

Abstract
The spleen plays a critical role in post-infarct myocardial remodeling. However, the role of the spleen in exacerbating myocardial infarction (MI) during acute ischemia/reperfusion (I/R) injury is unknown. The present study tests the hypothesis that splenic leukocytes are activated by substances released from ischemic myocardium to subsequently exacerbate myocardial injury during reperfusion. The left coronary artery in C57BL/6 mice underwent various durations of occlusion followed by 60 minutes of reperfusion (denoted as min/min of I/R) with or without splenectomy prior to I/R injury. Splenectomy significantly decreased myocardial infarct size (IS) in 40′/60′ and 50′/60′ groups (p<0.05); however, it had no effect on IS in 10′/60′, 20′/60′ and 30′/60′ groups (p=NS). In the 20′/60′ group, infusion of 40-minute ischemic heart homogenate (40-IHH) upon reperfusion increased IS by >3-fold versus infusion of 10-IHH (p<0.05). Splenectomy abolished the infarct exacerbating effect of 40-IHH, which was restored by splenic leukocyte adoptive transfer (SPAT). Furthermore, depletion of HMGB1 in the 40-IHH group abolished its infarct exacerbating effect (p<0.05), and 40-IHH failed to increase IS in both RAGE−/− mice and splenectomized wild-type mice with SPAT from RAGE−/− mice. The injection of 40-IHH significantly increased formyl peptide receptor 1 (FPR1) expression in sham spleens when compared to 10-IHH-treated sham & control mice. cFLFLF, a specific FPR1 antagonist, reduced myocardial neutrophil infiltration and abrogated the infarct exacerbating effect of 40-IHH during reperfusion.
Conclusions
A cardio (HMGB1) – splenic (RAGE receptor) signaling axis exists and contributes to myocardial infarct exacerbation during reperfusion after prolonged ischemic insults by activating splenic leukocytes. The FPR1 is a potential therapeutic target for inhibiting the cardio-splenic axis that augments infarct size during post-ischemic reperfusion.
Keywords: myocardial ischemia, reperfusion injury, spleen, HMGB1/RAGE pathway, neutrophils
INTRODUCTION
Ischemic heart disease remains the single leading cause of death in the United States, accounting for fully one out of every four deaths. Myocardial infarction (MI) and heart failure account for the vast majority of the morbidity and mortality associated with ischemic heart disease[31]. Current therapy for acute myocardial infarction is directed toward rapid restoration of perfusion to salvage as much of the jeopardized myocardium as possible[4, 5, 8]. This may be accomplished through interventional or surgical means, such as by PCI or CABG. Although reperfusion is a primary therapeutic goal, the reversal of ischemia entails additional damage, which is defined as reperfusion injury, and can paradoxically reduce the beneficial effects of reperfusion therapy[5, 20]. Numerous cardioprotective strategies, such as ischemic preconditioning, post-conditioning and remote ischemic preconditioning, have been developed to address myocardial reperfusion injury by acting on several molecular signal transduction pathways[14, 15]. However, clinical trials based on these translational strategies have largely yielded disappointing outcomes, perhaps due to our incomplete understanding of the pathogenesis of ischemia/reperfusion (I/R) injury.
Considerable evidence implicates the acute inflammatory response as a contributor to reperfusion injury[3, 5, 52, 53]. Studies from our group and others have demonstrated that inflammatory responses play a pivotal role in mediating acute I/R injury[49, 52, 53]. However, exactly how these inflammatory responses are triggered and mediated remains unclear. The spleen has critical roles in regulating inflammatory responses under several disease conditions including post-MI left ventricular remodeling. Swirski and colleagues first reported that the spleen serves as a reservoir to release monocytes after MI in animal models[39]. They found that splenectomy performed either prior to coronary ligation or 3 days after could significantly preserve cardiac function and limit left ventricular remodeling 4 weeks after MI[39]. Recently, Ismahil and colleagues reported a cardio-splenic axis in a murine heart failure model during long term follow up (8 weeks) after MI. Their results demonstrated that splenic mononuclear cells mediate myocardial inflammation in failing hearts and promote ventricular remodeling. Splenectomy can thus reduce the inflammatory response and improve heart function[21]. Moreover, the spleen is also been implicated in human studies that suggest the spleen plays an important role in mediating inflammatory responses during post-MI cardiac remodeling[10, 43]. However, whether the spleen contributes importantly to acute myocardial necrosis remains to be established.
In the current study, we hypothesized that prolonged ischemic insults could produce more pro-inflammatory substances, such as damage associated molecular patterns (DAMPs), like high-mobility group box-1 protein (HMGB1), that activate splenic leukocytes (i.e., a cardio-splenic axis), which subsequently exacerbate myocardial IS during reperfusion. Upon addressing this hypothesis in an in vivo mouse model, we found that prolonged ischemia exacerbates myocardial necrosis and increases levels of circulating HMGB1 upon reperfusion. This HMGB1 then activates splenic leukocytes by binding to the receptor for advanced glycation end products (RAGE), which promotes inflammatory responses during reperfusion and exacerbates myocardial IS. Antagonism of formyl peptide receptor 1 (FPR1) proved sufficient to inhibit this reperfusion-induced and spleen-enabled inflammatory response, thus attenuating myocardial IS.
MATERIALS and METHODS
This study conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Eighth Edition, revised 2011) and was conducted under protocols approved by the University of Virginia’s Institutional Animal Care and Use Committee.
