

Abstract
Background
Duchenne muscular dystrophy (DMD) is caused by mutations in the DMD gene, which codes for the dystrophin protein. While progress has been made in defining the molecular basis and pathogenesis of DMD, major gaps remain in understanding mechanisms that contribute to the marked delay in cardiac compared to skeletal muscle dysfunction.
Methods
To address this question, we analyzed cardiac and skeletal muscle tissue microarrays from golden retriever muscular dystrophy (GRMD) dogs, a genetically and clinically homologous model for DMD. A total of 15 dogs, 3 each GRMD and controls at 6 and 12 months plus 3 older (47–93 months) GRMD dogs, were assessed.
Results
GRMD dogs exhibited tissue- and age-specific transcriptional profiles and enriched functions in skeletal but not cardiac muscle, consistent with a “metabolic crisis” seen with DMD microarray studies. Most notably, dozens of energy production-associated molecules, including all of the TCA cycle enzymes and multiple electron transport components, were down regulated. Glycolytic and glycolysis shunt pathway-associated enzymes, such as those of the anabolic pentose phosphate pathway, were also altered, in keeping with gene expression in other forms of muscle atrophy. On the other hand, GRMD cardiac muscle genes were enriched in nucleotide metabolism and pathways that are critical for neuromuscular junction maintenance, synaptic function and conduction.
Conclusions
These findings suggest differential metabolic dysfunction may contribute to distinct pathological phenotypes in skeletal and cardiac muscle.
Electronic supplementary material
The online version of this article (doi:10.1186/s12920-017-0257-2) contains supplementary material, which is available to authorized users.
Keywords: Duchenne, BDNF, Muscular dystrophy, Cardiac, Dystrophin, Metabolism
Background
Duchenne muscular dystrophy (DMD) and the genetically homologous mdx mouse and golden retriever muscular dystrophy (GRMD) dog models are caused by mutations in the DMD gene, resulting in severely reduced or absent dystrophin protein [1–5]. Despite being genetically homologous, the three diseases demonstrate distinct phenotypes, with DMD and GRMD being more severe [4, 6]. Gene expression in mdx mice and DMD patients is also distinctive. By 16 weeks of age, the mdx transcriptome is relatively quiescent [7], while DMD expression profiles demonstrate a so-called “metabolic crisis” [8, 9]. Meta-analysis of gene expression datasets from six different studies of DMD skeletal muscle biopsies provide especially strong evidence that thematic metabolic disturbances are pathophysiologically relevant [10] and likely contribute to muscle atrophy [11]. In support of this concept, therapies that enhance metabolism, such as corticosteroids and coenzyme Q, are temporarily beneficial.
In contrast to the early progressive wasting seen with DMD skeletal muscle, the heart is relatively preserved, with onset of cardiomyopathy typically occurring in the second or third decade of life [12, 13]. How or why the heart is temporarily spared is not understood, and predictive markers for DMD cardiomyopathy are currently unavailable. As with skeletal muscle, the mdx and GRMD models are distinctive. As with DMD, GRMD dogs have progressive skeletal muscle weakness and late-onset cardiomyopathy [5, 14–16], while the mdx mouse exhibits subtle cardiac abnormalities [6]. We hypothesized that differences in cardiac and skeletal muscle metabolism contribute to this variable disease progression. To address this hypothesis, we studied a group of GRMD dogs that were previously shown to have altered expression of osteopontin (OPN) and brain-derived neurotropic factor (BDNF) in dystrophic skeletal and cardiac muscles, respectively [17]. These findings were subsequently translated to boys with DMD, demonstrating that circulating levels of OPN and BDNF correlated with skeletal muscle function or cardiac dysfunction [17]. Building on this work in the current study, we found strikingly different metabolic gene expression patterns in cardiac and skeletal tissue, providing further insight into potential molecular mechanisms underlying tissue-specific disease progression.
Methods
Animals
The study was approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill. All dogs were used and cared for according to principles outlined in the National Research Council Guide for the Care and Use of Laboratory Animals. The GRMD genotype was suspected based on elevated serum creatine kinase and confirmed by genotyping. Affected dogs subsequently developed characteristic clinical signs. Notably, the GRMD phenotype progresses dramatically over the 3 to 6 month age period and then tends to stabilize [18, 19]. Given the relative equivalency of the first year of a golden retriever’s life to the initial 20 years for a human [20], the 3 to 6 month period for GRMD corresponds to an analogous period of deterioration between ages 5 and 10 years in DMD [21–23]. A total of 15 dogs were included in the study: 3 each 6–7.5 and 12–13 month-old GRMD dogs and 6 age-matched (littermate) wild type controls, plus 3 older (47, 52, and 93 month-old) GRMD dogs. Samples from the medial head of the gastrocnemius (MHG) and left ventricular (LV) free wall were removed at necropsy and processed. Muscle sections were snap frozen in a Freon substitute, cooled in liquid nitrogen, stored at -80 °C and then shipped in cryovials to Vanderbilt for analysis.
Microarrays
Total RNA was isolated from the LV and MHG of the 15 dogs according to the manufacturer’s protocol (Qiagen, Germantown, MD). Quality assessment of RNA, further processing, and data acquisition were performed by the GSR Microarray Core at Vanderbilt. Affymetrix Canine Gene 1.0 Expression arrays (Affymetrix, Inc., Santa Clara, CA) were used, two arrays per animal (for paired LV and skeletal muscle samples), for a total of 30 arrays. Raw data were RMA normalized, followed by ANOVA with Benjamini & Hochberg correction, using Partek Genomics Suite 6.6 (Partek Incorporated, St. Louis, MO). Genes with a p value (with or without FDR) < 0.05 and fold-difference > 1.5 were considered significantly altered. Normalized and raw data generated from canine gene expression arrays were deposited under Accession Number GSE68626 in the Gene Expression Omnibus (GEO), available through NCBI (http://www.ncbi.nlm.nih.gov/geo/)GEO. Functional and pathway enrichment analyses were conducted using Ingenuity Pathway Analysis (IPA, Qiagen, Germantown, MD), Partek Genomics Suite 6.6 (Partek Incorporated, St. Louis, MO), DAVID Bioinformatics Resources 6.8 [24, 25],or Gene Set Enrichment Analysis (GSEA) desktop freeware version 2.2.3 (BROAD Institute). For GSEA using Partek software, normalized signal values were used. For GSEA using BROAD software, differentially expressed genes were first separated by directionality (up-regulated genes and down-regulated gene) and the subsequent lists ranked by p value, where ranked score = -log(p value).
Western blot analysis
Western blot analysis was performed as previously described [26]. Primary antibodies used were rabbit anti-AMPK (total and phosphorylated, Cell Signaling Technologies, Inc., Danvers, MA) and rabbit anti-p90 (Cell Signaling Technologies) as a loading control.
Statistics
All data are expressed as means ± SEM. Comparisons made between two variables were performed using a Student t test unless otherwise stated. P values of less than 0.05 were considered statistically significant. For gene microarray analysis, data were uploaded into Partek Genomics Suite version 6.6 (Partek Incorporated, St. Louis, MO) and RMA normalized before further analysis. Partek was used to perform pairwise comparisons of average group values and one-way ANOVA for the eight groups. Only probes that resulted in a fold-change of at least 1.5 and p value of less than 0.05 were considered significantly altered.
