphingosine-1-phosphate (DHS1P) (Fig. 3D), ceramides (Fig. 3E), dihydroceramides (Fig. 3F), sphingomyelins, and monohexosylceramides in the blood (Supp. Figure S2A, B). In cultured skin fibroblasts, S1P (Fig. 3G), SPH (Fig. 3H), DHS (Fig. 3I), and DHS1P (Fig. 3K) were increased in the patient compared to healthy controls. However, total ceramides or dihydroceramides were not significantly different between patient and controls (Supp. Figure S2C, D). Collectively, we show that a homozygous truncating SGPL1 mutation leads to significantly altered levels of sphingolipid metabolism intermediates.
Figure 2.

Outline of the metabolism of sphingolipids with emphasis on SGPL1.
Figure 3. The frameshift mutation in the SGPL1 gene alters blood and skin fibroblast sphingolipid levels.
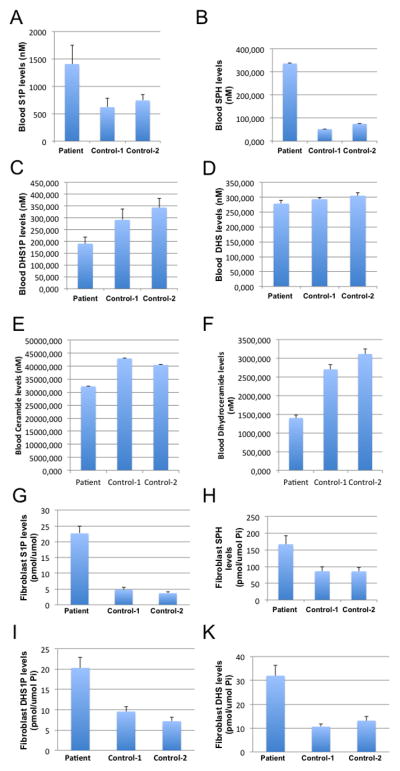
Blood samples were collected from healthy individuals (Control 1 and Control 2) and patient 3, and subjected to LC-MS/MS for the levels of sphingoid base phosphates, sphingoid bases, total ceramides, total dihydroceramides, total sphingomyelins, and total monohexosylceramides. Results for S1P (A), SPH (B), DHS1P (C), DHS (D), ceramide (E), dihydroceramide (F) are shown. The data represent mean values ± SD, n=3. *p<0.05. Skin fibroblasts from either healthy individuals (Control 1 and Control 2) and patient 3 were cultured to a 90% confluence in DMEM medium containing 20% FBS before being harvested for sphingolipid profiling by LC-MS/MS. The levels of sphingoid bases, sphingoid base phosphates, total ceramides, or total dihydroceramides were determined. In cultured skin fibroblasts, S1P (G), SPH (H), DHS1P (I), and DHS (K) were increased in the patient compared to healthy controls. For each sample, the data represent mean values from three measurements.
Discussion
Here we report that homozygous truncating SGPL1 mutations are present in unrelated families with CNS associated with adrenal calcifications and hypogonadism. We demonstrate significant changes in sphingolipid levels with high elevations in S1P and SPH in full blood and fibroblasts from one of these patients. Levels of other sphingolipids were variably affected in blood and fibroblasts indicating that the truncating mutation likely leads to a loss of the catalytic activity of SGPL1.
