

Abstract
miR-495 serves as an oncogenic miRNA or a tumor suppressor in different types of cancer. However, its role in the drug resistance of small cell lung cancer (SCLC) remains unidentified. In this study, we investigated whether miR-495 regulates the chemoresistance of SCLC through the epithelial-mesenchymal transition (EMT) via Epithelial and endothelial tyrosine kinase (Etk/BMX) using two drug-resistant cell lines. Loss- and gain-of-function experiments showed miR-495 regulated cell proliferation, tumor growth and drug resistance. miR-495 suppression or Etk/BMX elevation in SCLC specimens was correlated with poor pathologic stage and survival time. Etk/BMX was one of the directly targeted genes of miR-495. Ectopic expression of Etk/BMX obviously rescued the miR-495 elevation elevation-induced inhibition of drug resistance. Etk/BMX over-expression led to higher levels of EMT mesenchymal factors (Zeb-2, Twist, Vim) and lower levels of the epithelial molecule β-catenin, while suppression of Etk/BMX showed the opposite trend. Knockdown of Zeb-2 and Twist inhibited the chemoresistance of cells. Our study revealed that miR-495 promoted the chemoresistance of SCLC through the epithelial-mesenchymal transition via Etk/BMX. miR-495 re-expression or Etk/BMX depletion is a promising strategy for interfering with chemoresistance in SCLC.
Keywords: Chemoresistance, miR-495, EMT, Etk/BMX, SCLC
Introduction
Lung cancer is one of the leading causes of cancer death worldwide. Small cell lung cancer (SCLC) accounts for approximately 15% of all lung cancers [1,2]. The 2-year survival rate is generally less than 5% in patients with SCLC, and 90% of patients die within 5 years after the final diagnosis. Chemotherapy remains a major treatment for SCLC. Although it shows remarkable sensitivity to chemotherapeutic drugs, SCLC is characterized by high relapse rates and a subsequent poor prognosis. The aggressiveness of SCLC may, in part, be due to its intrinsic and extrinsic chemoresistance [3,4]. Therefore, it is increasingly challenging to understand the molecular mechanism of chemoresistance and develop effective therapeutic strategies to overcome the drug resistance of SCLC.
It is becoming clear that MicroRNA play a pivotal role in drug resistance, such as miR-214 in human ovarian and miR-216a/217 in hepatocellular carcinoma [5-8]. Our previous study also investigated the different miRNAs between SCLC-sensitive and -resistant cells, and we first reported that miR-495 was significantly lower in the resistant cells of SCLC [9]. miR-495 was first found in the brain tissues and can regulate neuronal plasticity by affecting the expression of brain-derived neurotropic factor [10]. The function of miR-495 remains controversial in different types of cancer. For example, it serves as a tumor suppressor in MLL-rearranged leukemia but functions as an oncogenic miRNA in breast cancer [11,12]. However, whether miR-495 serves as a tumor suppressor or an oncogene in SCLC has not been reported. We are also not completely certain whether miR-495 affects the chemoresistance of SCLC. Thus, more studies need to be performed to demonstrate the effect of miR-495 on the drug sensitivity of SCLC. Additionally, the mechanism of miR-495 in SCLC chemoresistance should be further studied.
To detect the target gene of miR-495, bioinformatics analysis was conducted, and the results indicated that Epithelial and endothelial tyrosine kinase (Etk), also known as bone marrow X kinase (BMX), may be a target gene of miR-495. Etk/BMX is an important member of the family of Tec non-receptor tyrosine kinases [13-16]. It has been reported that Etk/BMX is involved in various biological processes, including proliferation, differentiation, apoptosis and cell migration [17-19]. In addition, our previous studies found that Etk/BMX participated in the chemoresistance of SCLC. We knocked down Etk/BMX expression in multi-drug-resistant SCLC H69/AR cells by Etk/BMX-specific small interfering RNA, leading to the increased sensitivity of H69/AR cells to chemotherapeutic drugs [20]. Whether miR-495 directly influenced the expression of Etk/BMX is unknown.
Accumulating studies have revealed that EMT is linked to drug resistance in cancers such as bladder cancer and non-small cell lung cancer [21,22]. The hallmark of EMT is the loss of the epithelial markers, such as E-cadherin and β-catenin, and gain of mesenchymal molecules, such as N-cadherin and Vimentin (Vim). The factors containing Snail, Slug, Twist, and other transcription factors, which interact with E-box elements, are significant inducers of EMT by repressing E-cadherin expression [23,24]. Whether EMT plays an important role in the drug resistance of SCLC has not been reported. We analyzed the data from cDNA microarray in cellular models of SCLC that were widely used as sensitive (H69) and resistant (H69/AR) cell lines to chemotherapy in the previous study. The results showed that EMT-related markers (Zeb-2, Twist, Vim and β-catenin) were significantly different (> 3-fold) (Supplementary Figure 1). However, it remains unclear whether the aberrant expression of these EMT-associated genes is directly responsible for chemoresistance in SCLC.
As noted above, we proposed the hypothesis that the down-regulation of miR-495 promotes the chemoresistance of SCLC through the epithelial-mesenchymal transition via Etk/BMX. To investigate this hypothesis, we used two drug-resistant cell lines-H69/AR and H446/AR-in a series of assays.
Materials and methods
Tissue preparation
The experiments were undertaken with the understanding and written consent of each subject. The study methodologies conformed to the standards set by the Declaration of Helsinki. This study was approved by the Ethics Committee of Southern Medical University. Eighty-six SCLC and sixty normal lung tissue samples were obtained by biopsy from January 2008 to December 2011 from Zhujiang and Nanfang Hospitals (Southern Medical University, Guangzhou, China). All of the patients had not received chemotherapy before biopsy, and they received chemotherapy and radiation after biopsy if necessary. Each case was independently reviewed by two pathologists according to the WHO criteria.
Immunohistochemical staining
Tissues sections were deparaffinized and rehydrated routinely. Before the addition of primary antibody, antigen was retrieved by heating the sections. After blocking with 0.3% H2O2 and goat serum, the slides were then incubated with a primary antibody (Etk, 1:100; BD Biosciences, USA) at 4°C overnight. Following three washings in phosphate-buffered saline (PBS), an avidin-biotin complex (Vector Labs, Burlingame, USA) and alkaline phosphatase mixture were applied. The reaction products were visualized using 3’diaminobenzidine (DAB), 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) and 3-amino-9-ethylcarbazole (AEC) (MaximBiotech, Inc. CA, USA). All of the slides were subsequently counterstained with hematoxylin. Brown-yellow granules in the cytoplasm were considered positive staining for Etk/BMX. Negative controls were performed by replacing the primary antibodies noted above with PBS.
Cell culture and transfection
The human small lung cancer cell lines NCI-H446 and NCI-H69, as well as the corresponding drug-resistant subline H69/AR, were purchased from the American Type Culture Collection (ATCC) and were maintained in RPMI 1640 medium supplemented with 10% newborn bovine serum (GIBCO) at 37°C with 5% CO2. H446/AR cells were developed as described previously [25]. The two resistant cell lines were tested regularly for maintained resistance to the selected drugs. The growth and morphology of all cell lines were monitored on a weekly basis.
The plasmid encoding the full-length Etk was kindly provided by Dr. Hsing-Jien Kung [26]. The short hairpin RNA (shRNA) targeting Etk and small interfering RNAs (siRNAs) targeting Zeb-2 and Twist were all supplied by Shanghai GenePharma Co., Ltd. The mimics and antagomir of miR-495 were from Guangzhou RibiBio Co., Ltd. The transfection process was conducted according to the manufacturer’s instructions for Lipofectamine 2000 (Invitrogen).