Results
GRMD gene expression findings: an overview
A total of 30 samples from 15 dogs were included in the study, divided according to disease-state (GRMD or wild type) and tissue type (skeletal [MHG] muscle or cardiac [LV] muscle). We focused primarily on 12 dogs at ~6 months or 12 months, for which there were a total of 8 groups with 3 biological replicates each. Based on a non-stringent and straightforward analysis of age-matched and wild type samples, we compiled a single list of 5,835 probes that were significantly differential (p value < 0.05, fold-difference > 1.5) between GRMD and wild type tissues for at least one of the four comparisons (Additional file 1: Table S1). An overview of statistical results is shown in Fig. 1e. The most immediately obvious distinction among the tissues was the overall difference in gene expression profiles between the 6 and 12-month GRMD age groups. Whereas there were relatively few expression differences between LV samples at these two ages when compared to wild type control animals, MHG samples from these same dogs exhibited dramatic differences (Fig. 1). This is consistent with the more dramatic progression of skeletal muscle disease up to 6 months of age with subsequent stabilization, while cardiac muscle function is typically normal over this period. Hierarchical clustering of all 5,835 probes also indicated that the global transcriptional profile of GRMD MHG at 6 months was unique and distinct from all other samples, including GRMD MHG at 12 months (Additional file 2: Figure S1).
Fig. 1.
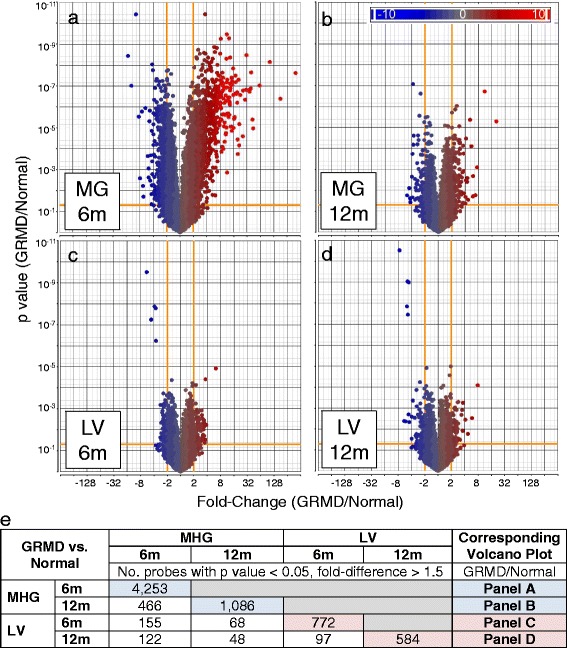
Volcano plots are shown highlighting gene expression differences between dystrophic medial head of the gastrocnemius (MHG) skeletal muscle of golden retriever muscular dystrophy (GRMD) dogs at 6 (a) and 12 (b) months (m), as well as GRMD left ventricle (LV) from 6 (c) and 12 (d) month-old dogs, each compared to their respective wild type controls. The y-axis displays p values, and fold-differences are plotted on the x-axis for GRMD versus control dogs for each of the four comparisons. Each circle represents an individual probe set (gene) and is colored based on fold-difference (blue for genes with lower expression in GRMD dogs, red for genes with higher expression values, and gray for no change), as shown in the legend. Orange lines mark significance cut-off values (p value < 0.05, fold-difference > 1.5). Panel (e) shows a matrix including the number (No.) of array signal intensity probes deemed as significantly differential (p value < 0.05, fold-difference > 1.5) between pairs of groups. Pairwise comparisons include GRMD versus wild-type (WT) dogs for matched skeletal muscle (MHG) and LV tissue of dogs aged 6 months and 12 months. Pairwise comparisons of the two age groups for each tissue are also shown
Similar to what has been reported elsewhere for DMD and animal models [27–29], up-regulated skeletal muscle genes included those associated with fibrosis (e.g., collagens, POSTN, TIMP1), inflammation (e.g., complement components, chemokines, cytokines), and muscle cytoskeletal proteins (e.g., myosin heavy chains, troponins, and actins). The most profoundly down-regulated genes were those associated with muscle growth and development (e.g., CLCN1, MYLK2, MYBPC2, and MSTN). There were surprisingly few genes (9.6%) that were differentially expressed between GRMD and wild type MHG tissue in both of the ages evaluated. The majority of these genes were altered more profoundly in the younger GRMD dogs relative to wild type controls. Embryonic and perinatal forms of myosin heavy chains (MYH3 and MYH8) for instance were up-regulated 180-fold and 61-fold in MHG at 6 months, compared to 6-fold and 7.4-fold in 12-month-old dogs, respectively. Up-regulated genes with a higher magnitude of increase in MHG from 12- versus 6- month-old GRMD dogs included those encoding adult myosin heavy chains, extracellular matrix components, parvalbumin, and β-1-syntrophin (SNTB1).
Cardiac expression differences included genes with disease-associated functions, such as muscular disorders, cardiovascular disease, and organismal injury. To identify non-directional (up or down) and non-age related (i.e., those altered at either age) functional patterns, we used Ingenuity Pathway Analysis software to analyze genes differentially expressed between GRMD versus control dogs. As shown in Additional file 3: Table S2, several over-represented functional categories were specific to genes altered in skeletal muscle (up- or down-regulated in GRMD versus wild type dogs at either age). For instance, multiple genes important for regulation of cell death (170 genes, p value = 0.0001) and membrane organization (85 genes, p value = 0.0041) were significantly perturbed in MHG but not LV tissues in GRMD. This functional signature supports the idea that tissue-dependent gene expression alterations drive differences in disease between the two muscle types. Genes important for metabolism and energy production were likewise restricted to skeletal muscle (Additional file 3: Table S2), consistent with the notion that a metabolic phenotype contributes to dystrophic skeletal muscle disease progression.
We also performed a more comprehensive gene set enrichment analysis (GSEA) of each relevant list of differential genes (age- and tissue-matched pairwise comparisons). The most significantly enriched biological processes for genes more highly expressed in 6 month GRMD MHG relative to controls included actin filament organization inflammation (36 genes, relative enrichment score (NES) = 1.9), ECM disassembly (20 genes, NES = 1.8), inflammation (100 genes, NES = 1.6, stem cell differentiation (24 genes, NES = 1.5), muscle structure development (51 genes, NES = 15), and angiogenesis (58 genes, NES = 1.4) (Table 1). Down-regulated genes (those more highly expressed in normal dogs compared to GRMD at 6 months) were almost exclusively associated with metabolism (Table 1), including oxidation reduction (98 genes, NES = -2.6), generation of precursor metabolites and energy (45 genes, NES = -2.5), cellular respiration (24 genes, NES = -2.4), mitochondrion organization (73 genes, NES = -2.4), and glucose metabolism (21 genes, NES = -2.1). For the most part, down-regulated functional categories were more significant (lower p value, higher magnitude of NES) than up-regulated ontologies. Most notably, however, there was a clear lack of “functional variety” in down-regulated genes (i.e., a catabolic theme) that was not observed for genes more highly expressed in MHG of 6 month-old GRMD (Table 1).
Table 1.