S1P represents an intermediate in the metabolism of the cell membrane components sphingomyelin and glycosphingolipids and is the ligand for a family of five differentially expressed extracellular G-protein-coupled receptors (Hla, 2004), which generate downstream signals that regulate angiogenesis, vascular maturation, cardiac development and immunity, and are important for directed cell movement. S1P exerts intracellular effects as well but those are incompletely understood (Hait et al., 2009; Alvarez et al., 2010). S1P is generated by phosphorylation of SPH by two sphingosine kinase isoenzymes (SphK1 and SphK2) (Maceyka et al., 2005), and can be dephosphorylated by S1P phosphatases, by nonspecific lipid phosphate phosphatases (Pyne et al., 2009), or cleaved irreversibly by SGPL1. Intracellular S1P levels are normally low and tightly regulated spatio-temporally (Lee et al., 1999; Kono et al., 2008). The European patient accumulated SPH, the precursor of S1P, in the blood to a greater extent than S1P, likely due to a feedback inhibition of SphK1 and SphK2 by their product S1P. Interestingly, total levels of ceramides, the precursors of SPH, were slightly but significantly decreased in the patient’s blood compared to the healthy controls, as were more complex sphingolipids sphingomyelins and monoglucosylceramides, which both are derived from ceramides. As ceramides are synthesized de-novo from serine and palmitoyl-CoA through multiple enzymatic steps (Merrill, 2002), we postulate that the decreases in the levels of ceramides and their complex sphingolipid derivatives may be due to a feedback inhibition in the de novo biosynthetic pathway of ceramides. This is supported by the findings that the levels of both DHS and dihydroceramides, intermediates in the de novo pathway for ceramide biosynthesis were also decreased in the patient’s blood. These results suggest that SGPL1 is a key enzyme in the homeostasis of sphingolipids in humans.
Ample evidence has suggested the possible involvement of sphingolipids in the development of various diseases. Indeed, the accumulation of SPH in patients with Niemann-Pick disease type C1 (NPC) might be responsible for the clinical effects (Lloyd-Evans et al., 2008) as SPH is a highly cytotoxic bioactive lipid that can induce apoptosis and necrosis (Cuvillier, 2002). Recent studies demonstrated that high levels of S1P could also lead to the death of certain cell types (Davaille et al., 2002; Le Stunff et al., 2002) although S1P in general is a pro-survival bioactive lipid. These results suggest that the symptoms observed in the patients might be caused by the accumulation of S1P and SPH in the blood, fibroblasts and potentially in other tissues. However, we cannot yet exclude the possibility that a lack of phosphoethanolamine production contributes in part to the mutant phenotype, such as was shown in the model organism Leishmania major (Zhang et al., 2007).
The renal changes seen in our patient might have resulted from a developmental signaling defect of angiogenic factors due to constantly elevated S1P levels. Indeed, EGF, PDGF, and VEGF, and other growth and pro-angiogenic factors stimulate and translocate SphK1 to the plasma membrane, resulting in local formation of S1P (Pitson, 2011). Binding of S1P to the S1PR1 receptor can increase PDGF and VEGF production, resulting in further S1P generation via positive feedback, and leads to transactivation of the respective growth factor receptors that in turn activate downstream signals that regulate vascular remodeling and cell movement (Liu et al., 2000; Hobson et al., 2001). Interestingly, homozygous Sgpl1 knockout mice developed proteinuria, swollen and hemorrhagic glomeruli. Homozygous Sgpl1 knockout mice failed to thrive after birth and died at the time of weaning. They had vascular abnormalities, which lead to fetal hemorrhage and anemia, and skeletal defects, improper palatal fusion, thoracic malformations of the sternum, ribs and vertebrae (Schmahl et al., 2007). Sgpl1 has a glomerular expression and most knockout mice showed a reduction in the number of smooth muscle actin-positive cells in the glomeruli, suggesting that these cells did not migrate into the glomerular space. This is supported by the observation that embryonic fibroblasts from Sgpl1 knockout mice show migration defects in vitro (Schmahl et al., 2007). In developing kidney glomeruli, PDGF-B is expressed by glomerular endothelium and drives the proliferation of mesangial cells contributing to the complex networks of capillaries, which create the large surfaces needed for filtration/excretion in the kidney (Lindahl et al., 1998).
Vascular and cell migration defects have been observed in Pdgfrb- and S1pr1-knockout mice as well. Although S1pr1−/− embryos developed a vascular network, they died in utero at E12.5–E14.5 due to defective coverage of large vessels by pericytes and vascular smooth muscle cells (VSMCs) (Liu et al., 2000). Together with the identification of Sgpl1 as a downstream target of PDGF signaling (Schmahl et al., 2007) these defects indicate that SGPL1 might have a role in the regulation of mammalian angiogenesis and other developmental processes.