Animals and experimental protocols
C57BL/6 wild type (WT) mice (9–13 weeks of age, purchased from The Jackson Laboratory) and RAGE knockout (RAGE−/−) mice[38] were randomly assigned to either I/R injury groups or sham surgery groups. RAGE−/− mice were backcrossed onto C57BL/6 for 10 generations and thus were congenic with C57BL/6. Acute splenectomy (SPLX) was performed 10 minutes before myocardial ischemia with or without adoptive transfer of splenic leukocytes after splenectomy. Isolation of splenic leukocytes was performed using the gentleMACS single splenocyte isolation system according to the manufacturer’s instructions (Miltenyi Biotec, San Diego, CA). Viability (>95%) of the splenocytes was confirmed with Trypan Blue staining. As described previously[40], splenic leukocyte adoptive transfer (SPAT) was performed by injecting 5×106 isolated splenocytes (in ~100μl PBS) into splenectomized mice via left external jugular vein 5 minutes after removing the spleen. Supernatant from ischemic heart tissue homogenate (IHH) was acquired from ischemic heart tissue at the end of 10 minutes (10-IHH) or 40 minutes (40-IHH) of ischemia. IHH was injected 5 minutes before reperfusion via left external jugular vein, and the dose of 10μg of total IHH protein per gram mouse body weight was selected based on pilot studies that examined the effect of 3 different doses on infarct size exacerbation (Online Resource Figure I). In cFLFLF-treated mice, cFLFLF was administered (0.5mg/kg, i.v.) 5 minutes before reperfusion.
Splenectomy
As described previously[40], mice were anesthetized with sodium pentobarbital (80 mg/kg i.p.) and orally intubated. Artificial respiration was maintained with a FiO2 of 0.80, 120 strokes per minute, and a 0.2 to 0.5mL stroke volume. A small midline abdominal incision was made and the peritoneal cavity entered. The spleen was located and brought to the incision. The hilum was clamped and ligated with 3-0 silk and the spleen excised. The incision was closed in two layers using 5-0 Prolene. The mice then underwent further procedures as described. The donor mice for splenic leukocytes were euthanized by cervical dislocation under deep anesthesia with death confirmed by absence of electric activity in ECG.
Myocardial I/R injury and measurement of infarct size
In previous work, we established that myocardial infarct size (IS) as measured by late-gadolinium enhanced (LGE) MRI at 60 minutes of reperfusion attains 95% of the size measured by the same method at 24 hours post-reperfusion in mice[40, 54]. We therefore used 60 minutes of reperfusion in the present study. The left coronary artery (LCA) of WT or RAGE−/− mice was ligated for durations of 10 to 50 minutes to impose graded levels of ischemia (I) followed by 60 minutes of reperfusion (R) as detailed previously[52–55]. The duration of I/R is denoted as minutes/minutes. Briefly, mice were anesthetized with sodium pentobarbital (80mg/kg, i.p.) and orally intubated. An additional dose of pentobarbital (40mg/kg, i.p.) was applied shortly after reperfusion. The adequacy of the anesthesia was confirmed by hind limb pinch reflex every 15 minutes. The heart was exposed through a left thoracotomy. The LCA was identified under a dissecting microscope. A 7-0 silk suture was placed around the LCA at a level 1 mm inferior to the left auricle. Ischemia was induced by securing a suture over a piece of PE-60 tubing placed parallel to the LCA, and reperfusion was achieved by removing the tube. Successful ligation of the LCA was confirmed by blanching in the ischemic zone and by ST segment elevation in the ECG. ECG was monitored perioperatively using PowerLab instrumentation (ADInstruments, Colorado Springs, CO). The mice were euthanized 60 minutes after reperfusion, and explanted hearts were cannulated through the ascending aorta for perfusion with 3ml of 1.0% TTC (Sigma-Aldrich, St. Louis, MO). The LCA was then re-occluded with the same suture used for coronary occlusion prior to 10% Phthalo blue perfusion to determine risk region (RR), which was statistically similar among all groups (Online Resource Figure II). The left ventricle was then cut into 5 to 7 transverse slices that were weighed and digitally photographed to determine IS as a percent of RR[52, 53].
Peripheral white blood cell counting
Blood samples (40μl per mouse) were obtained by puncturing the left external jugular vein at 60 minutes after reperfusion. Cell counts were performed using a HemaVet Hematology System (CDC Technologies, Oxford, CT).
Preparation of ischemic heart homogenate supernatant (IHH) and immunoprecipitation
At the end of 10 or 40 minutes of LCA occlusion or 10 min after thoracotomy without LCA occlusion (sham), hearts were harvested and placed on ice. The ischemic area was located on the anterolateral aspect of the left ventricle based on its pale color with clear demarcation. The full thickness of the ischemic zone was then cut, using a No. 11 scalpel blade, 0.5 mm inside of the demarcation line. The ischemic myocardium was then suspended in PBS (10μl/mg tissue) and homogenized with an electric homogenizer. The supernatant was acquired after centrifuging at 14,000rpm for 10 minutes at 4°C. Protein concentration of the IHH supernatant was determined by BCA protein assay kit for dilution to 5 mg/ml in PBS prior to storage at 4°C.
Western analysis
HMGB1 levels in heart homogenates and plasma were assessed by Western blot analysis as previous described[55]. Briefly, heart tissue was homogenized in PBS. Plasma samples were obtained by centrifuging blood samples at 1600g for 20 minutes. The total protein concentrations were determined by BCA protein assay (Thermo Scientific, Rockford, IL). Twenty micrograms of protein were separated by 10% SDS–PAGE. After transfer, nitrocellulose membranes (Bio-Rad, Hercules, CA) were probed with primary antibodies against HMGB1 (Abcam, Cambridge, MA) at a 1:2,000 dilution and secondary antibodies (Promega, Madison, WI) at a 1:5,000 dilution in blocking solution (0.5% BSA in TBS-T). Proteins were visualized with enhanced chemiluminescent substrate (Thermo Scientific, Rockford, IL) followed by densitometry using a Fluorchem 8900 imaging system (Alpha Innotech, Santa Clara, CA). β-actin was used as a loading control for HMGB1 measurements in IHH samples.