Real-time quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen) and was quantified with a NanoDrop2000. Reverse transcription reactions were performed for 15 min at 37°C, followed by 5 s at 85°C for complementary DNA synthesis. Real-time reactions were performed using the SYBR® PrimeScript™ RT-PCR Kit (Takara Biotechnology Co., Ltd. Dalian, China). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or U6 snRNA was used as the housekeeping gene. The mRNA expression levels of all samples were normalized to the housekeeping gene. The high value of miR-495/U6 indicates the low expression of miR-495.
Western blot analysis
Cell proteins were extracted with RIPA lysis buffer, and the concentrations were determined using the standard BCA method (BCA™ Protein Assay Kit, Pierce, USA). Each protein sample was homogenized in the loading buffer and boiled for 5 min and then separated by 8% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Etten-leur, Netherlands). The membranes were blocked with 5% non-fat dried milk for 2 h and then treated with primary antibodies, including Etk, Zeb-2, Twist, Vim, and β-catenin antibodies, overnight at 4°C. The membranes were washed again with TBST (10 mM Tris, pH 8.0, 150 mM NaCl, and 0.1% Tween 20), followed by incubation with horseradish peroxidase-labeled secondary antibody (Beijing Biosynthesis Biotechnology Co. Ltd.) for 45 min at room temperature. Finally, after treatment with super echochemiluminescence (ECL) plus detection reagents, the protein bands of the membranes were visualized by exposure to X-ray film. The protein band intensities were quantified using Quantity One software.
In vitro drug resistance assay
The ranges of drug concentrations were based on earlier studies and were aimed at obtaining the half-maximal (50%) inhibitory concentration of a substance (IC50) [20,27]. Three anti-cancer drugs-cisplatin (DDP; Shangdong, China), etoposide (VP-16; Jiangshu, China), and adriamycin (ADM; Jiangshu, China)-were obtained from commercial sources and were dissolved according to the manufacturer’s instructions. Anti-cancer drug-induced cell death was quantified using the Cell Counting Kit-8 assay (CCK-8). Cells were seeded in 96-well plates and then treated for 24 h in 200 μl of medium with anti-cancer drugs. CCK-8 reagent (Dojindo, Kumamoto, Japan) was then added, and the cells were incubated at 37°C for 2 h before the absorbency was read at 450 nm using a micro-plate reader.
Cell proliferation assay
The cells were seeded in 96-well plates at 2 × 103 cells/well. At the indicated times (0 h, 24 h, 48 h, 72 h, and 96 h), 10 μl of CCK-8 and 100 μl of RPMI 1640 were added to each well. The cells were incubated for 2 h, and the absorbance at 450 nm was measured to calculate the cell growth rates. Growth rate = (absorbance at 450 nm at × h- absorbance at 450 nm at 0 h)/(absorbance at 450 nm at 0 h).
Cell scratch-wound healing assay
The cells were seeded on 24-well plates and grown until monolayer formation. Wound areas were scraped using 100-μl plastic tips. At the indicated times (0 h, 12 h, and 24 h), the wound areas were photographed and the wound healing rate was calculated. Healing rate = (width of wound at × h- width of the wound at 0 h)/width of wound at 0 h.
Cell invasion assay
Using 24-well transwell units with 8-μm-pore-size polycarbonate inserts, Matrigel (50 μl) as a basement membrane was spread on the polycarbonate membrane and allowed to solidify for 1 h at room temperature. Cells suspended in RPMI 1640 containing 5 g/l BSA were added to each upper compartment of the transwell units. After culture for 48 h, cells migrating through the Matrigel-coated polycarbonate membrane were fixed with paraformaldehyde, stained with crystal violet and counted in five different fields randomly.
Flow cytometry assay for cell apoptosis
The cells treated with or without chemotherapeutic drugs including DDP, VP-16 and ADM were harvested, washed with PBS, and resuspended in binding buffer containing 7-AAD for 10 minutes, followed by the addition of Annexin V-PE. Cell apoptosis analysis was carried out using a flow cytometer (BD Biosciences, Oxford, United Kingdom).
In vivo tumorigenicity assay
Male nude mice (six weeks old, weighing 18-20 g, from the Medical Experimental Animal Center of Guangdong Province, China) were used for in vivo assays. The mice were raised under pathogen-free conditions. All of the procedures were performed according to the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International.
The miR-495/antagomir cells were subcutaneously inoculated into the legs of nude mice to establish the tumor model. After 10 days, the 5 nmol miR-495/antagomir was inoculated into the tumor every 3 days. The tumor volume was determined three times per week by direct measurement with a sliding caliper and was calculated with the following equation: V = (4/3) × π × R12 × R2, where R1 is radius 1 and R2 is radius 2 and R1 < R2. Growth curves of the tumors were constructed. Twenty-two days later, 5 mice from each group were sacrificed and the tumors were excised. The tumors were homogenized to extract protein for detecting the expression of Etk/BMX, Twist, Zeb-2, Vim and β-catenin. The other mice of each group were continually raised to record the survival time.
Luciferase reporter assay
The wild-type and mutated 3’UTR segments of Etk/BMX predicted to interact with miR-495 were amplified by PCR from human genomic DNA and were inserted into the psiCHECK-2 vector immediately downstream from the stop codon of luciferase (Promega) to develop psiCHECK2-Etk/BMX-3’UTR and psiCHECK2-Etk/BMX-mut-3’UTR, respectively. Cells in 24-well plates were transfected with psiCHECK2-Etk/BMX-3’UTR, psiCHECK2-Etk/BMX-mut-3’UTR or psiCHECK-2, respectively. Additionally, miR-495 mimics and antagomir were co-transfected into the cells. Luciferase activity was assayed at 48 h post-transfection using a dual-luciferase reporter assay system (Promega).
Statistical analysis
Data were analyzed using SPSS20.0 software. The results are presented as the means ± SD (standard deviation). Comparison of the means between two samples was performed using Student’s t test. Statistical comparisons of more than two groups were performed using one-way analysis of variance (ANOVA). The associations between Etk/BMX expression and clinical features were analyzed with a Pearson chi-squared test. Survival curves were obtained by Kaplan-Meier analysis. In all cases, P < 0.05 was considered statistically significant.
Results
The role of miR-495 and Etk/BMX in the clinicopathological characteristics of SCLC patients
To investigate the role of miR-495 expression in patients with SCLC, we examined the expression level of miR-495 in 86 SCLC tissues and 60 normal lung tissues. The results revealed that the average expression level of miR-495 was obviously lower in the SCLC samples than in the normal tissues (Figure 1A). Furthermore, the relationships between miR-495 expression and clinicopathological characteristics in SCLC patients are summarized in Table 1. No significant association was observed in any of the 86 SCLC cases between miR-495 expression and the patient’s sex or age. However, the expression of miR-495 was positively correlated with the tumor pathologic stage. Kaplan-Meier analysis revealed that miR-495 expression was closely associated with the overall survival rate of the SCLC patients (Figure 1B).
Figure 1.