Adrenal calcification and function have not been addressed in Sgpl1 knockout mice. However, the importance of sphingolipids in steroid-hormone biosynthesis is known. Intriguingly, the balance between levels of SPH and S1P might act as a switch to turn-on or - off steroid-hormone production in response to ACTH in the adrenal cortex (Ozbay et al., 2006; Urs et al., 2006; Urs et al., 2007). Tight control of sphingolipid intermediates has also been shown to be required for normal growth and development in D. melanogaster, as revealed through a genetic epistasis analysis (Herr et al., 2003).
The three different steroidogenic cell types in the testis, ovary, and adrenal are all under endocrine control from the pituitary and male and female Sgpl1 knockout mice are sterile, and the genital abnormalities seen in our patients might represent primary defects of SGPL1 deficiency. In contrast to Sgpl1 knockout mice (Borowsky et al., 2012), the young patients described here did neither present with lymphopenia, nor with neurological or hepatic symptoms, and there were none of the marked changes seen in levels of non-sphingolipid metabolites, serum and liver triglycerides, cholesterol, and phospholipids.
The SGPL1 enzyme is conserved from yeast to humans and its loss of activity had detrimental effects in several model organisms, i.e. abnormalities of growth regulation and carbon metabolism were revealed in Saccharomyces cerevisiae (Gottlieb et al., 1999), developmental and migration defects in Dictyostelium discoideum (Li et al., 2001), reproductive defects in Caenorhabditis elegans (Mendel et al., 2003), and defects of reproductive organs and muscles in Drosophila melanogaster (Herr et al., 2003) following SGPL1 inactivation. These observations are in support of our conclusion, that truncating SGPL1 mutations that inactivate the catalytic function of the enzyme lead to an accumulation of S1P and SPH in blood, fibroblasts and probably in many other tissues, which may be responsible for the developmental and functional effects observed in the patients.
As roles for S1P-mediated signaling were also implicated in pathological angiogenesis, cancer, inflammation, allergy, autoimmune diseases, diabetes, and the response to ischemic injury of the heart, kidney and brain (Aguilar and Saba, 2012), an SPH analogue and S1P receptor antagonist, FTY720 (fingolimod), and a humanized form of the monoclonal S1P-specific antibody LT1009 (O’Brien et al., 2009) are available, and might represent means to lower S1P levels in patients with SGPL1 deficiency.
Supplementary Material
Acknowledgments
We thank Izolda Mileva in the Lipidomics Core Facility at Stony Brook University for performing sphinglipidomic analyses.
Contract Grant Sponsors: Österreichische Nationalbank (15627). This work was supported in part by National Institutes of Health (USA) Grants R01CA163825 and P01CA097132.
Footnotes
The authors declare no conflict of interest.