Immunofluorescence staining
Spleens and hearts were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 1 hour at room temperature and then incubated in 30% sucrose overnight at 4°C before freezing in OCT. Frozen sections were cut and blocked with CD16/32 (eBiosciences, San Diego, CA) in 10% normal serum for 1 hour at room temperature. Spleen sections were co-incubated with goat anti-FPR (Santa Cruz, Dallas, Texas), rat Alexa Fluor-594 labeled anti-CD11b (BioLegend San Diego, CA) and rat FITC-labeled anti-Ly6G antibodies (Cat. 11-9668-80, eBiosciences, San Diego, CA) overnight at 4°C. The anti-FPR sections were then incubated with donkey anti-goat Alexa Fluor 660-conjugated secondary antibodies for 1 hour at room temperature. Alexa Fluor 594- and FITC-labeled rat IgG (Serotec, Raleigh, NC) were used as negative controls for CD11b and Ly6G staining, respectively. Goat IgG (Santa Cruz, Dallas, Texas) was used as a negative control for FPR staining. Heart sections were incubated with anti-Ly6G for neutrophil staining. ProLong Gold anti-fade reagent with DAPI (Life Technologies, Grand Island, NY) was used to mount the specimens. All images were acquired under the same parameters for each fluorochrome using an Olympus BX-41 Microscope (Olympus, America, Inc., Center Valley, PA) with a Retiga-2000R camera (QImaging, Surrey, BC). Neutrophils were counted on 10 high-power fields per section. Fluorescence intensity of FPR expression in at least 100 nuclei per sample was determined with ImageJ software (NIH).
Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) analysis
Splenic leukocytes and bone marrow cells were harvested from mice with 40-IHH or vehicle pre-treatment. mRNA levels of IL-1β, INF-γ, TNF-α and MCP-1 were assessed by qRT-PCR. In brief, total RNA was isolated from cells using the RNeasy Mini Kit (QIAGEN China Co., Ltd, Shanghai) according to the manufacturer’s instructions. cDNAs were synthesized using RevertAidTM First Strand cDNA Synthesis Kit (Fermentas China Co., Ltd. Shenzhen). qPCR was performed with Maxima® SYBR Green qPCR Master Mix (Fermentas China Co., Ltd. Shenzhen). Actin was used as a housekeeping gene. mRNA levels were quantified using the 2(-ΔΔCt) relative quantification method as previously described[42]. The primer sequences are provided in Online Resource Table I.
Immunoprecipitation
Depletion of HMGB1 protein in IHH was achieved by immunoprecipitation with a Dynabeads® Protein A Immunoprecipitation Kit according to the manufacturer’s instructions (Life Technologies, Grand Island, NY). Briefly, the HMGB1 antibody (10μg in 200ul binding buffer) was pre-incubated with Dynabeads® Protein A for 15 minutes at room temperature. Then, 100μl of 40-IHH was mixed with the Dynabeads for 1 hour at room temperature, and the supernatant was collected for use. Western blot analysis was used to confirm efficient HMGB1 depletion. Rabbit IgG was used as a negative control for this analysis.
Statistical analysis
All data are presented as mean ± SEM (standard error of the mean). Changes in peri-operative white blood cell counts and heart rates were analyzed using repeated measures ANOVA followed by Bonferroni pairwise comparisons. All other data were compared using one-way ANOVA followed by t-test for unpaired data with Bonferroni correction.
RESULTS
Splenectomy attenuates myocardial I/R injury resulting from prolonged ischemic insult
C57BL/6 mice underwent various durations of LAD occlusion followed by 60 min of reperfusion with or without acute splenectomy (SPLX) prior to myocardial I/R. There were no differences in risk region (RR) size among these groups (Online resources Figure IIA). As shown in the Figure 1A, SPLX significantly decreased myocardial infarct size (IS) in the 40′/60′ group (I/R, min/min; 49.6 ± 1.4% vs. 35.7±1.9%, p<0.05) and the 50′/60′ group (56.4±1.3% vs. 44±2.0%, p<0.05). However, SPLX had no effect on IS in the 10′/60′, 20′/60′ and 30′/60′ groups (p=NS). The sham procedure of laparotomy without splenectomy had no effect on IS in any of the ischemic groups (data not shown). Acute splenic leukocyte adoptive transfer (SPAT) completely restored IS in splenectomized mice after 40′/60′ IR injury (50.9±1.2% vs. 35.7±1.9%, p<0.05) to the level of control mice (50.9±1.2% vs. 49.6±1.4%, p=NS) (Figure 1B and 1C). However, SPAT did not increase IS in SPLX+20′/60′ mice (7.0±1.6% vs. 7.3±1.4, p=NS, Figure 1C).
Figure 1. Splenectomy attenuates myocardial ischemia/reperfusion injury after prolonged ischemic insult.

A: Splenectomy significantly decreased myocardial infarct size in 40′/60′ and 50′/60′ groups. It had no effect on 10′/60′, 20′/60′ and 30′/60′ groups. B: Experimental protocol for the adoptive transfer of splenic leukocytes. C: Acute adoptive transfer of splenic leukocytes restored the IS of splenectomized mice in the 40′/60′ group to the level of intact control mice. However, it did not increase the IS of splenectomized mice in the 20′/60′ group. N = ≥5 mice/group. I/R: ischemia/reperfusion injury; SPLX: splenectomy; RR: risk region; SPAT: acute splenic leukocyte adoptive transfer.
Homogenates from hearts subject to prolonged ischemia exacerbate IS in mice subjected to brief ischemia by acting on splenic leukocytes
Based on the above results, we hypothesized that substance(s) derived from hearts subjected to prolonged ischemia might be released to serve as messengers between the heart and spleen during reperfusion by activating splenocytes. To test this hypothesis, supernatants from ischemic heart homogenates (IHH) were acquired from mouse hearts undergoing either 10 (10-IHH) or 40 (40-IHH) minutes of ischemia without reperfusion. Recipient WT mice underwent 20′/60′ I/R, and IHH at a dose of 10 μg protein per g mouse weight was injected IV into the recipient mice immediately before reperfusion. In the recipient mice, IHH had no effect on heart rates after reperfusion (Table 1). The risk regions (RRs) were similar among all groups. Treatment with 40-IHH significantly exacerbated myocardial IS in the 20′/60′ group (7.3±1.3% versus 33.3±2.5, p<0.05) (Figure 2). However, treatment with 10-IHH had no effect on IS (7.3±1.3% versus 9.6±2.3%, p=NS) (Figure 2). The exacerbating effect of 40-IHH on IS disappeared in splenectomized versus normal 20′/60′ mice (9.9±2.6% versus 33.3±2.5%, p<0.05) but was restored by SPAT (Figure 2).