Functional analysis results of GRMD-induced gene alterations
| GO Biological Process/Pathway | MHG at 6 m | LV at 6 m | ||||
|---|---|---|---|---|---|---|
| No | NES | p val | No | NES | p val | |
| Actin filament organization | 36 | 1.91 | 0.001 | - | ||
| Regulation of extrinsic apoptotic pathway | 37 | 1.70 | 0.001 | - | ||
| Regulation of cytoskeleton organization | 31 | 1.79 | 0.0021 | - | ||
| ECM disassembly | 20 | 1.78 | 0.0064 | - | ||
| Endothelial cell differentiation | 15 | 1.77 | 0.0078 | - | ||
| Regulation of actin filament organization | 57 | 1.64 | 0.0081 | - | ||
| Protein localization to organelle | 55 | 1.58 | 0.006 | - | ||
| Inflammatory response | 100 | 1.55 | 0.002 | - | ||
| Stem cell differentiation | 24 | 1.54 | 0.0305 | - | ||
| Positive regulation of cell-substrate adhesion | 24 | 1.54 | 0.0369 | - | ||
| Regulation of ion homeostasis | 32 | 1.52 | 0.0327 | - | ||
| Immune system process | 369 | 1.50 | 0.001 | - | ||
| Endocytosis | 111 | 1.50 | 0.006 | - | ||
| Regeneration | 36 | 1.50 | 0.0369 | - | ||
| Muscle structure development | 51 | 1.46 | 0.0433 | - | ||
| Regulation of cell-cell adhesion | 74 | 1.45 | 0.025 | - | ||
| Angiogenesis | 58 | 1.44 | 0.0351 | - | ||
| Cell motility | 167 | 1.43 | 0.008 | - | ||
| Regulation of cell proliferation | 233 | 1.39 | 0.012 | 19 | ns | |
| Regulation of cell differentiation | 206 | 1.38 | 0.013 | 20 | ns | |
| Cellular macromolecule localization | 149 | 1.35 | 0.028 | - | ||
| Oxidation reduction | 98 | −2.82 | 0.001 | 16 | ns | |
| Carboxylic acid catabolism | 29 | −2.58 | 0.001 | - | ||
| Generation of precursor metabolites and energy | 45 | −2.45 | 0.001 | - | ||
| Cellular respiration | 24 | −2.44 | 0.001 | - | ||
| Mitochondrion organization | 73 | −2.35 | 0.001 | - | ||
| Organonitrogen compound metabolism | 153 | −2.24 | 0.001 | 28 | ns | |
| Coenzyme metabolism | 39 | −2.23 | 0.002 | - | ||
| Small molecule metabolism | 176 | −2.20 | 0.001 | 30 | ns | |
| Catabolic process | 142 | −2.18 | 0.001 | 19 | ns | |
| Hexose metabolism | 22 | −2.12 | 0.0042 | - | ||
| Glucose metabolism | 21 | −2.05 | 0.0058 | - | ||
| Regulation of ketone metabolism | 26 | −2.05 | 0.002 | - | ||
| Cofactor metabolism | 46 | −1.93 | 0.0038 | - | ||
| Macroautophagy | 27 | −1.93 | 0.0039 | - | ||
| Nitrogen compound transport | 31 | −1.93 | 0.0118 | - | ||
| Protein ubiquitination | 63 | −1.88 | 0.0062 | - | ||
| Organophosphate metabolism | 84 | −1.86 | 0.0117 | - | ||
| Small molecule biosynthesis | 48 | −1.85 | 0.0096 | - | ||
| NTP metabolism | 21 | −1.82 | 0.0081 | - | ||
| Autophagy | 33 | −1.79 | 0.022 | - | ||
| Cellular amino acid catabolism | 21 | −1.78 | 0.0135 | - | ||
| Alpha amino acid catabolism | 17 | −1.74 | 0.0259 | - | ||
| Macromolecule metabolism | 60 | −1.71 | 0.0279 | - | ||
| Anion transmembrane transport | 21 | −1.63 | 0.041 | |||
| Carbohydrate metabolism | 63 | −1.62 | 0.0299 | - | ||
| Acyl-CoA metabolism | 18 | −1.62 | 0.0433 | - | ||
| Regulation of transport | 120 | ns | 19 | −2.11 | 0.001 | |
| MHG at 12 m | LV at 12 m | |||||
| No | NES | p val | No | NES | p val | |
| DNA metabolism | 18 | 1.80 | 0.0045 | - | ||
| Lipid biosynthesis | 15 | 1.66 | 0.0216 | - | ||
| Cell projection organization | 37 | 1.62 | 0.0249 | - | ||
| Cellular response to stress | 38 | 1.62 | 0.0227 | - | ||
| Cell proliferation | 19 | 1.53 | 0.0473 | - | ||
| Regulation of cellular protein localization | 22 | 1.53 | 0.0378 | - | ||
| Regulation of organelle organization | 33 | 1.50 | 0.041 | - | ||
| Regulation of transport | - | 19 | −1.76 | 0.013 | ||
Functional analysis was performed using GSEA software, as detailed in the methods section. Positive and negative NES values respectively represent pathways enriched or down-regulated in GRMD animals compared to healthy counterparts. Nominal p values (p val) are shown, as are overlapping categories in skeletal (MHG) and left ventricular (LV) tissues for animals aged 6 months (6 m, top part of table) and 12 m (lowermost art of table). GO = gene ontology. No = number of differentially expressed genes in the category listed. NES = normalized (or relative) enrichment score. ns = not significant
Surprisingly, these enriched functions were not recapitulated in 12 month-old animals. The most significantly enriched functional categories of genes up-regulated in MHG from these older animals were DNA metabolism and lipid biosynthesis (Table 1). Significantly enriched functional categories observed in MHG of 6 month-old dogs were likewise not observed in LV at either age (Table 1). Only one biological process, regulation of transport, was significantly enriched in LV of GRMD at either age (19 genes, NES = -2.1 and -1.8 at 6 months and 12 months, respectively).
Skeletal muscle-specific transcriptional profile: the metabolic phenotype
A direct comparison of age-matched MHG and LV tissues resulted in large lists of differentially expressed genes consistent with known differences in these two tissue types, as expected (Additional file 4: Figure S2). To eliminate those gene expression changes driven primarily by tissue specificity, we instead compared GRMD to normal for LV and MG tissues and used those separately generated lists to find GRMD driven differences between the two tissue types. To identify GRMD skeletal- and cardiac muscle-specific gene expression profiles, we compared the most significantly altered genes in GRMD MHG and LV at 6 months of age. For this analysis, we chose a more stringent significance cutoff (p value < 0.01 and 2-fold) to eliminate potentially false positive differences between GRMD-mediated changes in the two tissue types, such as p value = 0.049 for the MHG comparison and p value = 0.051 for the same comparison in LV for example). Based on these criteria, there were a total of 1,172 annotated genes that were altered in MHG from young GRMD dogs which were unchanged in matched LV samples. The majority of these (893 transcripts) were up-regulated in GRMD dogs relative to wild type controls, with only 279 more highly expressed in the wild type control animals.