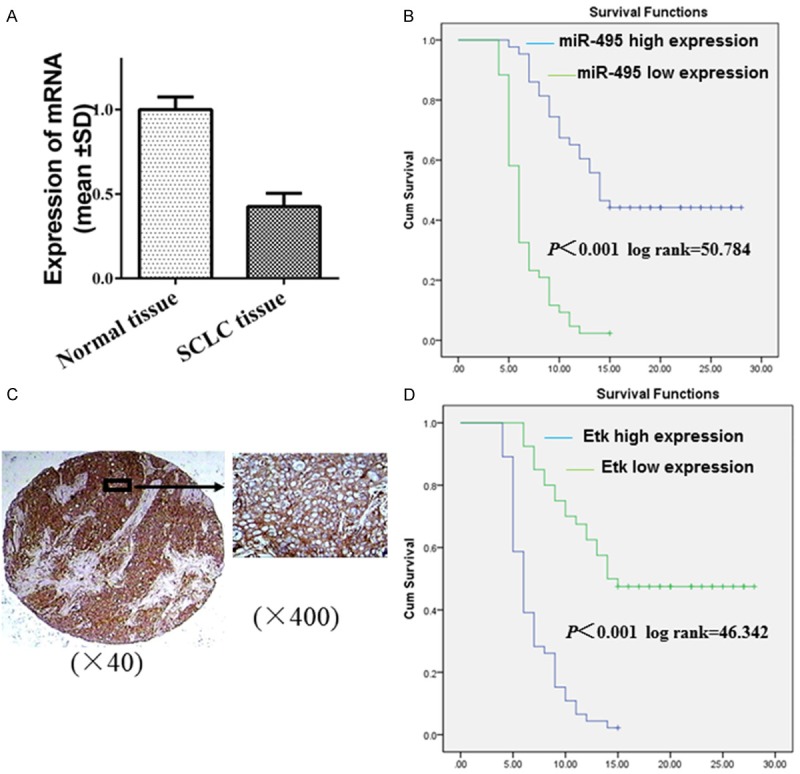
The role of miR-495 and Etk/BMX in the clinicopathological characteristics of SCLC patients was studied. A: Expression of miR-495 in SCLC and normal lung tissues by qRT-PCR. B: Kaplan-Meier analysis of the overall survival of 86 patients with SCLC based on miR-495 expression. C: The brown-yellow granules in the cytoplasm were considered positive staining for Etk/BMX. D: The relationship between Etk/BMX and survival was studied by Kaplan-Meier analysis in patients with SCLC.
Table 1.
Association of miR-495 expression and clinicopathological characteristic in SCLC patients
| Characteristics | No. | Expression of miR-495/U6 | P |
|---|---|---|---|
| Gender | |||
| Male | 69 | 0.88±0.19 | 0.306 |
| Female | 17 | 0.83±0.16 | |
| Age (years) | |||
| ≤ 59 years | 49 | 0.83±0.15 | 0.824 |
| > 59 years | 37 | 0.85±0.19 | |
| Clinical stage | |||
| LD | 66 | 0.75±0.14 | 0.000 |
| ED | 20 | 1.03±0.12 | |
| Status | |||
| Survival | 20 | 0.66±0.10 | 0.000 |
| Death | 66 | 00.90±0.15 |
Significant differences are shown in bold.
Table 2 summarizes the relationship between Etk/BMX expression and clinical characteristics in SCLC patients. Of the 66 cases with limited stage SCLC, 42 (63.6%) showed Etk/BMX expression, while all 20 (100.0%) cases were positive for Etk/BMX expression in patients with extensive stage. The expression of Etk/BMX revealed an obvious correlation with the pathologic stage (P = 0.001). The negative expression of Etk/BMX was more frequent in cases with a survival rate > 15 months (19/20, 95.0%) than in cases with a survival rate < 15 months (21/66, 31.8%) (P < 0.001). The expression of Etk/BMX exhibited no significant relationship with gender (P = 0.448) or age (P = 0.106). Kaplan-Meier analysis revealed that Etk/BMX expression was related to the overall survival rate of the SCLC patients (Figure 1C, 1D).
Table 2.
Association of ETK/BMX expression and clinicopathological characteristic in SCLC patients
| Characteristics | Total | ETK/BMX expression | x2 | P value | |
|---|---|---|---|---|---|
|
| |||||
| Negative expression | Positive expression | ||||
| Gender | 0.575 | 0.448 | |||
| Male | 69 | 18 | 51 | ||
| Female | 17 | 6 | 11 | ||
| Age (years) | 2.608 | 0.106 | |||
| ≤ 59 years | 49 | 17 | 32 | ||
| > 59 years | 37 | 7 | 30 | ||
| Clinical stage | 10.088 | 0.001 | |||
| LD | 66 | 24 | 42 | ||
| ED | 20 | 0 | 20 | ||
| Status | 24.629 | < 0.001 | |||
| Survival | 20 | 19 | 1 | ||
| Death | 66 | 21 | 45 | ||
Significant differences are shown in bold.
miR-495 was involved in the chemoresistance of SCLC through inhibiting apoptosis induced by chemotherapeutic drugs
The sensitivity of the H69/AR cells to anti-cancer drugs was greatly increased following transfection with the miR-495 mimics as described previously [9]. To better understand the roles of miR-495 in the drug sensitivity of SCLC cells, both H69/AR and H446/AR cells were used to quantify the expression of miR-495 by qRT-PCR. The H69/AR and H446/AR cells presented a lower level of miR-495 than their corresponding control groups (Figure 2A). Next, the miR-495 expression was down-regulated in H69 and H446 cells by antagomir (Figure 2B) and was up-regulated in H69/AR and H446/AR cells by miR-495 mimics (Figure 2C). Furthermore, the effect of miR-495 on the drug sensitivity of SCLC cells was analyzed. We found that H69 and H446 cells treated with miR-495 antagomir exhibited higher IC50 values to chemotherapeutic drugs, including ADM, DDP and VP-16. In contrast, the IC50 values were obviously decreased in H69/AR and H446/AR cells by miR-495 mimics (Table 3). These findings demonstrated that miR-495 was related to the chemoresistance of SCLC cells.
Figure 2.

miR-495 affected the chemoresistance and apoptosis induced by chemotherapeutic drugs of SCLC cells. A: Expression of miR-495 in SCLC drug-resistant and -sensitive cells. B: miR-495 expression was down-regulated in H69 and H446 cells by antagomir. C: miR-495 expression was up-regulated in H69/AR and H446/AR cells by miR-495 mimics. D: The apoptotic rate was analyzed by flow cytometry following treatment of the cells with chemotherapeutic drugs (ADM) by flow cytometry.
Table 3.
Effect of miR-495 on drug sensitivity (X̅±s, n = 3, µg/ml)
| Cells | IC50 | ||
|---|---|---|---|
|
|
|||
| ADM | DDP | VP-16 | |
| H69 | 7.61±0.81 | 18.96±1.00 | 53.14±1.70 |
| H69/NC | 8.03±1.20 | 18.37±1.62 | 51.58±1.08 |
| H69/antagomir | 91.99±3.90* | 114.38±4.54* | 176.01±5.04* |
| H446 | 6.77±0.98 | 23.37±1.26 | 58.26±2.84 |
| H446/NC | 6.8±1.48 | 23.16±1.57 | 57.75±2.83 |
| H446/antagomir | 81.74±2.11* | 127.73±3.62* | 189.09±4.61* |
| H69/AR | 150.28±6.02 | 155.50±11.51 | 216.76±4.29 |
| H69/AR/NC | 149.89±4.88 | 154.57±12.82 | 217.97±6.50 |
| H69/AR/mimics | 38.38±2.81* | 51.85±3.67* | 83.33±4.04* |
| H446/AR | 135.25±2.52 | 130.10±4.62 | 219.11±4.01 |
| H446/AR/NC | 135.07±3.63 | 130.47±3.84 | 218.57±3.52 |
| H446/AR/mimics | 49.63±3.25* | 39.81±6.39* | 85.66±3.948* |
P < 0.05 (compared with corresponding control groups).