Table 1.
Heart rates in IHH treated mice (N = ≥5 mice/group)
| Group | Baseline | 10 min after ischemia | 10 min after reperfusion |
|---|---|---|---|
| 20′/60-I/R | 424±14 | 468±12* | 484±14* |
| 20′/60′-I/R+10-IHH | 423±8 | 461±6* | 496±4* |
| 20′/60′-I/R+40-IHH | 429±10 | 476±4* | 496±3* |
p<0.05 compared with baseline.
Figure 2. Substances elaborated from the myocardium after prolonged ischemia exacerbate myocardial IS by acting on splenic leukocytes.

IHH from 40 minute ischemic myocardium (40-IHH) significantly exacerbated myocardial IS in 20′/60′ mice. However, 10-IHH had no effect on IS. The IS exacerbating effect of 40-IHH disappeared in splenectomized mice, but IS could be restored in splenectomized mice by SPAT. N = ≥5 mice/group. IHH: ischemic heart homogenate.
HMGB1 from 40-IHH activates splenocytes during reperfusion
Consistent with previous reports[2], we found that HMGB1 levels in mice treated with 40-IHH were significantly increased compared with 10-IHH or sham IHH; whereas there was no difference between 10-IHH and sham IHH (Figure 3A). Depletion of HMGB1 in 40-IHH by immunoprecipitation, as confirmed by Western blot analysis (Figure 3B), completely abrogated the exacerbating effect of 40-IHH on IS (Figure 3C). The immunoprecipitation negative control using rabbit IgG did not negate the IS exacerbating effect of 40-IHH.
Figure 3. HMGB1 from ischemic myocardium and RAGE receptors on splenocytes mediate the IS exacerbating effects of 40-IHH.

A: Western blot analysis showing that 40-IHH had significantly higher HMGB1 levels in heart homogenates than in sham or 10-IHH. B: Immunoprecipitation was used to deplete HMGB1 in heart homogenates, which was confirmed by Western blots. C: Depletion of HMGB1 in 40-IHH completely abrogated its IS exacerbating effect. D: 40-IHH completely failed to increase IS in either RAGE−/− mice or splenectomized WT mice with adoptive transfer of RAGE−/− splenocytes . N = ≥5 mice/group. IP: immunoprecipitation.
The infarct exacerbating effect of 40-IHH is lost in RAGE−/− mice
It has been reported that the HMGB1-RAGE pathway plays important roles in myocardial reperfusion injury[2]. Hence, we next investigated whether the presence of RAGE on splenocytes was required for the HMGB1-induced exacerbation of I/R injury. As shown in Figure 3D, 40-IHH failed to increase IS in RAGE−/− mice. Moreover, 40-IHH failed to exacerbate IS in splenectomized WT mice reconstituted with RAGE−/− splenocytes.
40-IHH activates splenic leukocytes and induces neutrophilia during reperfusion
40′/60′ I/R injury caused significant neutrophilia and induced more myocardial neutrophil infiltration than 20′/60′ I/R or sham surgery (Figure 4). Neutrophilia in the 40′/60′ group was significantly attenuated by splenectomy (Figure 4A). 40-IHH significantly increased IL-1β, INF-γ, TNF-α and MCP-1 gene expression in splenic leukocytes but not in bone marrow cells (Online Resource Figure III). The injection of 40-IHH significantly augmented neutrophilia and myocardial neutrophil infiltration in the 20′/60′ I/R group, which was attenuated by splenectomy. Conversely, neutrophil accumulation in the 40′/60′ I/R group was significantly attenuated by splenectomy (Figure 4B). Immunofluorescence staining revealed that splenic leukocytes in the marginal zone of spleens from 40-IHH treated mice displayed elevated levels of FPR1 expression, which is a key receptor that regulates neutrophil chemoattraction (Figure 5). Confocal imaging further showed that the elevated FPR1 expression was primarily localized to Ly6G+ cells (Figure 5). These experiments were conducted with the Ly6G monoclonal antibody produced by clone 1A8, which has been reported to be specific for neutrophils since it does not cross-react with Ly6C[9].
Figure 4. 40-IHH significantly increases both circulating and myocardial tissue neutrophils after IR injury.

A: Circulating neutrophils were significantly increased in 40′/60′ and 40-IHH treated 20′/60′ I/R injury mice as compared to sham or 20′/60′ I/R mice. This neutrophilia could be attenuated by splenectomy. B: Myocardial neutrophil infiltration was significantly increased in 40′/60′ and 40-IHH treated 20′/60′ I/R mice. The increase in neutrophil infiltration could also be attenuated by splenectomy. N = ≥5 mice/group. Arrows indicate neutrophils. Scale bar: 50μm.
Figure 5. Splenic FPR1 expression levels are significantly increased in 40-IHH treated mice.

Immunofluorescence staining indicated that 40-IHH significantly increases FPR1 expression levels (purple staining) in the splenic marginal zone. Confocal imaging showed that the elevations in FPR1 expression were mainly localized to Ly6G+ cells (stained in green). N = ≥5 mice/group. Arrows indicate Ly6G+ cells with elevated FPR1 expression levels. Scale bar: 20μm.