Multiple pathways were associated with genes more highly expressed in 6 month GRMD MHG, including those related to inflammation (e.g., leukocyte migration, chemokine signaling, complement and coagulation cascades, phagocytosis, Toll-like receptor signaling), cytoskeletal regulation, ECM interaction/adhesion, cell cycle, platelet activation, growth and survival signaling pathways (e.g., NF-κB, p53, PI3K- Akt) and catabolic metabolism (e.g.,. lysosome, glycan degradation) (Additional file 5: Figure S3). Pathways for down-regulated genes (those more highly expressed in MHG of 6 month-old normal dogs relative to GRMD), on the other hand were almost exclusively metabolic (e.g., TCA cycle, fatty acid biosynthesis and degradation, AMPK signaling, glycolysis/gluconeogenesis, amino acid metabolism, insulin signaling) (Additional file 6: Figure S4). MHG from 12 month-old GRMD included pathways that were similar to, albeit fewer than, those found in younger dogs for both up- and down-regulated genes (Additional file 7: Figure S5).
Consistent with functional and pathway analyses, manual examination of MHG-specific genes revealed a metabolic pattern consistent with a general perturbation of multiple metabolism and energy production pathways. For example, three out of four of the pyruvate dehydrogenase subunits were down-regulated, along with most of the TCA cycle enzymes and several electron transport chain components (Fig. 2). Conversely, the gene encoding ATP citrate lyase (ACLY), which functions counter to the other TCA cycle enzymes by synthesizing acetyl-CoA, was up-regulated, as was the transcript for the mitochondrial membrane potential de-coupler 2 (UCP2) that un-couples oxidative phosphorylation from ATP synthesis with energy instead dissipated as heat (Fig. 2).
Fig. 2.
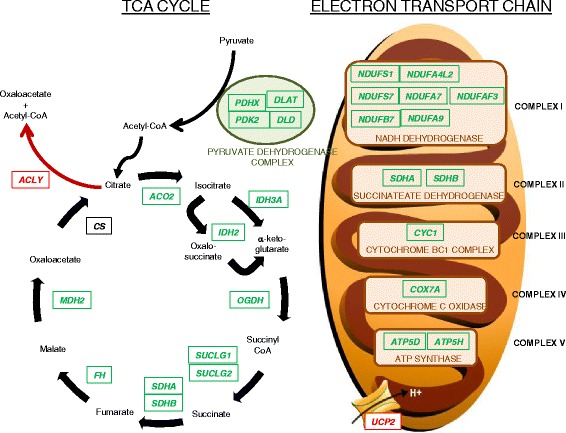
Diagram of TCA cycle and electron transport chain is shown, overlaid with gene expression results from GRMD versus wild type skeletal muscle samples. All listed genes, except for citrate synthase (CS), were differential between GRMD and wild type dogs, and each is colored according to direction of difference. Green genes represent those that were more highly expressed in wild type relative to GRMD dogs. Only two genes (ACLY and UCP2, colored in red) were more highly expressed in GRMD relative to control dogs
Multiple lysosome enzymes were also up-regulated in GRMD skeletal muscle (Fig. 3), possibly as a re-salvage energy source, and the glycolytic pathway and associated shunts were altered (Fig. 4) suggesting conversion to alternative energy sources instead of, or in addition to, glycolysis. Our findings were consistent with dysregulation of the AMPK pathway, which when activated in muscle contributes to a slower, more oxidative phenotype. Indeed, the gene encoding the catalytic subunit of AMPK (PRKAA2, p value 3.7 × 10−6, -2.3-fold) was down-regulated in GRMD MHG, as was the gene encoding the important downstream effector, PGC-1α (PPARGC1A, p value 9.6 × 10−3, -2.0-fold). We confirmed down-regulation of AMPK in GRMD skeletal muscle by Western blot analysis (Fig. 5) but did not find any difference in the phosphorylation status of AMPK in these same tissues. The AMPK levels in GRMD dogs were significantly lower than those from respective normal controls. AMPK levels were lower in the older (47–93 months) versus younger GRMD dogs, although this difference was not significant. Surprisingly, protein levels of AMPK were undetectable in matched cardiac tissue from these same animals (data not shown).
Fig. 3.
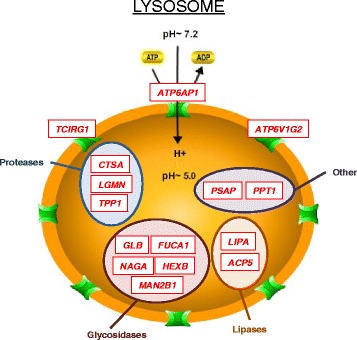
Diagram of lysosome genes that were more highly expressed in GRMD relative to wild type dogs is shown. All listed genes were more highly expressed in GRMD versus wild type skeletal muscle (red for up-regulation). Genes are grouped (colored circles) based on substrate type: proteases (blue), glycosidases (maroon), lipases (dark orange/brown), and other (purple). Genes encoding membrane proteins are displayed on the lysosomal periphery
Fig. 4.
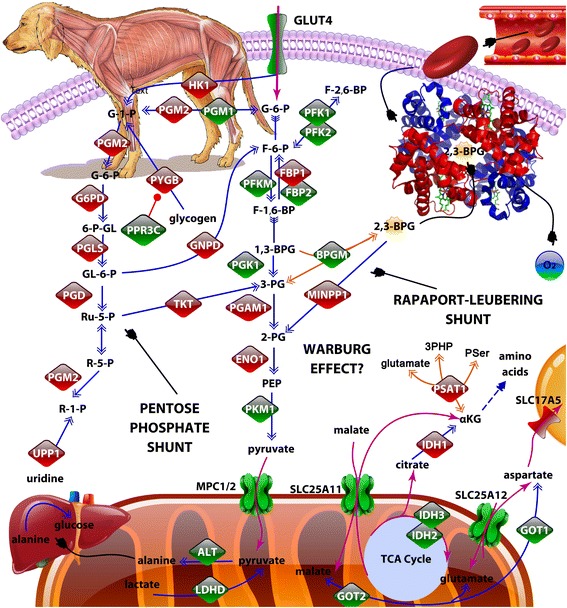
Diagram of glycolysis is shown, including enzymes whose transcripts were more highly expressed in GRMD (red) or wild type dogs (green). Metabolic transporters are also shown. The information was compiled using various public databases, and the figure was produced using Protein Lounge pathway builder software (ePath3D). The hemoglobin molecule was obtained from the Protein Databank (www.rcsb.org). The embedded dog illustration showing dystrophic musculature (top left) is an original work of art produced by Dave Carlson (Carlson-Art.com). Note: Common protein symbols are shown, some of which are represented by several different synonyms or have different names for their respective transcripts. For instance, MCP1 and 2 are also called BP44 and BP44L. PFK2 is encoded by PFKFB3
Fig. 5.