Subsequently, the apoptotic rate was analyzed following treatment of the cells with chemotherapeutic drugs, including ADM, DDP and VP-16, using flow cytometry. Cell apoptosis was significantly suppressed in H69/antagomir and H446/antagomir cells. However, it was obviously increased in H69/AR mimic and H446/AR mimic cells (Figure 2D and Supplementary Figure 2). These data provided strong evidence that miR-495 was involved in the chemoresistance of SCLC by inhibiting apoptosis induced by chemotherapeutic drugs.
miR-495 promoted cancer cell proliferation, migration, invasion, and tumor growth and shortened the survival time
To investigate the biological roles of miR-495 in SCLC cells, cell proliferation was assessed with the CCK-8 assay. Compared with the results from the control cells, H69/antagomir and H446/antagomir cells presented a significant increase in the cell growth rates (Figure 3A), while H69/AR mimic and H446/AR mimic cells showed obviously decreased cell growth rates (Figure 3B). We also detected the effects of miR-495 on migration and invasion ability using the scratch-wound healing and transwell-invasion assay. As shown in Figure 3C and 3D, miR-495 over-expression inhibited the healing rates of H446/AR and H446/AR cells (Figure 3C). However, knockdown of miR-495 in H446 cells enhanced the healing rates (Figure 3D). For the transwell-invasion assay, more H446/antagomir cells penetrated the Matrigel-coated membrane than the control H446 cells. By contrast, H69/AR mimic and H446/AR mimic cells showed a notably reduced number of cells penetrating the Matrigel-coated membrane (Figure 3E).
Figure 3.
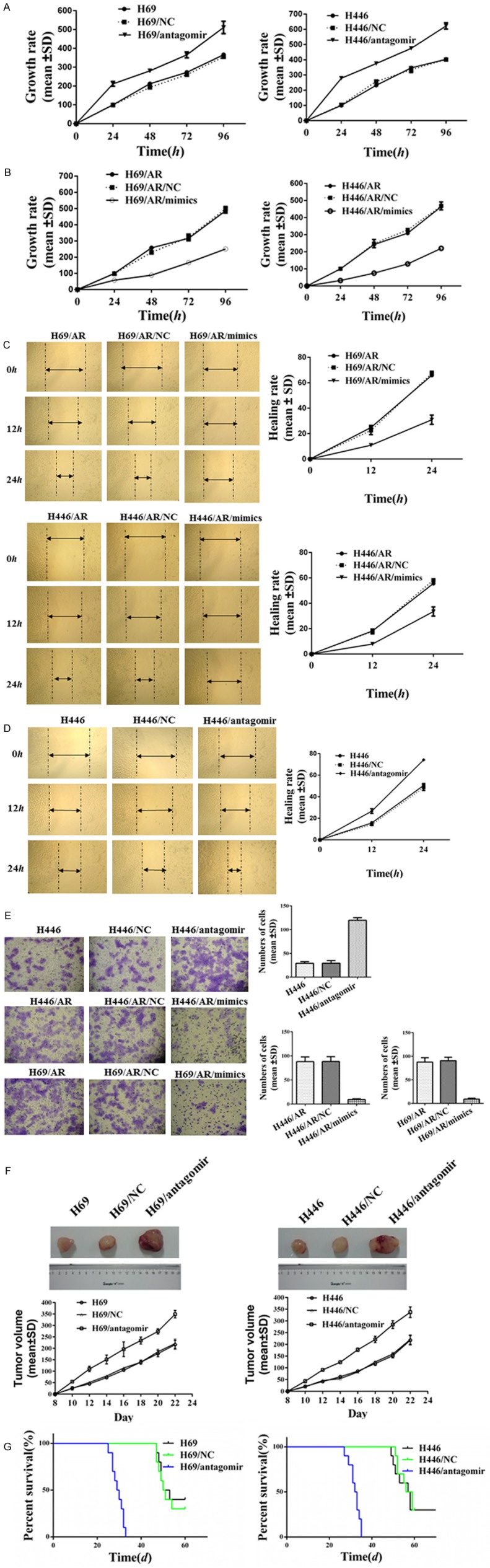
miR-495 influenced cancer cell proliferation, migration, invasion, tumor growth and survival time. A: The effect of miR-495 suppression on cell growth rates in H69 and H446 was assessed with the CCK-8 assay. B: Elevation of miR-495 in H69/AR and H446/AR affected the cell growth rates. C, D: The regulation of miR-495 in the migration ability of SCLC cells was evaluated with the scratch-wound healing assay. E: The transwell-invasion assay was performed to detect the invasion ability induced by miR-495. F, G: The regulation of miR-495 suppression in the tumor growth and survival of the mice was studied.
The tumorigenic properties of miR-495 in vivo were investigated in nude mice xenografts. The nude mice were subcutaneously inoculated with miR-495 down-expressing SCLC cells. The tumors of H69/antagomir and H446/antagomir cells grew significantly rapidly compared with those of H69 and H446 cells (Figure 3F). The null mice presented a shorter survival time accompanied by the suppression of miR-495 (Figure 3G).
Taken together, both loss- and gain-of-function experiments demonstrated that the down-regulation of miR-495 promoted SCLC proliferation, migration, invasion and tumor growth but shortened the survival time.
Down-regulation of the EMT-related inducer Zeb-2 or Twist inhibited the chemoresistance of SCLC
Our previous studies had shown that EMT-related markers such as the (epithelial molecule β-catenin and mesenchymal markers such as Zeb-2, Twist, and Vim were expressed at significantly different levels between H69 and H69/AR cells by cDNA microarray (Supplementary Figure 1). In the current study, we further examined the expression of the related markers in H69/AR and H446/AR cells at the mRNA and protein levels. The results were consistent with the data from cDNA microarray. The expression of Zeb-2, Twist, and Vim was higher in H69/AR and H446/AR cells than in H69 and H446 cells, while β-catenin showed an opposite trend (Figure 4A, 4B). Zeb-2 and Twist have been documented as important transcriptional repressors of EMT [21]. Knockdown of Zeb-2 and Twist can cause a reversal of EMT [28,29]. Next, Zeb-2 and Twist expression levels were inhibited with Zeb-2 siRNA (Figure 4C, 4D) and Twist siRNA, respectively (Figure 4E, 4F), in H69/AR and H446/AR cells. The expression of Vim was subsequently suppressed while that of β-catenin was increased accompanied by decreased Zeb-2 and Twist. The results indicated that the down-regulation of Zeb-2 or Twist reversed the EMT process. We further investigated whether reversing EMT can influence the drug sensitivity of SCLC cells. The results showed that the IC50 values of cells with down-expression of Zeb-2 and Twist were notably reduced when cells were exposed to chemotherapeutic drugs, including ADM, DDP and VP-16 (Table 4). The results suggested that the drug resistance to chemotherapeutic drugs was inhibited by reversing EMT in SCLC.
Figure 4.

Expression of Zeb-2, Twist, Vim and β-catenin at the mRNA and protein levels after H69/AR and H446/AR cells were treated with Zeb-2 siRNA and Twist siRNA. A, B: The different expression levels of mRNA and protein of Etk/BMX, Zeb-2, Twist, Vim and β-catenin in SCLC cells. C: Down-regulation of Zeb-2 by siRNA in H69/AR influenced the expression of Vim and β-catenin. D: Suppression of Zeb-2 by siRNA in H446/AR cells regulated the expression of Vim and β-catenin. E: Decreased Twist by siRNA in H69/AR modulated the expression of Vim and β-catenin. F: Inhibition of Twist by siRNA in H446/AR cells affected the expression of Vim and β-catenin.
Table 4.