The formyl peptide receptor (FPR) antagonist, cFLFLF, abrogates the infarct exacerbating effect of 40-IHH
We next examined whether cFLFLF, a selective FPR1 antagonist[12], could reduce myocardial I/R injury. cFLFLF was administered at a dose of 0.5mg/kg as an i.v. bolus 5 minutes before reperfusion. This dose was selected because it had been used in a previous study for in vivo tracking of activated neutrophils[47]. cFLFLF significantly reduced circulating neutrophils and myocardial neutrophil infiltration in 40-IHH treated 20′/60′ I/R mice (Figures 6A and 6B). Myocardial infarct sizes in 40-IHH treated 20′/60′ I/R mice and 40′/60′ I/R mice were significantly reduced by pretreatment with cFLFLF (Figure 6C and Online Resource Figure IV).
Figure 6. FPR1 receptor blockade attenuates the IS exacerbating effect of 40-IHH.

A: cFLFLF significantly decreased myocardial neutrophil infiltration in 40-IHH treated mice (n=3 per group). B: cFLFLF significantly decreased circulating neutrophils in 40-IHH treated mice (N = ≥5 mice/group). C: cFLFLF significantly attenuated myocardial IS in 40-IHH treated mice (n=8 per group). Arrows indicate neutrophils. Scale bar: 50μm.
DISCUSSION
The present study represents the first demonstration, to the best of our knowledge, that the cardio-splenic axis plays a pivotal role in exacerbating myocardial infarct size during post-ischemic reperfusion. Our results show that prolonged ischemia elevates myocardial HMGB1 levels, and that HMGB1 is released into the bloodstream during reperfusion to activate splenocytes via RAGE stimulation. The activation of splenic leukocytes subsequently activates neutrophils, which transmigrate to the myocardium during reperfusion to exacerbate myocardial infarct size. Blocking FPR1, a key receptor regulating neutrophil chemoattraction, upon reperfusion blocks this cardio-splenic signaling axis and abrogates the reperfusion–induced exacerbation of infarct size.
The spleen exacerbates myocardial infarct size during reperfusion after prolonged ischemic insult
A role for the spleen in mediating I/R injury has been reported in the kidney[46] and brain[56]. Leuschner and colleagues[26] reported recently that splenectomy limited myocardial inflammatory responses and reduced IS after I/R in ApoE−/− mice, which have significantly larger infarct sizes than wild-type mice due to chronic inflammatory stimulation. Recently, we found that splenectomy had no effect on myocardial infarct size after 30 min of ischemia followed by reperfusion; however, it was sufficient to abolish the hyperglycemia-induced exacerbation of myocardial infarct size [40]. Collectively, these findings indicate that the spleen may not contribute importantly to myocardial reperfusion injury unless a strong pro-inflammatory stimulus is present. In this study, we found that 40-IHH significantly increased IL-1β, INF-γ, TNF-α and MCP-1 gene expression in splenic leukocytes but not in bone marrow cells in mice without I/R injury (Online Resource Figure III). Our current study clearly demonstrates that the spleen contributes to acute myocardial post-ischemic reperfusion injury; however, its role becomes significant in mice only after relatively long ischemic insults (>30 minutes, Figure 1).
HMGB1 from ischemic myocardium is necessary for spleen-mediated infarct exacerbation during reperfusion
Historically, the immune system is understood to protect the body against foreign insults; however, recent evidence has shown that the innate immune system is alerted following tissue injury with the release of endogenous ligands. An increasing body of evidence supports the contention that innate immune responses contribute importantly to myocardial I/R injury[11, 17, 24, 52, 53]. I/R injury results in the release of endogenous ‘danger’ signals referred to as danger associated molecular patterns (DAMPs) by necrotic or activated cells that trigger innate immune responses[29]. DAMPs include mediators such as heat shock proteins[23], HMGB1[48], hyaluronic acid[22], S100[35], DNA fragments[51] and fibronectin fragments[36]. As an important DAMP, HMGB1 has been shown to play critical roles in sterile tissue injury such as stroke[25] and I/R injury of the liver[30], kidney[45] and heart[2, 18]. HMGB1 can mediate the chemotaxis of leukocytes[19, 50]. Andrassy and colleagues reported that myocardial HMGB1 levels were elevated during ischemia[2]. Consistent with these results, we found that HMGB1 levels were significantly higher in supernatants of 40-IHH versus 10-IHH. The gentle homogenization process employed in our study was conducted without membrane lysis buffer to avoid destroying the nuclear or mitochondrial membranes of viable cardiomyocytes, and thus the elevated HMGB1 in IHH is primarily derived from necrotic cardiomyocytes (Figure 3). We found that 40-IHH tripled the infarct size in mice as compared to 20′/60′ I/R injury. More importantly, depletion of HMGB1 in 40-IHH supernatants completely abolished its infarct exacerbating effect (Figures 2 and 3). Moreover, 40-IHH significantly increased cytokine gene expressions in splenic leukocytes, but not in bone marrow cells (Online Resource Figure III). These results strongly suggest that HMGB1 is released from the myocardium after prolonged ischemia to activate splenocytes (but not bone marrow cells), which in turn exacerbate inflammation and myocardial infarct size during reperfusion. It has been reported that HMGB1 can be actively secreted by inflammatory cells, but that process is usually delayed by 6–8 hours after the initiation of injury[44]. Thus, the HMGB1 released by ischemic myocardium is most likely passively released from necrotic cardiomyocytes.
Myocardial HMGB1 signaling through splenic RAGE creates a cardio-splenic axis that activates splenic leukocytes
RAGE has been reported to play an important role in HMGB1-induced inflammatory responses[19]. An HMGB1-RAGE pathway has previously been implicated in mediating myocardial I/R injury[1, 2]. In the present study, infusion of 40-IHH upon reperfusion failed to exacerbate myocardial infarct size (IS) in RAGE−/− mice or in splenectomized wild-type mice after reconstitution with RAGE−/− splenocytes (Figure 3D). In contrast, 40-IHH effectively increased IS in splenectomized mice after reconstitution of wild-type splenocytes (Figure 1C). However, depletion of HMGB1 from 40-IHH negated its infarct exacerbating effect (Figure 3). These results demonstrate that HMGB1 released from ischemic myocardium activates splenic leukocytes during reperfusion by binding to splenocyte RAGE. Of note, it has been reported that RAGE−/− cardiomyocytes have significantly reduced hypoxia-reoxygenation injury in a cell culture model[37] and in a perfused isolated heart model[7]. However, the current study clearly demonstrates that, in intact mice, it is the RAGE present on splenocytes and the cardio-splenic axis that mediates HMGB1-induced I/R injury in vivo.