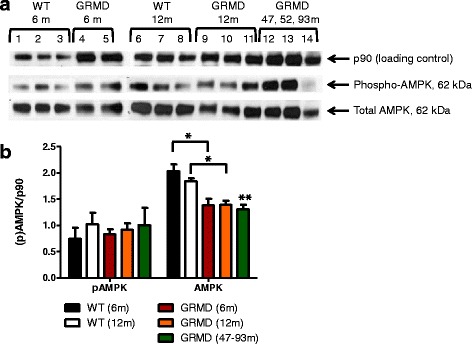
a Western blots for p-AMPK, total AMPK and p90 (loading control) in skeletal muscle tissues of GRMD and normal (wild type) dogs, including the three older GRMD dogs (47, 52, and 93 months-old) [17], are shown. b Bar graph showing semi-quantification of Western blot results for p-AMPK and AMPK, as calculated by densitometry, for skeletal muscle tissues from normal dogs aged 6 (black bars) or 12 (white bars) months, and GRMD dogs aged 6 (red bars), 12 (orange bars), or 47–93 (green striped bars) months. Asterisks indicate significance (*p < 0.05, **p < 0.01)
Interestingly, GLUT4, which encodes an insulin-regulated facilitative glucose transporter, was also significantly down-regulated in MHG from 6 month-old dogs. This is likely an important finding, given the previously suggested role of GLUT4 alterations in insulin resistance in DMD patients [30].
Cardiac muscle-specific transcriptional profile: the peroxisomal bypass
In contrast to skeletal muscle findings, the expression levels of the genes encoding AMPK and PGC-1α were unaltered in LV tissues from 6 and 12-month-old GRMD dogs when compared to age-matched controls, despite alteration in 5 genes associated with the AMPK signaling pathway in the older animals (Additional file 8: Figure S6). Similarly, mass down-regulation of critical energy production genes was not observed for LV tissue, nor was there up-regulation of multiple lysosomal genes. En bloc down-regulation of genes involved in peroxisome-specific beta-oxidation and uric acid production was, instead, seen in parallel with the up-regulation of key entry points for mitochondrial-based energy production (Fig. 6). Interestingly, the NT5C gene encoding cytosolic 5′,3′-pyrimidine nucleotidase (also called deoxy-5′-nucleotidase, or dNT-1) was up-regulated in GRMD versus wild type dogs, concomitant with down-regulation of genes encoding enzymes that function up-stream and downstream of NT5C (Fig. 7). While interpretation of these enzymatic changes would require further experimentation, two possible consequences might include accumulation of adenosine in the dystrophic heart (but not skeletal muscle) and up-regulation of uric acid production in dystrophic skeletal muscle (but not in the heart).
Fig. 6.
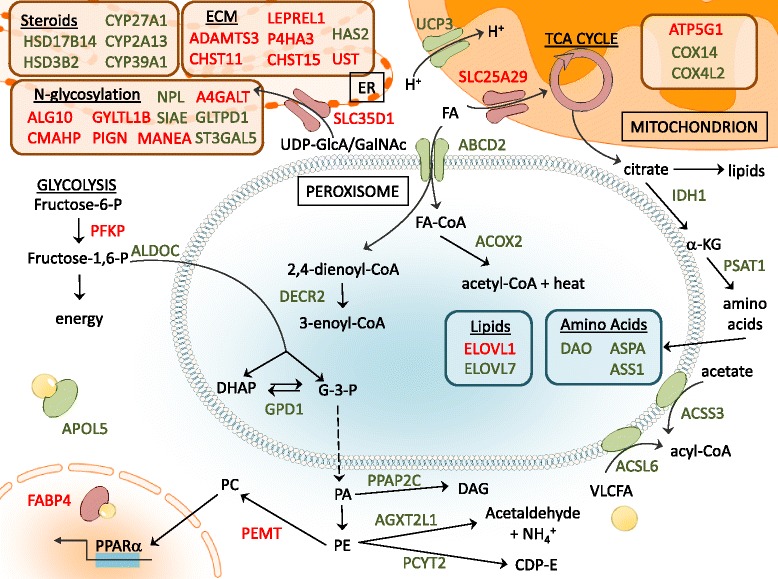
Diagram of peroxisomal beta-oxidation and other metabolic changes observed in GRMD LV tissues is shown. Proteins shown represent up-regulation (red) or down-regulation (green) of associated transcripts in dystrophic dogs as compared to normal controls
Fig. 7.
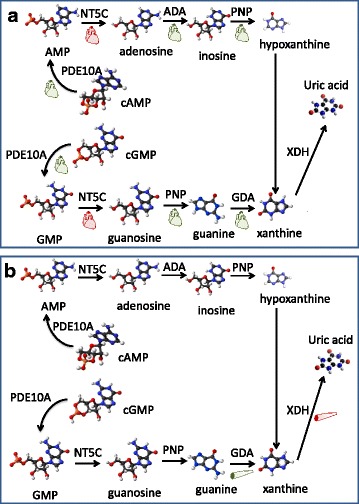
Diagram of nucleic acid gene expression differences in GRMD versus wild type dogs is shown. Panel a, Small red heart icons are shown below the names of enzymes whose transcripts were more highly expressed in LV of GRMD compared to wild type dogs. Green heart icons accompany those encoded by genes that were more highly expressed in wild type versus GRMD dogs. Panel b, Small skeletal muscle icons are shown below the names of enzymes whose transcripts were more highly expressed in MHG of GRMD compared to wild type dogs (red) or in wild type versus GRMD dogs (green)
Discussion
Duchenne muscular dystrophy is characterized by striking temporal differential involvement of cardiac and skeletal muscles [31], despite shared dystrophin protein deficiency and similar physiological functions in the two muscle types. The GRMD model provides an excellent platform for studying human DMD, largely because of the remarkable level of similarity between dogs and humans in general (e.g., in body structure) as well as likenesses in DMD/GRMD disease features [16]. In this study, we endeavored to discern whether the distinct cardiac and skeletal phenotypes in GRMD (and, by extension, DMD) are associated with differential gene expression within the respective muscle groups. By evaluating distinct ages representing different stages of GRMD disease, and two different tissue types historically observed to show contrasting levels of disease over this period, we were able to compare expression profiles in context with disease state [19].
The global transcriptome of MHG but not LV from 6 month-old GRMD dogs was profoundly altered. However, consistent with subsequent relative stabilization of skeletal muscle disease, these early expression changes were not sustained at 12 months. Looking more specifically at metabolic pathways in MHG from 6-month-old GRMD dogs, we identified a gene expression signature consistent with a “metabolic crisis” in DMD studies [9]. This canonical expression signature was entirely absent from matched GRMD LV tissues. Thus, our findings suggest differential metabolic dysfunction may contribute to distinct pathological phenotypes in these two muscle types. The most obvious transcriptional alterations that could contribute to metabolic dysfunction were down-regulation of dozens of energy production-associated molecules, most notably all of the TCA cycle enzymes and multiple electron transport components. Likewise, the majority of glycolytic pathway-associated enzymes were altered in a manner consistent with shunting of glycolytic molecule intermediates into alternative pathways, such as the anabolic pentose phosphate pathway. This is consistent with generalized muscle atrophy of any cause [11] and DMD in particular [29].
Gene expression alterations in MHG muscle from GRMD dogs also suggested that the Rappaport-Leubering Shunt was interrupted. For example, there was down-regulation of the gene encoding bisphosphoglycerate mutase (BPGM), the enzyme that produces 2,3 bisphosphoglycerate (2,3-BPG), an allosteric effector which enhances release of oxygen from hemoglobin to the tissues. Multiple inositol-polyphosphate phosphatase 1 (MINP1), whose product converts 2,3-BPG to 2-phosphoglycerate, was instead up-regulated. Decrease in 2,3-BPG levels could lead to impaired tissue oxygenation, such as that observed in iron deficient anemia or chronic respiratory disease with hypoxia. Consistent with this hypothesis, we also observed a small but significant reduction (1.4-fold, p value = 0.006) in the myoglobin gene in MHG (but not LV) tissues of GRMD animals.