Effect of Zeb-2 and Twist on drug sensitivity (X̅±s, n = 3, µg/ml)
| Cells | IC50 | ||
|---|---|---|---|
|
|
|||
| ADM | DDP | VP-16 | |
| H69/AR | 148.57±7.80 | 155.48±12.49 | 217.39±4.43 |
| H69/AR/NC | 147.17±3.85 | 155.02±12.46 | 216.95±5.63 |
| H69/AR/Zeb-2 si1 | 55.47±4.91* | 51.13±2.01* | 98.63±2.77* |
| H69/AR/Zeb-2 si2 | 21.32±1.67* | 19.66±2.53* | 35.06±3.53* |
| H446/AR | 134.88±3.78 | 129.09±3.38 | 220.34±8.29 |
| H446/AR/NC | 133.74±5.94 | 129.81±2.26 | 219.11±8.55 |
| H446/AR/Zeb-2 si1 | 41.21±4.62* | 38.60±1.29* | 100.46±5.56* |
| H446/AR/Zeb-2 si2 | 18.17±3.14* | 21.12±2.17* | 36.55±3.13* |
| H69/AR | 152.03±6.93 | 162.73±2.41 | 217.28±5.07 |
| H69/AR/NC | 152.17±9.03 | 160.68±2.96 | 216.70±5.51 |
| H69/AR/Twist si1 | 52.07±2.57* | 51.75±2.92* | 97.25±4.18* |
| H69/AR/Twist si2 | 24.07±0.93* | 22.30±2.88* | 33.06±5.24* |
| H446/AR | 131.58±3.37 | 135.68±1.61 | 215.14±4.40 |
| H446/AR/NC | 132.11±3.77 | 135.18±1.71 | 215.88±4.63 |
| H446/AR/Twist si1 | 44.59±3.18* | 40.67±3.54* | 97.31±2.52* |
| H446/AR/Twist si2 | 18.07±2.77* | 25.63±3.85* | 43.42±4.19* |
P < 0.05 (compared with corresponding control groups).
Etk/BMX contributed to decreased drug resistance by reversing the EMT of SCLC
The drug-resistant H69/AR and H446/AR cells showed greatly elevated mRNA and protein expression of Etk/Bmx compared with their parental cells by qRT-PCR and Western blot assay (Figure 4A). To further study the role of Etk/Bmx in SCLC cells, we developed Etk/Bmx stably over-expressed cells (H69/Etk and H446/Etk) (Figure 5A, 5B) and down-expressed cells (H69/AR/sh and H446/AR/sh) (Figure 5C, 5D). Next, we analyzed the viabilities of SCLC cells when exposed to chemotherapeutic drugs. The IC50 values with chemotherapeutic drugs were greatly decreased in H69/AR/sh and H446/AR/sh cells but were notably increased in H69/Etk and H446/Etk cells (Table 5). These data indicated that Etk/Bmx modulated SCLC cells to chemotherapeutic drugs.
Figure 5.
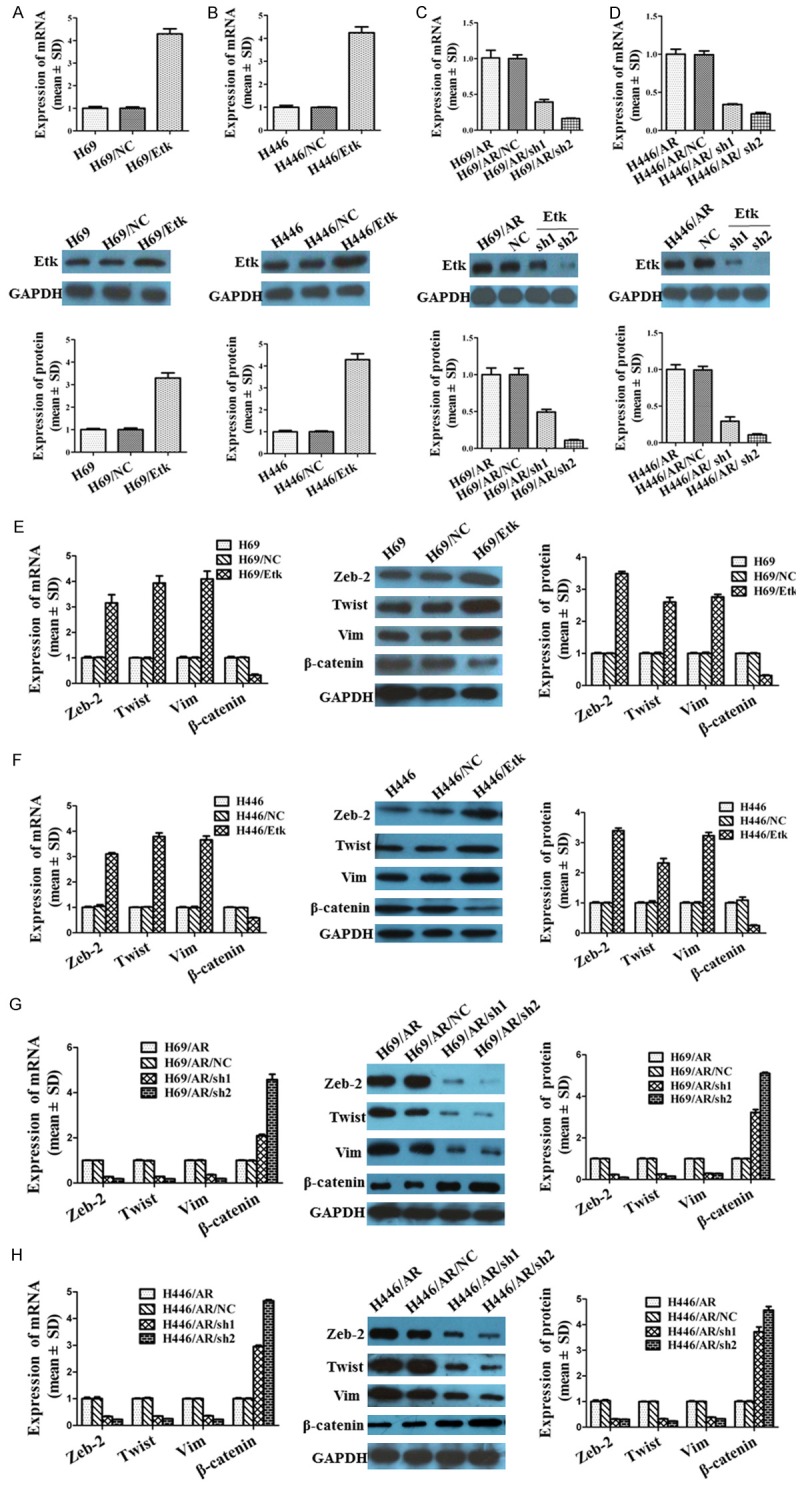
Up- or down-regulation of Etk/Bmx affected the expression of Zeb-2, Twist, Vim and β-catenin. A, B: The H69/Etk and H446/Etk cells stably expressing high Etk/Bmx were developed. C, D: The down-expression of Etk/Bmx cells (H69/AR/sh including H69/AR/sh1, H69/AR/sh2; H446/AR/sh including H446/AR/sh1 and H446/AR/sh2) were developed. E, F: Elevation of Etk/Bmx regulated the mRNA and protein expression of EMT related markers in H69 and H446 cells. G, H: Inhibition of Etk/Bmx influenced the mRNA and protein of EMT related markers in H69/AR and H446/AR cells.
Table 5.
Effect of Etk/BMX on drug sensitivity (X̅±s, n = 3, µg/ml)
| Cells | IC50 | ||
|---|---|---|---|
|
|
|||
| ADM | DDP | VP-16 | |
| H69 | 7.93±1.22 | 17.03±1.96 | 54.00±3.58 |
| H69/NC | 7.89±1.49 | 17.73±2.71 | 53.82±4.85 |
| H69/Etk | 85.37±5.75* | 100.14±4.17* | 190.28±6.56* |
| H446 | 6.65±1.07 | 21.87±1.47 | 58.54±2.31 |
| H446/NC | 6.63±1.55 | 21.50±1.64 | 58.84±3.56 |
| H446/Etk | 72.39±3.74* | 107.97±2.24* | 204.80±4.58* |
| H69/AR | 146.93±3.86 | 154.45±7.04 | 214.4±5.65 |
| H69/AR/NC | 149.26±5.76 | 155.53±4.44 | 213.34±6.27 |
| H69/AR/sh1 | 57.48±2.40* | 59.14±3.97* | 108.18±2.35* |
| H69/AR/sh2 | 26.33±2.57* | 27.34±1.48* | 45.38±3.62* |
| H446/AR | 135.95±5.40 | 133.03±3.95 | 207.62±10.08 |
| H446/AR/NC | 136.46±5.89 | 132.21±2.81 | 209.39±9.76 |
| H446/AR/sh1 | 45.27±4.70* | 45.57±2.44* | 102.22±2.79* |
| H446/AR/sh2 | 24.13±1.51* | 22.31±1.18* | 41.95±1.68* |
P < 0.05 (compared with corresponding control groups).