Neutrophil infiltration is critical in the acute phase of myocardial infarction[16, 32]. A recent study demonstrated that activation of the HMGB1-RAGE pathway triggers recruitment of neutrophils, but not macrophages, to sites of necrosis, and that the newly recruited neutrophils mediate subsequent amplification of injury[19]. We found that 40-IHH significantly increased FPR1 expression on splenic leukocytes (Figure 5) and facilitated both the exodus of neutrophils into the bloodstream and their infiltration into injured myocardium (Figure 4). Increased concentrations of HMGB1 (and possibly other FPR1 ligands) in the necrotic myocardium secondary to prolonged ischemia may exert strong chemotactic signals to attract circulating neutrophils[6, 19, 33]. These results document considerable cross-talk between the heart and spleen during the acute phase of myocardial I/R injury: 1) cardiomyocytes subjected to prolonged ischemia release HMGB1 into the circulation, 2) HMGB1 activates splenic leukocytes by binding to RAGE, 3) activation of RAGE triggers leukocyte activation in the spleen leading to stimulation of neutrophils and neutrophilia, and 4) circulating neutrophils migrate to the ischemic myocardium during reperfusion, facilitated by elevated myocardial HMGB1 and/or chemokine release (Figure 7).
Figure 7. Schematic mechanism of the cardio-splenic signaling axis in acute myocardial I/R injury.
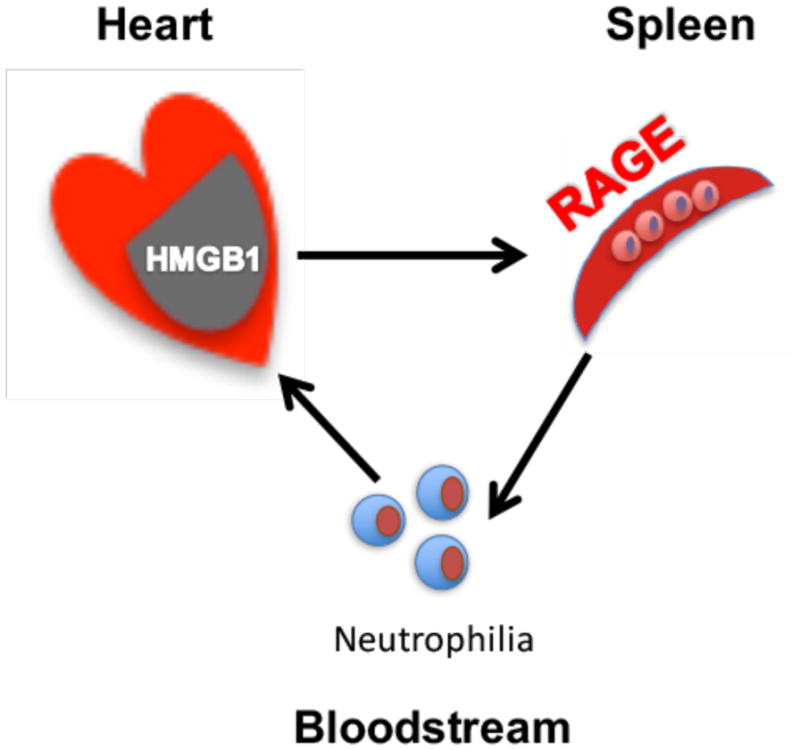
1) Necrotic cardiomyocytes release HMGB1 into the bloodstream upon reperfusion, 2) HMGB1 activates splenic neutrophils by stimulating RAGE, 3) activated splenic neutrophils enter the bloodstream, and 4) circulating splenic neutrophils transmigrate to the ischemic myocardium guided by HMGB1 and/or other chemokine gradients.
FPR1 antagonism blocks crosstalk between the heart and spleen
Previous work has demonstrated that neutrophils are the first cell type to accumulate to significant levels in previously ischemic myocardium after I/R, and that these cells impose additional tissue injury via oxidative damage and production of pro-inflammatory cytokines. It has also been reported that the HMGB1-RAGE pathway promotes neutrophil migration[19, 33] in a formyl-methionyl-leucyl-phenyl-alanine (fMLP)-dependent manner[6], and that fMLP is a well-known ligand of FPR. FPRs are members of the seven-transmembrane, G-protein coupled receptor superfamily and are highly expressed on neutrophils and mononuclear phagocytes. FPRs are involved in host defense and in sensing cellular dysfunction[34], and activated monocytes and neutrophils overexpress FPRs[28]. By using a selective FPR1 receptor antagonist, cFLFLF[12, 47], we demonstrated that cFLFLF blocks the downstream arm of the cardio-splenic axis by limiting neutrophil transmigration to the heart and abrogating IHH-induced infarct exacerbation (Figure 6).