References
- Aguilar A, Saba JD. Truth and consequences of sphingosine-1-phosphate lyase. Adv Biol Regul. 2012;52:17–30. doi: 10.1016/j.advenzreg.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, Milstien S, Spiegel S. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–8. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RA, Byrum RS, Coates PM, Sando GN. Mutations at the Lysosomal Acid Cholesteryl Ester Hydrolase Gene Locus in Wolman-Disease. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:2718–2722. doi: 10.1073/pnas.91.7.2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39:82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Borowsky AD, Bandhuvula P, Kumar A, Yoshinaga Y, Nefedov M, Fong LG, Zhang M, Baridon B, Dillard L, de Jong P, Young SG, West DB, et al. Sphingosine-1-phosphate lyase expression in embryonic and adult murine tissues. J Lipid Res. 2012;53:1920–31. doi: 10.1194/jlr.M028084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvillier O. Sphingosine in apoptosis signaling. Biochim Biophys Acta. 2002;1585:153–62. doi: 10.1016/s1388-1981(02)00336-0. [DOI] [PubMed] [Google Scholar]
- Davaille J, Li L, Mallat A, Lotersztajn S. Sphingosine 1-phosphate triggers both apoptotic and survival signals for human hepatic myofibroblasts. J Biol Chem. 2002;277:37323–30. doi: 10.1074/jbc.M202798200. [DOI] [PubMed] [Google Scholar]
- Gottlieb D, Heideman W, Saba JD. The DPL1 gene is involved in mediating the response to nutrient deprivation in Saccharomyces cerevisiae. Mol Cell Biol Res Commun. 1999;1:66–71. doi: 10.1006/mcbr.1999.0109. [DOI] [PubMed] [Google Scholar]
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, Spiegel S. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–7. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr DR, Fyrst H, Phan V, Heinecke K, Georges R, Harris GL, Saba JD. Sply regulation of sphingolipid signaling molecules is essential for Drosophila development. Development. 2003;130:2443–53. doi: 10.1242/dev.00456. [DOI] [PubMed] [Google Scholar]
- Hla T. Physiological and pathological actions of sphingosine 1-phosphate. Semin Cell Dev Biol. 2004;15:513–20. doi: 10.1016/j.semcdb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Hobson JP, Rosenfeldt HM, Barak LS, Olivera A, Poulton S, Caron MG, Milstien S, Spiegel S. Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motility. Science. 2001;291:1800–3. doi: 10.1126/science.1057559. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Kihara A, Igarashi Y. Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochem Biophys Res Commun. 2004;325:338–43. doi: 10.1016/j.bbrc.2004.10.036. [DOI] [PubMed] [Google Scholar]
- Indumathi CK, Dinakar C, Lewin S, Phadke KD. Congenital Nephrotic Syndrome with adrenal calcification and cardiac malformation. Indian J Pediatr. 2005;72:1049–51. doi: 10.1007/BF02724410. [DOI] [PubMed] [Google Scholar]
- Kono M, Allende ML, Proia RL. Sphingosine-1-phosphate regulation of mammalian development. Biochim Biophys Acta. 2008;1781:435–41. doi: 10.1016/j.bbalip.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Stunff H, Galve-Roperh I, Peterson C, Milstien S, Spiegel S. Sphingosine-1-phosphate phosphohydrolase in regulation of sphingolipid metabolism and apoptosis. J Cell Biol. 2002;158:1039–49. doi: 10.1083/jcb.200203123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha’afi RI, Hla T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–12. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- Li G, Foote C, Alexander S, Alexander H. Sphingosine-1-phosphate lyase has a central role in the development of Dictyostelium discoideum. Development. 2001;128:3473–83. doi: 10.1242/dev.128.18.3473. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl P, Hellstrom M, Kalen M, Karlsson L, Pekny M, Pekna M, Soriano P, Betsholtz C. Paracrine PDGF-B/PDGF-Rbeta signaling controls mesangial cell development in kidney glomeruli. Development. 1998;125:3313–22. doi: 10.1242/dev.125.17.3313. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, Liu CH, Hla T, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106:951–61. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–55. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH, Jr, Milstien S, Spiegel S. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005;280:37118–29. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- Mendel J, Heinecke K, Fyrst H, Saba JD. Sphingosine phosphate lyase expression is essential for normal development in Caenorhabditis elegans. J Biol Chem. 2003;278:22341–9. doi: 10.1074/jbc.M302857200. [DOI] [PubMed] [Google Scholar]