Conclusions
When considered together, these various gene expression alterations provide a plausible mechanism for metabolic changes observed in the clinical setting. Lowered glucose uptake and glycolytic changes may contribute to insulin resistance. Impaired tissue oxygenation, along with a reduction in TCA and electron transport energy production, would support a shift from aerobic to anaerobic respiration, causing in essence a Warburg effect. In concert, these metabolic events, paired with the high-energy demands of regenerating muscle, almost certainly contribute to disease progression. In summary, this study provides novel insight into the metabolic gene expression profile of dystrophic cardiac tissue, which is unique from matched skeletal muscle of GRMD dogs. Further, these findings suggest a plausible mechanistic explanation for the chronological pattern of disease in these two different muscle types.
Additional files
Statistically significant gene list. Column A = Affymetrix unique probe set ID, Column B = Gene ID, Column C = gene symbol, Column D = RefSeq ID, Column E = gene assignment, Column F = gene title, Column G = gene symbol, Column H = ANOVA-generated p values for all groups, Column I = ANOVA-generated p values for comparison of left ventricular (LV) tissues from 6 month-old (6 m) Golden Retriever Muscular Dystrophy (GRMD) dogs versus LV from 6 m normal controls, Column J = Fold difference for [LV 6 m GRMD/LV 6 m normal], Column K = ANOVA-generated p values for comparison of LV 12 m GRMD versus LV 12 m normal, Column L = Fold difference for [LV 12 m GRMD/LV 12 m normal], Column M = ANOVA-generated p values for comparison of medial head of the gastrocnemius (MG) from 6 m GRMD versus MG 6 m normal, Column N = Fold difference for [MG 6 m GRMD/MG 6 m normal], Column O = ANOVA-generated p values for comparison of MG 12 m GRMD versus MG 12 m normal, Column P = Fold difference for [MG 12 m GRMD/MG 12 m normal]. Gene annotations were obtained within Partek (Columns B-C) and using Affymetrix NetAffx database information (Columns D-G). (XLSX 995 kb)
Hierarchical clustering of 5,835 significantly altered transcripts is shown. Rows represent individual samples, labeled according to group, and columns represent individual transcripts. Bright red, bright blue and gray represent highest, lowest and median normalized and scaled signal intensity values, respectively. (DOCX 116 kb)
Functional analysis results of GRMD-induced gene alterations. MHG = medial head of the gastrocnemius, LV = left ventricle, B-H p val = Benjamini & Hochberg-corrected p value, No. = number of differentially expressed genes in the given functional category, ns = not significant, Dash (“-”) indicates not identified in the analysis. (DOCX 16 kb)
Volcano plot is shown highlighting gene expression differences between dystrophic left ventricle (LV) and medial head of the gastrocnemius (MHG) skeletal muscle of golden retriever muscular dystrophy (GRMD) dogs at 6 months. The y-axis displays p values, and fold-differences are plotted on the x-axis for comparison of GRMD LV versus MHG (LV/MHG). Each circle represents an individual probe set (gene) and is colored based on fold-difference (blue for genes with lower expression in GRMD MHG and higher expression in LV, red for genes with higher expression values in LV and lower in MHG, and gray for no change), as shown in the legend. Orange lines mark cut-off values (p value < 0.05, fold-difference > 2). Official gene symbols are indicated to highlight major expression differences between LV and MHG. (PPTX 136 kb)
Plot shows over-represented pathways for genes up-regulated in 6 m GRMD MHG. Statistical significance (smaller p values = larger numbers = greater significance) is shown on the x-axis, and numbers of genes altered per pathway is shown to the right of each bar. (DOCX 32 kb)
Plot shows over-represented pathways for genes down-regulated in 6 m GRMD MHG. Statistical significance (smaller p values = larger numbers = greater significance) is shown on the x-axis, and numbers of genes altered per pathway is shown to the right of each bar. (DOCX 35 kb)
Plot shows over-represented pathways for genes up- (red bars) or down-regulated (green bars) in 12 m GRMD MHG. Statistical significance (smaller p values = larger numbers = greater significance) is shown on the x-axis, and numbers of genes altered per pathway is shown to the right of each bar. (DOCX 30 kb)
Plot shows over-represented pathways for genes altered in 6 m (light blue bars, top) or 12 m (dark blue bars, bottom) GRMD LV. Statistical significance (smaller p values = larger numbers = greater significance) is shown on the x-axis, and numbers of genes altered per pathway is shown to the right of each bar. AMPK signaling pathway genes are shown below the corresponding pathway bar, colored based on directionality (red for up-regulation and green for down-regulation, respectively in GRMD versus normal dogs). (DOCX 26 kb)
Acknowledgements
The authors would like to thank Dave Carlson (Carlson-Art.com) for the design and production of the embedded dog illustration showing dystrophic musculature (Fig. 4).
Funding
This project was supported in part by the Fighting Duchenne Foundation and the Fight DMD/Jonah & Emory Discovery Grant (Nashville, TN) (Markham) and was supported by the following grants: American Heart Association Grant 13CRP14530007 (Soslow) (Dallas, TX); the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number K23HL123938 (Bethesda, MD) (Soslow), Award Number K01HL121045 (Galindo), and U01 HL100398 (Sawyer); National Center for Research Resources, Grant UL1 RR024975-01 now at the National Center for Advancing Translational Sciences, Grant 2 UL1 TR000445-06 (Bethesda, MD). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Availability of data and materials
Raw and normalized microarray data were deposited into the NIH Gene Expression Omnibus (GEO) public repository under the Accession number GSE68626.
Authors’ contributions
LM contributed to study design, interpretation of data, and writing and editing the manuscript. CB contributed to study design, interpretation of microarray data, and edited the manuscript. JS contributed to study design, interpretation of data, and writing and editing the manuscript. MG isolated protein and performed Western blot analyses. DS contributed to study design, interpretation of data, and writing and editing the manuscript. JK contributed to the study design, provided the GRMD tissue samples, and edited the manuscript. CG performed microarray data analysis and interpretation, analyzed Western blots, and contributed to writing and editing the manuscript. All authors read and approved the final manuscript.