Study limitations
The HMGB1-TLR (Toll like receptor) pathway was not investigated in the current study, but it is known to participate in sterile inflammatory responses including myocardial I/R injury. It has been reported that there is crosstalk between RAGE and TLRs in mediating HMGB1-induced inflammatory responses. Thus the relative contributions of the RAGE and TLR signaling pathways to myocardial I/R injury will require further study. Also, our experiments employing the adoptive transfer of splenic leukocytes are not neutrophil-specific and thus other types of splenic immune cells may also participate in the cardio-splenic axis. HMGB1 activates a broad spectrum of splenic leukocytes via RAGE, including splenic dendritic cells. Our previous work showed that myocardial I/R injury is mediated by CD4+ T cells[52], and activated CD4+ T cells likely enhance splenic neutrophil activation by secreting interferon gamma. The relative contribution of T cell signaling and the roles of splenic monocytes and dendritic cells remain to be defined. Thus, it is entirely possible that other circulating and spleen-resident immune cells may function upstream to enhance the activation of neutrophils. Other DAMPs, such as S100[35], HSP60/70[27] and mtDNA[51] may also contribute importantly to myocardial I/R injury. Their potential roles in activating splenic leukocytes remain to be evaluated. Finally, the FPR gene family is comprised of multiple members; of which FPR1 is the most prominent in activated neutrophils. Our study focused on FPR1 to evaluate splenocyte activation, and cFLFLF has marked selectivity for FPR1, but it can also activate FPR2 at higher concentrations.
In summary, by using a mouse model of acute myocardial I/R injury, we discovered a novel cardio-splenic axis that exacerbates myocardial infarct size during reperfusion injury after prolonged ischemic insult (>30 minutes). Prolonged ischemia is required to achieve the threshold of myocardial necrosis necessary to release high levels of HMGB1, which then enter the bloodstream during reperfusion. The circulating HMGB1 activates splenic leukocytes by engaging RAGE, which ultimately induces the stimulation of neutrophils and neutrophilia. The circulating neutrophils then transmigrate to the previously ischemic myocardium to exacerbate myocardial infarction. Selective blockade of FPR1 upon reperfusion interrupts this cardio-splenic axis, thus suppressing a major component of reperfusion injury. This study has important clinical implications because it demonstrates that selective FPR1 antagonists have the potential to reduce infarct size in patients with acute myocardial infarction during percutaneous coronary intervention or CAGB. Finally, recent work showing that treatment of the spleen with pulsed ultrasound can reduce renal[13] and myocardial[41] I/R injury opens the possibility that non-invasive imaging could provide a clinically-relevant, drug-free option for reducing the size of myocardial infarction.
Supplementary Material
Acknowledgments
FUNDING SOURCES
This study was funded in part by a University of Virginia School of Medicine Collaborative Science Pilot Grant and a NIH R01 HL130082 to ZY and a National Natural Science Foundation of China Grant (81400213) to YT.
Footnotes
DISCLOSURES
None.
References
- 1.Aleshin A, Ananthakrishnan R, Li Q, Rosario R, Lu Y, Qu W, Song F, Bakr S, Szabolcs M, D’Agati V, Liu R, Homma S, Schmidt AM, Yan SF, Ramasamy R. RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am J Physiol Heart Circ Physiol. 2008;294:H1823–1832. doi: 10.1152/ajpheart.01210.2007. [DOI] [PubMed] [Google Scholar]
- 2.Andrassy M, Volz HC, Igwe JC, Funke B, Eichberger SN, Kaya Z, Buss S, Autschbach F, Pleger ST, Lukic IK, Bea F, Hardt SE, Humpert PM, Bianchi ME, Mairbaurl H, Nawroth PP, Remppis A, Katus HA, Bierhaus A. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008;117:3216–3226. doi: 10.1161/CIRCULATIONAHA.108.769331. [DOI] [PubMed] [Google Scholar]
- 3.Appleyard RF, Cohn LH. Myocardial stunning and reperfusion injury in cardiac surgery. J Card Surg. 1993;8:316–324. doi: 10.1111/j.1540-8191.1993.tb01332.x. [DOI] [PubMed] [Google Scholar]
- 4.Bagai A, Dangas GD, Stone GW, Granger CB. Reperfusion strategies in acute coronary syndromes. Circ Res. 2014;114:1918–1928. doi: 10.1161/CIRCRESAHA.114.302744. [DOI] [PubMed] [Google Scholar]
- 5.Bainey KR, Armstrong PW. Clinical perspectives on reperfusion injury in acute myocardial infarction. Am Heart J. 2014;167:637–645. doi: 10.1016/j.ahj.2014.01.015. [DOI] [PubMed] [Google Scholar]
- 6.Berthelot F, Fattoum L, Casulli S, Gozlan J, Marechal V, Elbim C. The effect of HMGB1, a damage-associated molecular pattern molecule, on polymorphonuclear neutrophil migration depends on its concentration. J Innate Immun. 2012;4:41–58. doi: 10.1159/000328798. [DOI] [PubMed] [Google Scholar]
- 7.Bucciarelli LG, Kaneko M, Ananthakrishnan R, Harja E, Lee LK, Hwang YC, Lerner S, Bakr S, Li Q, Lu Y, Song F, Qu W, Gomez T, Zou YS, Yan SF, Schmidt AM, Ramasamy R. Receptor for advanced-glycation end products: key modulator of myocardial ischemic injury. Circulation. 2006;113:1226–1234. doi: 10.1161/CIRCULATIONAHA.105.575993. [DOI] [PubMed] [Google Scholar]
- 8.Cannon CP, Gibson CM, Lambrew CT, Shoultz DA, Levy D, French WJ, Gore JM, Weaver WD, Rogers WJ, Tiefenbrunn AJ. Relationship of symptom-onset-to-balloon time and door-to-balloon time with mortality in patients undergoing angioplasty for acute myocardial infarction. JAMA. 2000;283:2941–2947. doi: 10.1001/jama.283.22.2941. [DOI] [PubMed] [Google Scholar]