- Merrill AH., Jr De novo sphingolipid biosynthesis: a necessary, but dangerous, pathway. J Biol Chem. 2002;277:25843–6. doi: 10.1074/jbc.R200009200. [DOI] [PubMed] [Google Scholar]
- Miller WL. Disorders in the initial steps of steroid hormone synthesis. J Steroid Biochem Mol Biol. 2016 doi: 10.1016/j.jsbmb.2016.03.009. [DOI] [PubMed] [Google Scholar]
- O’Brien N, Jones ST, Williams DG, Cunningham HB, Moreno K, Visentin B, Gentile A, Vekich J, Shestowsky W, Hiraiwa M, Matteo R, Cavalli A, et al. Production and characterization of monoclonal anti-sphingosine-1-phosphate antibodies. J Lipid Res. 2009;50:2245–57. doi: 10.1194/jlr.M900048-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbay T, Rowan A, Leon A, Patel P, Sewer MB. Cyclic adenosine 5′-monophosphate-dependent sphingosine-1-phosphate biosynthesis induces human CYP17 gene transcription by activating cleavage of sterol regulatory element binding protein 1. Endocrinology. 2006;147:1427–37. doi: 10.1210/en.2005-1091. [DOI] [PubMed] [Google Scholar]
- Pitson SM. Regulation of sphingosine kinase and sphingolipid signaling. Trends in Biochemical Sciences. 2011;36:97–107. doi: 10.1016/j.tibs.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Powers RJ, Cohen ML, Williams J. Adrenal calcification and congenital nephrotic syndrome in three American Indians. Pediatr Nephrol. 1990;4:29–31. doi: 10.1007/BF00858433. [DOI] [PubMed] [Google Scholar]
- Pyne S, Lee SC, Long J, Llyne NJ. Role of sphingosine kinases and lipid phosphate phosphatases in regulating spatial sphingosine 1-phosphate signalling in health and disease. Cellular Signalling. 2009;21:14–21. doi: 10.1016/j.cellsig.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ, Tan W, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26:1279–89. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmahl J, Raymond CS, Soriano P. PDGF signaling specificity is mediated through multiple immediate early genes. Nat Genet. 2007;39:52–60. doi: 10.1038/ng1922. [DOI] [PubMed] [Google Scholar]
- Schreyer-Shafir N, Sukenik-Halevy R, Tepper R, Arnon S, Litmanovitch I, Eliakim A, Pommeranz A, Ludman MD, Raas-Rothschild A. Prenatal bilateral adrenal calcifications, hypogonadism, and nephrotic syndrome: beyond Wolman disease. Prenat Diagn. 2014;34:608–11. doi: 10.1002/pd.4344. [DOI] [PubMed] [Google Scholar]
- Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, Anarat A, Caliskan S, Emma F, Gellermann J, Oh J, Baskin E, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol. 2015;10:592–600. doi: 10.2215/CJN.06260614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs AN, Dammer E, Kelly S, Wang E, Merrill AH, Jr, Sewer MB. Steroidogenic factor-1 is a sphingolipid binding protein. Mol Cell Endocrinol. 2007;265–266:174–8. doi: 10.1016/j.mce.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs AN, Dammer E, Sewer MB. Sphingosine regulates the transcription of CYP17 by binding to steroidogenic factor-1. Endocrinology. 2006;147:5249–58. doi: 10.1210/en.2006-0355. [DOI] [PubMed] [Google Scholar]
- Van Veldhoven PP, Gijsbers S, Mannaerts GP, Vermeesch JR, Brys V. Human sphingosine-1-phosphate lyase: cDNA cloning, functional expression studies and mapping to chromosome 10q22(1) Biochim Biophys Acta. 2000;1487:128–34. doi: 10.1016/s1388-1981(00)00079-2. [DOI] [PubMed] [Google Scholar]
- van Veldhoven PP, Mannaerts GP. Sphingosine-phosphate lyase. Adv Lipid Res. 1993;26:69–98. [PubMed] [Google Scholar]
- Zhang K, Pompey JM, Hsu FF, Key P, Bandhuvula P, Saba JD, Turk J, Beverley SM. Redirection of sphingolipid metabolism toward de novo synthesis of ethanolamine in Leishmania. EMBO J. 2007;26:1094–104. doi: 10.1038/sj.emboj.7601565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Saba JD. Identification of the first mammalian sphingosine phosphate lyase gene and its functional expression in yeast. Biochem Biophys Res Commun. 1998;242:502–7. doi: 10.1006/bbrc.1997.7993. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.