Authors’ information
In addition to their research activities, LM and JS are both pediatric cardiologists who are specialized in (and regularly treat) Duchenne patients at Vanderbilt Children’s Hospital. JK is a doctor of veterinary medicine who is also specialized in muscular dystrophy, including the GRMD dog model.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
The study was approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill. All dogs were used and cared for according to principles outlined in the National Research Council Guide for the Care and Use of Laboratory Animals.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- 2,3-BPG
2,3 bisphosphoglycerate
- ACLY
ATP citrate lyase
- AMPK
5′ AMP-activated protein kinase
- ANOVA
Analysis of Variance
- BDNF
Brain-derived neurotropic factor
- B-H
Benjamini and Hochberg
- BPGM
bisphosphoglycerate mutase
- CLCN1
Chloride Voltage-Gated Channel 1
- DMD
Dystrophin Muscular Dystrophy
- GRMD
Golden Retriever Muscular Dystrophy
- LV
Left ventricle
- mdx
X chromosome-linked muscular dystrophy (in the mouse)
- MHG
Medial head of gastrocnemius
- MINP1
Multiple inositol-polyphosphate phosphatase 1
- MSTN
Myostatin
- MYBPC2
Myosin Binding Protein C, fast type
- MYLK2
Myosin Light Chain Kinase 2
- NT5C
5′, 3′-Nucleotidase, cytosolic
- OPN
Osteopontin
- PGC-1α/PPARGC1A
Peroxisome Proliferator-activated Receptor-γ Coactivator
- POSTN
Periostin
- PRKAA2
Protein Kinase AMP-Activated Catalytic Subunit Alpha 2
- SEM
Standard Error of the Mean
- SNTB1
Syntrophin beta 1
- TCA
Tricarboxylic Acid (citric acid cycle, Krebs cycle)
- TIMP1
Tissue Inhibitor Of Metalloproteinases 1
- UCP2
Uncoupling Protein 2
Contributor Information
Larry W. Markham, Email: larry.markham@Vanderbilt.Edu
Candice L. Brinkmeyer-Langford, Email: CBrinkmeyer@cvm.tamu.edu
Jonathan H. Soslow, Email: jonathan.h.soslow@vanderbilt.edu
Manisha Gupte, Email: manisha.gupte@Vanderbilt.Edu.
Douglas B. Sawyer, Email: dsawyer@mmc.org
Joe N. Kornegay, Email: JKornegay@cvm.tamu.edu
Cristi L. Galindo, Phone: 1-615 875-3072, Email: Cristi.L.Galindo@Vanderbilt.edu
References
- 1.Moser H. Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum Genet. 1984;66(1):17–40. doi: 10.1007/BF00275183. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–28. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 3.Kunkel LM, Monaco AP, Middlesworth W, Ochs HD, Latt SA. Specific cloning of DNA fragments absent from the DNA of a male patient with an X chromosome deletion. Proc Natl Acad Sci U S A. 1985;82(14):4778–82. doi: 10.1073/pnas.82.14.4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dangain J, Vrbova G. Muscle development in mdx mutant mice. Muscle Nerve. 1984;7(9):700–4. doi: 10.1002/mus.880070903. [DOI] [PubMed] [Google Scholar]
- 5.Sharp NJ, Kornegay JN, Van Camp SD, Herbstreith MH, Secore SL, Kettle S, Hung WY, Constantinou CD, Dykstra MJ, Roses AD, et al. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics. 1992;13(1):115–21. doi: 10.1016/0888-7543(92)90210-J. [DOI] [PubMed] [Google Scholar]
- 6.McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech. 2015;8(3):195–213. doi: 10.1242/dmm.018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tseng BS, Zhao P, Pattison JS, Gordon SE, Granchelli JA, Madsen RW, Folk LC, Hoffman EP, Booth FW. Regenerated mdx mouse skeletal muscle shows differential mRNA expression. J Appl Physiol. 2002;93(2):537–45. doi: 10.1152/japplphysiol.00202.2002. [DOI] [PubMed] [Google Scholar]
- 8.Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R, Hoffman EP. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. 2005;65(6):826–34. doi: 10.1212/01.wnl.0000173836.09176.c4. [DOI] [PubMed] [Google Scholar]
- 9.Chen YW, Zhao P, Borup R, Hoffman EP. Expression profiling in the muscular dystrophies: identification of novel aspects of molecular pathophysiology. J Cell Biol. 2000;151(6):1321–36. doi: 10.1083/jcb.151.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baron D, Magot A, Ramstein G, Steenman M, Fayet G, Chevalier C, Jourdon P, Houlgatte R, Savagner F, Pereon Y. Immune response and mitochondrial metabolism are commonly deregulated in DMD and aging skeletal muscle. PLoS One. 2011;6(11):e26952. doi: 10.1371/journal.pone.0026952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calura E, Cagnin S, Raffaello A, Laveder P, Lanfranchi G, Romualdi C. Meta-analysis of expression signatures of muscle atrophy: gene interaction networks in early and late stages. BMC Genomics. 2008;9:630. doi: 10.1186/1471-2164-9-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boland BJ, Silbert PL, Groover RV, Wollan PC, Silverstein MD. Skeletal, cardiac, and smooth muscle failure in Duchenne muscular dystrophy. Pediatr Neurol. 1996;14(1):7–12. doi: 10.1016/0887-8994(95)00251-0. [DOI] [PubMed] [Google Scholar]
- 13.Perloff JK, de Leon AC, Jr O’Doherty D. The cardiomyopathy of progressive muscular dystrophy. Circulation. 1966;33(4):625–48. doi: 10.1161/01.CIR.33.4.625. [DOI] [PubMed] [Google Scholar]
- 14.Valentine BA, Cummings JF, Cooper BJ. Development of Duchenne-type cardiomyopathy. Morphologic studies in a canine model. Am J Pathol. 1989;135(4):671–8. [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper BJ, Winand NJ, Stedman H, Valentine BA, Hoffman EP, Kunkel LM, Scott MO, Fischbeck KH, Kornegay JN, Avery RJ, et al. The homologue of the Duchenne locus is defective in X-linked muscular dystrophy of dogs. Nature. 1988;334(6178):154–6. doi: 10.1038/334154a0. [DOI] [PubMed] [Google Scholar]
- 16.Howell JM, Fletcher S, Kakulas BA, O’Hara M, Lochmuller H, Karpati G. Use of the dog model for Duchenne muscular dystrophy in gene therapy trials. Neuromuscul Disord. 1997;7(5):325–8. doi: 10.1016/S0960-8966(97)00057-6. [DOI] [PubMed] [Google Scholar]
- 17.Galindo CL, Soslow JH, Brinkmeyer-Langford CL, Gupte M, Smith HM, Sengsayadeth S, Sawyer DB, Benson DW, Kornegay JN, Markham LW. Translating golden retriever muscular dystrophy microarray findings to novel biomarkers for cardiac/skeletal muscle function in Duchenne muscular dystrophy. Pediatr Res. 2016;79(4):629–36. doi: 10.1038/pr.2015.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Li J, Nghiem P, Detwiler DA, Larsen CA, Grange RW, Bhavaraju-Sanka RK, et al. Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Mamm Genome. 2012;23(1–2):85–108. doi: 10.1007/s00335-011-9382-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kornegay J, Childers M. Canine inherited dystrophinopathies and centronuclear myopathies. In: Childers M, editor. Regenerative Medicine for Degenerative Muscle Diseases. New York: Humana Press; 2016. pp. 309–29. [Google Scholar]
- 20.Patronek GJ, Waters DJ, Glickman LT. Comparative longevity of pet dogs and humans: implications for gerontology research. J Gerontol A Biol Sci Med Sci. 1997;52(3):B171–8. doi: 10.1093/gerona/52A.3.B171. [DOI] [PubMed] [Google Scholar]
- 21.Vignos PJ, Jr, Spencer GE, Jr, Archibald KC. Management of progressive muscular dystrophy in childhood. Jama. 1963;184:89–96. doi: 10.1001/jama.1963.03700150043007. [DOI] [PubMed] [Google Scholar]
- 22.McDonald CM, Abresch RT, Carter GT, Fowler WM, Jr, Johnson ER, Kilmer DD, Sigford BJ. Profiles of neuromuscular diseases. Duchenne muscular dystrophy. Am J Phys Med Rehabil. 1995;74(5 Suppl):S70–92. doi: 10.1097/00002060-199509001-00003. [DOI] [PubMed] [Google Scholar]
- 23.Nicholson LV, Johnson MA, Bushby KM, Gardner-Medwin D, Curtis A, Ginjaar IB, den Dunnen JT, Welch JL, Butler TJ, Bakker E, et al. Integrated study of 100 patients with Xp21 linked muscular dystrophy using clinical, genetic, immunochemical, and histopathological data. Part 1. Trends across the clinical groups. J Med Genet. 1993;30(9):728–36. doi: 10.1136/jmg.30.9.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 25.da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galindo CL, Kasasbeh E, Murphy A, Ryzhov S, Lenihan S, Ahmad FA, Williams P, Nunnally A, Adcock J, Song Y, et al. Anti-remodeling and anti-fibrotic effects of the neuregulin-1beta glial growth factor 2 in a large animal model of heart failure. J Am Heart Assoc. 2014;3(5):e000773. doi: 10.1161/JAHA.113.000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffman EP, Gorospe JRM: Chapter 8 The Animal Models of Duchenne Muscular Dystrophy: Windows on the Pathophysiological Consequences of Dystrophin Deficiency. In: Current Topics in Membranes, editors. Mark SM, Jon SM, vol. 38. Cambridge: Academic Press; 1991: 113–154.