As noted above, the EMT process participated in the regulation of the drug resistance of SCLC. We further studied whether Etk/BMX modulated the sensitivity of SCLC cells to chemotherapeutic drugs through EMT. The expression of Zeb-2, Twist, Vim and β-catenin was evaluated after up- or down-regulation of Etk/BMX. The results revealed that the expression of Zeb-2, Twist and Vim at the mRNA and protein levels was remarkably increased, while β-catenin was inhibited by the over-expression of Etk/BMX in H69 (Figure 5E) and H446 cells (Figure 5F). In contrast, the suppression of Etk/BMX reduced the expression of Zeb-2, Twist and Vim but promoted the expression of β-catenin in H69/AR (Figure 5G) and H446/AR cells (Figure 5H). Our observations provided valid evidence that Etk/BMX may influence SCLC chemoresistance through the EMT process.
miR-495 modulates chemoresistance by targeting Etk/BMX
Considering Etk/BMX as a potential target gene of miR-495 by bioinformatics analysis (Figure 6G), we examined Etk/BMX expression after the modulation of miR-495 by qRT-PCR and western blotting. The results showed that the Etk/BMX mRNA and protein expression levels were elevated in H69 (Figure 6A) and H446 cells (Figure 6B) by the inhibition of miR-495 but were decreased in H69/AR (Figure 6C) and H446/AR (Figure 6D) cells as a result of the up-regulation of miR-495. We also analyzed the expression of the Etk/BMX and EMT markers in above-noted xenograft tumors. The results were consistent with the data from the cell lines. As shown in Figure 6, in the H69/antagomir (Figure 6E) and H446/antagomir (Figure 6F) xenografts, the expression of Etk/BMX, Zeb-2, Twist and Vim was lower than that in the control groups, but the opposite was true in the H69/AR/mimics (Figure 6G) and H446/AR/mimics groups (Figure 6H).
Figure 6.
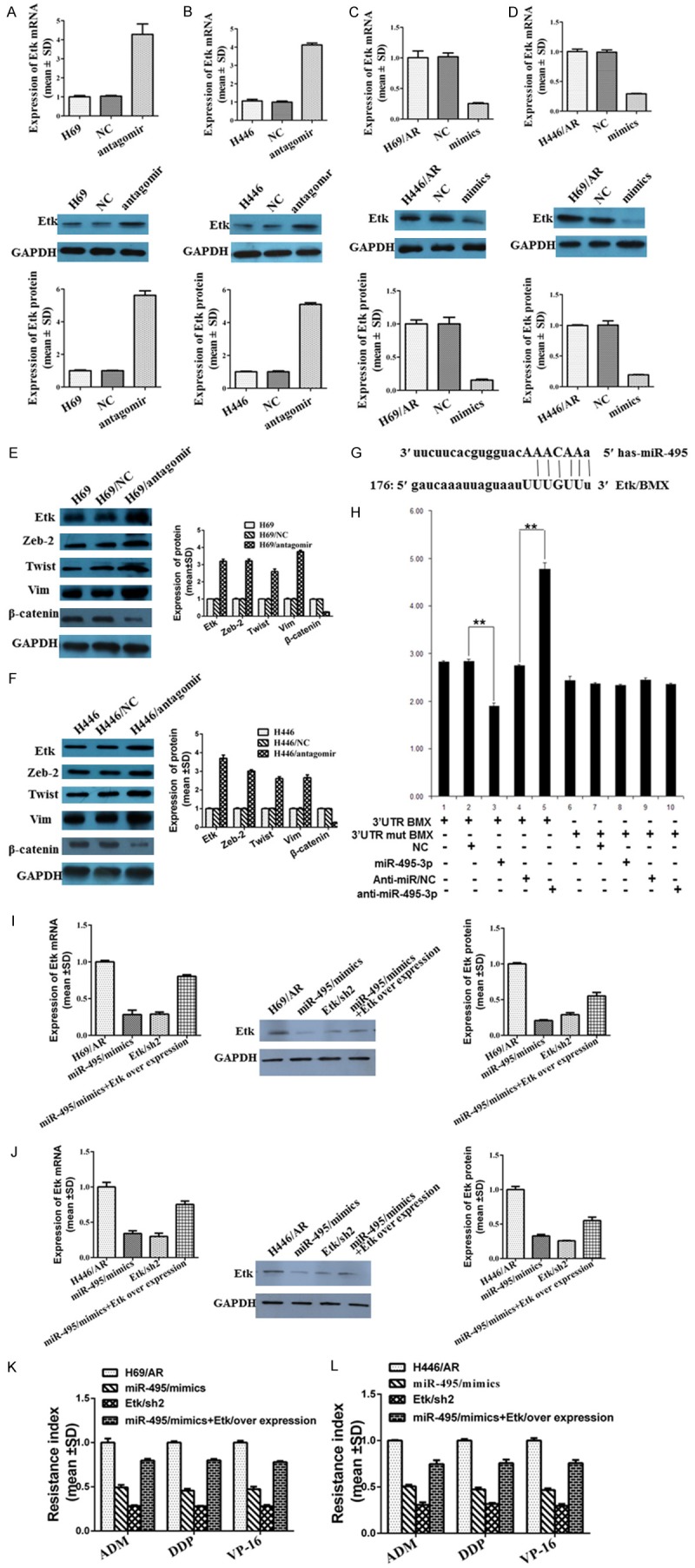
miR-495 modulated chemoresistance by targeting Etk/BMX. A, B: miR-495 antagomir affected the mRNA and protein expression of Etk/BMX in H69 and H446 cells. C, D: The expression of Etk/BMX mRNA and protein was regulated by miR-495 mimics in H69/AR and H446/AR cells. E, F: The regulation of miR-495 antagomir regarding the Etk/BMX and EMT markers was analyzed in the xenograft tumors noted above, and the results were consistent with the data from the cell lines. G, H: Whether Etk/BMX was one of the directly targeted genes of miR-495 was detected by bioinformatics analysis and the luciferase reporter assay. I: H69/AR cells were transfected with miR-495 mimics and the plasmid encoding the full-length Etk. J: H446/AR cells were transfected with miR-495 mimics and the plasmid encoding full-length Etk. K: The resistance index was detected when H69/AR cells were transfected with miR-495 mimics and the plasmid encoding the full-length Etk. L: The resistance index was detected when H446/AR cells were transfected with miR-495 mimics and plasmid encoding full-length Etk.
The correlation between miR-495 and Etk/BMX was further detected by luciferase reporter assay. The target sequence of Etk/BMX 3’UTR (3’UTR wt BMX) or the mutant sequence (3’UTR mut BMX) was cloned into a luciferase reporter vector. H69/AR cells were then transfected with wt or mut BMX vector and miR-495 mimics or inhibitor. The results revealed that the Etk/BMX luciferase activity was significantly decreased in H69/AR cells treated with miR-495 mimics and 3’UTR wt BMX vector (Figure 6H, lanes 2 and 3; P < 0.001) but was obviously elevated by treatment with the miR-495 inhibitor and 3’UTR wt BMX vector (Figure 6H, lanes 4 and 5; P < 0.001). The Etk/BMX luciferase activity was not affected by miR-495 mimics or the miR-495 inhibitor when the cells were treated with 3’UTR mut BMX vector (Figure 6H, lanes 6-9).
Subsequently, we evaluated whether the over-expression of Etk/BMX could rescue the suppressed drug resistance induced by miR-495 mimics. H69/AR and H446/AR cells were transfected with miR-495 mimics and the plasmid encoding the full-length Etk (Figure 6I, 6J). Ectopic expression of Etk/BMX obviously rescued miR-495 mimics-induced inhibition of drug resistance (Figure 6K, 6L). Thus, all these results suggested that the influence of miR-495 on SCLC chemoresistance may occur through Etk/BMX.
Discussion
Our previous studies had shown that the suppression of Etk/BMX expression reduced the chemoresistance of H69/AR cells [30]. In this study, we further confirmed the effect of Etk/BMX on the chemoresistance of SCLC using two multi-drug-resistant H69/AR and H446/AR cell lines. The H69/AR/sh and H446/AR/sh cells showed much greater sensitivity to chemotherapeutic drugs. In contrast, the over-expression of Etk/BMX in H69 and H446 cells reduced the drug sensitivity. Thus, we provided more evidence to demonstrate that the suppression of Etk/Bmx decreased the SCLC drug resistance.
We have first reported that the transfection of the H69/AR cells with the miR-495 mimics significantly reduced chemoresistance [31]. In the present study, two drug-resistant cell lines were used to further investigate the role of miR-495 in the drug sensitivity of SCLC. Up- or down-regulation of miR-495 can modulate IC50 values to chemotherapeutic drugs, indicating that miR-495 was involved in the chemoresistance of SCLC cells. In addition, our research revealed that the cell apoptosis induced by chemotherapeutic drugs was significantly decreased after the down-regulation of miR-495, but it was increased obviously with the elevation of miR-495. There was a positive correlation between miR-495 and cell apoptosis. The elevation of miR-495 resulted in the decreased expression of Etk/BMX. By contrast, Etk/BMX was over-expressed following the reduced expression of miR-495. The result from the luciferase reporter assay confirmed that Etk/BMX was one of the directly targeted genes of miR-495. The over-expression of Etk/BMX could rescue the suppressed drug resistance induced by miR-495 mimics. These results provided sufficient proof to demonstrate that miR-495 affected the chemoresistance of SCLC by regulating Etk/Bmx.
It has been reported that miR-495 is associated with various cellular processes. Studies have shown that miR-495 serves as a tumor suppressor or an oncogene in multi-models of cancer. In this study, we investigated the influence of miR-495 on the biological behaviors of SCLC by loss- and gain-of-function experiments. The proliferation, migration and invasion abilities were increased after the suppression of miR-495, while they were all decreased following the up-regulation of Etk/BMX. The in vivo data revealed that the down-regulation of miR-495 can inhibit tumor growth and shorten the survival time. By clinical samples, we observed that the expression of miR-495 showed a close relationship with poor pathologic stage and survival time. In summary, our findings indicated that miR-495 played an important role in the biological behaviors of SCLC, suggesting that miR-495 may serve as a predictor for the poor prognosis in SCLC.
Studies have shown that the EMT process confers resistance to chemotherapy in ovarian carcinoma, colorectal cancer and other cancers [32-34]. Based on our previous study, the role of EMT in SCLC chemoresistance was first clarified in this study. The inhibition of Zeb-2 and Twist in H69/AR and H446/AR cells led to declined Vim but elevated β-catenin. The results indicated that the suppression of Zeb-2 and Twist can reverse the EMT process. Furthermore, the IC50 values for treatment with chemotherapeutic drugs were suppressed after the knockdown of Zeb-2 and Twist. These findings suggested that EMT was associated with the chemoresistance of SCLC. Subsequently, the relationship between Etk/BMX and EMT was explored. The H69/AR/sh and H446/AR/sh cells exhibited a significant decrease in the expression of mesenchymal markers but an increase in epithelial markers. However, the H69 and H446 cells presented opposing results by the elevation of Etk/BMX. The expression of EMT markers noted above in xenograft tumors was consistent with the data from the cell lines. Taken together, Etk/BMX modulated the chemoresistance of SCLC possibly through the EMT process.
Taken together, our study supported, for the first time, that the elevation of miR-495 can decrease the drug resistance of SCLC by the regulation of Etk/Bmx through the EMT process. Therefore, our research provides new insight into the mechanism of SCLC chemoresistance. miR-495 or Etk/Bmx can be a useful predictor for drug sensitivity in SCLC, raising the possibility of miR-495 re-expression or Etk/BMX depletion as a promising strategy for interfering with chemoresistance in SCLC.
Acknowledgements
This paper was supported by the National Natural Science Foundation of China (81172241) and (81302031); the National Natural Science Foundation of Guangdong Province, China (S2011040003563).
Disclosure of conflict of interest
None.
Authors’ contribution
Linlang Guo and Jian Zhang designed this study. Ting Wei and Weilliang Zhu performed most of the experiments. The flow cytometry assay for cell apoptosis in Figure 2C and Supplementary Figure 2 was performed by Shun Fang. The drug resistance assay in Table 4 was performed by Xiangpin Zeng. The cell scratch-wound healing assay was performed by Jie Huang. The cell invasion assay was performed by Jie Yang. Experiments were performed under the supervision of Linlang Guo and Jian Zhang. Linlang Guo and Ting Wei wrote the manuscript.
Supporting Information
References
- 1.Govindan R, Page N, Morgensztern D, Read W, Tierney R, Vlahiotis A, Spitznagel EL, Piccirillo J. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J. Clin. Oncol. 2006;24:4539–4544. doi: 10.1200/JCO.2005.04.4859. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 3.Samson DJ, Seidenfeld J, Simon GR, Turrisi AT 3rd, Bonnell C, Ziegler KM, Aronson N American College of Chest Physicians. Evidence for management of small cell lung cancer: ACCP evidence-based clinical practice guidelines (2nd edition) Chest. 2007;132:314S–323S. doi: 10.1378/chest.07-1384. [DOI] [PubMed] [Google Scholar]
- 4.Sorensen M, Felip E ESMO Guidelines Working Group. Small-cell lung cancer: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20(Suppl 4):71–72. doi: 10.1093/annonc/mdp133. [DOI] [PubMed] [Google Scholar]
- 5.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 6.Sun L, Yao Y, Liu B, Lin Z, Lin L, Yang M, Zhang W, Chen W, Pan C, Liu Q, Song E, Li J. MiR-200b and miR-15b regulate chemotherapy-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene. 2012;31:432–445. doi: 10.1038/onc.2011.263. [DOI] [PubMed] [Google Scholar]
- 7.Yang H, Kong W, He L, Zhao JJ, O’Donnell JD, Wang J, Wenham RM, Coppola D, Kruk PA, Nicosia SV, Cheng JQ. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008;68:425–433. doi: 10.1158/0008-5472.CAN-07-2488. [DOI] [PubMed] [Google Scholar]