- 28.Porter JD, Merriam AP, Leahy P, Gong B, Khanna S. Dissection of temporal gene expression signatures of affected and spared muscle groups in dystrophin-deficient (mdx) mice. Hum Mol Genet. 2003;12(15):1813–21. doi: 10.1093/hmg/ddg197. [DOI] [PubMed] [Google Scholar]
- 29.Kornegay JN, Spurney CF, Nghiem PP, Brinkmeyer-Langford CL, Hoffman EP, Nagaraju K. Pharmacologic management of Duchenne muscular dystrophy: target identification and preclinical trials. ILAR J. 2014;55(1):119–49. doi: 10.1093/ilar/ilu011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez-Cruz M, Sanchez R, Escobar RE, Cruz-Guzman Odel R, Lopez-Alarcon M, Bernabe Garcia M, Coral-Vazquez R, Matute G, Velazquez Wong AC. Evidence of Insulin Resistance and Other Metabolic Alterations in Boys with Duchenne or Becker Muscular Dystrophy. Int J Endocrinol. 2015;2015:867273. doi: 10.1155/2015/867273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ergul Y, Ekici B, Nisli K, Tatli B, Binboga F, Acar G, Ozmen M, Omeroglu RE. Evaluation of the North Star Ambulatory Assessment scale and cardiac abnormalities in ambulant boys with Duchenne muscular dystrophy. J Paediatr Child Health. 2012;48(7):610–6. doi: 10.1111/j.1440-1754.2012.02428.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistically significant gene list. Column A = Affymetrix unique probe set ID, Column B = Gene ID, Column C = gene symbol, Column D = RefSeq ID, Column E = gene assignment, Column F = gene title, Column G = gene symbol, Column H = ANOVA-generated p values for all groups, Column I = ANOVA-generated p values for comparison of left ventricular (LV) tissues from 6 month-old (6 m) Golden Retriever Muscular Dystrophy (GRMD) dogs versus LV from 6 m normal controls, Column J = Fold difference for [LV 6 m GRMD/LV 6 m normal], Column K = ANOVA-generated p values for comparison of LV 12 m GRMD versus LV 12 m normal, Column L = Fold difference for [LV 12 m GRMD/LV 12 m normal], Column M = ANOVA-generated p values for comparison of medial head of the gastrocnemius (MG) from 6 m GRMD versus MG 6 m normal, Column N = Fold difference for [MG 6 m GRMD/MG 6 m normal], Column O = ANOVA-generated p values for comparison of MG 12 m GRMD versus MG 12 m normal, Column P = Fold difference for [MG 12 m GRMD/MG 12 m normal]. Gene annotations were obtained within Partek (Columns B-C) and using Affymetrix NetAffx database information (Columns D-G). (XLSX 995 kb)
Hierarchical clustering of 5,835 significantly altered transcripts is shown. Rows represent individual samples, labeled according to group, and columns represent individual transcripts. Bright red, bright blue and gray represent highest, lowest and median normalized and scaled signal intensity values, respectively. (DOCX 116 kb)
Functional analysis results of GRMD-induced gene alterations. MHG = medial head of the gastrocnemius, LV = left ventricle, B-H p val = Benjamini & Hochberg-corrected p value, No. = number of differentially expressed genes in the given functional category, ns = not significant, Dash (“-”) indicates not identified in the analysis. (DOCX 16 kb)
Volcano plot is shown highlighting gene expression differences between dystrophic left ventricle (LV) and medial head of the gastrocnemius (MHG) skeletal muscle of golden retriever muscular dystrophy (GRMD) dogs at 6 months. The y-axis displays p values, and fold-differences are plotted on the x-axis for comparison of GRMD LV versus MHG (LV/MHG). Each circle represents an individual probe set (gene) and is colored based on fold-difference (blue for genes with lower expression in GRMD MHG and higher expression in LV, red for genes with higher expression values in LV and lower in MHG, and gray for no change), as shown in the legend. Orange lines mark cut-off values (p value < 0.05, fold-difference > 2). Official gene symbols are indicated to highlight major expression differences between LV and MHG. (PPTX 136 kb)
Plot shows over-represented pathways for genes up-regulated in 6 m GRMD MHG. Statistical significance (smaller p values = larger numbers = greater significance) is shown on the x-axis, and numbers of genes altered per pathway is shown to the right of each bar. (DOCX 32 kb)
Plot shows over-represented pathways for genes down-regulated in 6 m GRMD MHG. Statistical significance (smaller p values = larger numbers = greater significance) is shown on the x-axis, and numbers of genes altered per pathway is shown to the right of each bar. (DOCX 35 kb)
Plot shows over-represented pathways for genes up- (red bars) or down-regulated (green bars) in 12 m GRMD MHG. Statistical significance (smaller p values = larger numbers = greater significance) is shown on the x-axis, and numbers of genes altered per pathway is shown to the right of each bar. (DOCX 30 kb)
Plot shows over-represented pathways for genes altered in 6 m (light blue bars, top) or 12 m (dark blue bars, bottom) GRMD LV. Statistical significance (smaller p values = larger numbers = greater significance) is shown on the x-axis, and numbers of genes altered per pathway is shown to the right of each bar. AMPK signaling pathway genes are shown below the corresponding pathway bar, colored based on directionality (red for up-regulation and green for down-regulation, respectively in GRMD versus normal dogs). (DOCX 26 kb)
Data Availability Statement
Raw and normalized microarray data were deposited into the NIH Gene Expression Omnibus (GEO) public repository under the Accession number GSE68626.