- 8.Xia H, Ooi LL, Hui KM. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013;58:629–641. doi: 10.1002/hep.26369. [DOI] [PubMed] [Google Scholar]
- 9.Guo LL, Liu YG, Bai YF, Sun YQ, Xiao F, Guo Y. Gene expression profiling of drug-resistant small cell lung cancer cells by combining microRNA and cDNA expression analysis. Eur J Cancer. 2010;46:1692–1702. doi: 10.1016/j.ejca.2010.02.043. [DOI] [PubMed] [Google Scholar]
- 10.Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang X, Huang H, Li Z, He C, Li Y, Chen P, Gurbuxani S, Arnovitz S, Hong GM, Price C, Ren H, Kunjamma RB, Neilly MB, Salat J, Wunderlich M, Slany RK, Zhang Y, Larson RA, Le Beau MM, Mulloy JC, Rowley JD, Chen J. MiR-495 is a tumor-suppressor microRNA down-regulated in MLL-rearranged leukemia. Proc Natl Acad Sci U S A. 2012;109:19397–19402. doi: 10.1073/pnas.1217519109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao M, Nie W, Li J, Zhang Y, Yan X, Guan X, Chen X, Zen K, Zhang CY, Jiang X, Hou D. MicroRNA-495 induces breast cancer cell migration by targeting JAM-A. Protein Cell. 2014;5:862–872. doi: 10.1007/s13238-014-0088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai B, Chen H, Guo S, Yang X, Linn DE, Sun F, Li W, Guo Z, Xu K, Kim O, Kong X, Melamed J, Qiu S, Chen H, Qiu Y. Compensatory upregulation of tyrosine kinase Etk/BMX in response to androgen deprivation promotes castration-resistant growth of prostate cancer cells. Cancer Res. 2010;70:5587–5596. doi: 10.1158/0008-5472.CAN-09-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Semaan N, Alsaleh G, Gottenberg JE, Wachsmann D, Sibilia J. Etk/BMX, a Btk family tyrosine kinase, and Mal contribute to the cross-talk between MyD88 and FAK pathways. J Immunol. 2008;180:3485–3491. doi: 10.4049/jimmunol.180.5.3485. [DOI] [PubMed] [Google Scholar]
- 15.Qiu Y, Kung HJ. Signaling network of the Btk family kinases. Oncogene. 2000;19:5651–5661. doi: 10.1038/sj.onc.1203958. [DOI] [PubMed] [Google Scholar]
- 16.Salzer GM, Muller LC, Huber H. Further evidence of the benefits of surgery in the combined management of small cell lung cancer. Thorac Cardiovasc Surg. 1988;36:161. doi: 10.1055/s-2007-1020067. [DOI] [PubMed] [Google Scholar]
- 17.Paz K, Brennan LA, Iacolina M, Doody J, Hadari YR, Zhu Z. Human single-domain neutralizing intrabodies directed against Etk kinase: a novel approach to impair cellular transformation. Mol Cancer Ther. 2005;4:1801–1809. doi: 10.1158/1535-7163.MCT-05-0174. [DOI] [PubMed] [Google Scholar]
- 18.Abassi YA, Rehn M, Ekman N, Alitalo K, Vuori K. p130Cas Couples the tyrosine kinase Bmx/Etk with regulation of the actin cytoskeleton and cell migration. J Biol Chem. 2003;278:35636–35643. doi: 10.1074/jbc.M306438200. [DOI] [PubMed] [Google Scholar]
- 19.Chau CH, Chen KY, Deng HT, Kim KJ, Hosoya K, Terasaki T, Shih HM, Ann DK. Coordinating Etk/Bmx activation and VEGF upregulation to promote cell survival and proliferation. Oncogene. 2002;21:8817–8829. doi: 10.1038/sj.onc.1206032. [DOI] [PubMed] [Google Scholar]
- 20.Guo LL, Zhou YY, Sun YQ, Zhang F. Non-receptor tyrosine kinase Etk regulation of drug resistance in small-cell lung cancer. Eur J Cancer. 2010;46:636–641. doi: 10.1016/j.ejca.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 21.McConkey DJ, Choi W, Marquis L, Martin F, Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, Siefker-Radtke A, Dinney C. Role of epithelial-to-mesenchymal transition (EMT) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev. 2009;28:335–344. doi: 10.1007/s10555-009-9194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hung JJ, Yang MH, Hsu HS, Hsu WH, Liu JS, Wu KJ. Prognostic significance of hypoxia-inducible factor-1alpha, TWIST1 and Snail expression in resectable non-small cell lung cancer. Thorax. 2009;64:1082–1089. doi: 10.1136/thx.2009.115691. [DOI] [PubMed] [Google Scholar]
- 23.Lin CY, Tsai PH, Kandaswami CC, Lee PP, Huang CJ, Hwang JJ, Lee MT. Matrix metalloproteinase-9 cooperates with transcription factor Snail to induce epithelial-mesenchymal transition. Cancer Sci. 2011;102:815–827. doi: 10.1111/j.1349-7006.2011.01861.x. [DOI] [PubMed] [Google Scholar]
- 24.Hugo HJ, Kokkinos MI, Blick T, Ackland ML, Thompson EW, Newgreen DF. Defining the E-cadherin repressor interactome in epithelial-mesenchymal transition: the PMC42 model as a case study. Cells Tissues Organs. 2011;193:23–40. doi: 10.1159/000320174. [DOI] [PubMed] [Google Scholar]
- 25.Zhou YY, Sun YY, Guo LL. Establishment of an adriamycin multi-drug resistant human small cell lung cancer cell line and its correlationship with the expression of Bcl-2 family proteins. Shandong Medical Journal. 2009;49:31–34. [Google Scholar]
- 26.Qiu Y, Robinson D, Pretlow TG, Kung HJ. Etk/Bmx, a tyrosine kinase with a pleckstrin-homology domain, is an effector of phosphatidylinositol 3’-kinase and is involved in interleukin 6-induced neuroendocrine differentiation of prostate cancer cells. Proc Natl Acad Sci U S A. 1998;95:3644–3649. doi: 10.1073/pnas.95.7.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo LL, Chen PL, Zhou YY, Sun YQ. Non-receptor tyrosine kinase Etk is involved in the apoptosis of small cell lung cancer cells. Exp Mol Pathol. 2010;88:401–406. doi: 10.1016/j.yexmp.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Zhuo WL, Wang Y, Zhuo XL, Zhang YS, Chen ZT. Short interfering RNA directed against TWIST, a novel zinc finger transcription factor, increases A549 cell sensitivity to cisplatin via MAPK/mitochondrial pathway. Biochem Biophys Res Commun. 2008;369:1098–1102. doi: 10.1016/j.bbrc.2008.02.143. [DOI] [PubMed] [Google Scholar]
- 29.Adam L, Zhong M, Choi W, Qi W, Nicoloso M, Arora A, Calin G, Wang H, Siefker-Radtke A, McConkey D, Bar-Eli M, Dinney C. miR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clin Cancer Res. 2009;15:5060–5072. doi: 10.1158/1078-0432.CCR-08-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo LL, Zhou YY, Sun YQ, Zhang F. Non-receptor tyrosine kinase Etk regulation of drug resistance in small-cell lung cancer. Eur J Cancer. 2010;46:636–641. doi: 10.1016/j.ejca.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Guo LL, Liu YG, Bai YF, Sun YQ, Xiao F, Guo Y. Gene expression profiling of drug-resistant small cell lung cancer cells by combining microRNA and cDNA expression analysis. Eur J Cancer. 2010;46:1692–1702. doi: 10.1016/j.ejca.2010.02.043. [DOI] [PubMed] [Google Scholar]
- 32.Rosano L, Cianfrocca R, Spinella F, Di Castro V, Nicotra MR, Lucidi A, Ferrandina G, Natali PG, Bagnato A. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin a receptor pathway in ovarian carcinoma cells. Clin Cancer Res. 2011;17:2350–2360. doi: 10.1158/1078-0432.CCR-10-2325. [DOI] [PubMed] [Google Scholar]
- 33.Hoshino H, Miyoshi N, Nagai K, Tomimaru Y, Nagano H, Sekimoto M, Doki Y, Mori M, Ishii H. Epithelial-mesenchymal transition with expression of SNAI1-induced chemoresistance in colorectal cancer. Biochem Biophys Res Commun. 2009;390:1061–1065. doi: 10.1016/j.bbrc.2009.10.117. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Lingala S, Khoobyari S, Nolta J, Zern MA, Wu J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J Hepatol. 2011;55:838–845. doi: 10.1016/j.jhep.2010.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.