- 9.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 10.Emami H, Singh P, MacNabb M, Vucic E, Lavender Z, Rudd JH, Fayad ZA, Lehrer-Graiwer J, Korsgren M, Figueroa AL, Fredrickson J, Rubin B, Hoffmann U, Truong QA, Min JK, Baruch A, Nasir K, Nahrendorf M, Tawakol A. Splenic metabolic activity predicts risk of future cardiovascular events: demonstration of a cardiosplenic axis in humans. JACC Cardiovasc Imaging. 2015;8:121–130. doi: 10.1016/j.jcmg.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frenkel D, Pachori AS, Zhang L, Dembinsky-Vaknin A, Farfara D, Petrovic-Stojkovic S, Dzau VJ, Weiner HL. Nasal vaccination with troponin reduces troponin specific T-cell responses and improves heart function in myocardial ischemia-reperfusion injury. JACC Cardiovasc Imaging. 2009;21:817–829. doi: 10.1093/intimm/dxp051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge L, Zhou X, Ji WJ, Lu RY, Zhang Y, Zhang YD, Ma YQ, Zhao JH, Li YM. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol. 2015;308:H500–509. doi: 10.1152/ajpheart.00381.2014. [DOI] [PubMed] [Google Scholar]
- 13.Gigliotti JC, Huang L, Ye H, Bajwa A, Chattrabhuti K, Lee S, Klibanov AL, Kalantari K, Rosin DL, Okusa MD. Ultrasound prevents renal ischemia-reperfusion injury by stimulating the splenic cholinergic anti-inflammatory pathway. J Am Soc Nephrol. 2013;24:1451–1460. doi: 10.1681/ASN.2013010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116:674–699. doi: 10.1161/CIRCRESAHA.116.305348. [DOI] [PubMed] [Google Scholar]
- 15.Heusch G, Botker HE, Przyklenk K, Redington A, Yellon D. Remote ischemic conditioning. J Am Coll Cardiol. 2015;65:177–195. doi: 10.1016/j.jacc.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hiroi T, Wajima T, Negoro T, Ishii M, Nakano Y, Kiuchi Y, Mori Y, Shimizu S. Neutrophil TRPM2 channels are implicated in the exacerbation of myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2013;97:271–281. doi: 10.1093/cvr/cvs332. [DOI] [PubMed] [Google Scholar]
- 17.Homma T, Kinugawa S, Takahashi M, Sobirin MA, Saito A, Fukushima A, Suga T, Takada S, Kadoguchi T, Masaki Y, Furihata T, Taniguchi M, Nakayama T, Ishimori N, Iwabuchi K, Tsutsui H. Activation of invariant natural killer T cells by alpha-galactosylceramide ameliorates myocardial ischemia/reperfusion injury in mice. J Mol Cell Cardiol. 2013;62:179–188. doi: 10.1016/j.yjmcc.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Hu X, Fu W, Jiang H. HMGB1: a potential therapeutic target for myocardial ischemia and reperfusion injury. Int J Cardiol. 2012;155:489. doi: 10.1016/j.ijcard.2011.12.066. [DOI] [PubMed] [Google Scholar]
- 19.Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, Loike JD, Jenkins RE, Antoine DJ, Schwabe RF. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2014;125:539–550. doi: 10.1172/JCI76887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ibanez B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65:1454–1471. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 21.Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res. 2014;114:266–282. doi: 10.1161/CIRCRESAHA.113.301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 23.Jin C, Cleveland JC, Ao L, Li J, Zeng Q, Fullerton DA, Meng X. Human myocardium releases heat shock protein 27 (HSP27) after global ischemia: the proinflammatory effect of extracellular HSP27 through toll-like receptor (TLR)-2 and TLR4. Mol Med. 2014;20:280–289. doi: 10.2119/molmed.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kain V, Prabhu SD, Halade GV. Inflammation revisited: inflammation versus resolution of inflammation following myocardial infarction. Basic Res Cardiol. 2014;109:444. doi: 10.1007/s00395-014-0444-7. [DOI] [PubMed] [Google Scholar]
- 25.Kikuchi K, Tancharoen S, Ito T, Morimoto-Yamashita Y, Miura N, Kawahara K, Maruyama I, Murai Y, Tanaka E. Potential of the angiotensin receptor blockers (ARBs) telmisartan, irbesartan, and candesartan for inhibiting the HMGB1/RAGE axis in prevention and acute treatment of stroke. Int J Mol Sci. 2013;14:18899–18924. doi: 10.3390/ijms140918899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leuschner F, Panizzi P, Chico-Calero I, Lee WW, Ueno T, Cortez-Retamozo V, Waterman P, Gorbatov R, Marinelli B, Iwamoto Y, Chudnovskiy A, Figueiredo JL, Sosnovik DE, Pittet MJ, Swirski FK, Weissleder R, Nahrendorf M. Angiotensin-converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Circ Res. 2010;107:1364–1373. doi: 10.1161/CIRCRESAHA.110.227454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Si R, Feng Y, Chen HH, Zou L, Wang E, Zhang M, Warren HS, Sosnovik DE, Chao W. Myocardial ischemia activates an injurious innate immune signaling via cardiac heat shock protein 60 and Toll-like receptor 4. J Biol Chem. 2011;286:31308–31319. doi: 10.1074/jbc.M111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandal P, Novotny M, Hamilton TA. Lipopolysaccharide induces formyl peptide receptor 1 gene expression in macrophages and neutrophils via transcriptional and posttranscriptional mechanisms. J Immunol. 2005;175:6085–6091. doi: 10.4049/jimmunol.175.9.6085. [DOI] [PubMed] [Google Scholar]
- 29.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 30.McDonald KA, Huang H, Tohme S, Loughran P, Ferrero K, Billiar T, Tsung A. The TLR4 Antagonist Eritoran Tetrasodium Attenuates Liver Ischemia and Reperfusion Injury Through Inhibition of HMGB1 Signaling. Mol Med. 2014 doi: 10.2119/molmed.2014.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2010. Natl Vital Stat Rep. 2013;61:1–117. [PubMed] [Google Scholar]
- 32.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, Chavakis T. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007;26:1129–1139. doi: 10.1038/sj.emboj.7601552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panaro MA, Acquafredda A, Sisto M, Lisi S, Maffione AB, Mitolo V. Biological role of the N-formyl peptide receptors. Immunopharmacol Immunotoxicol. 2006;28:103–127. doi: 10.1080/08923970600625975. [DOI] [PubMed] [Google Scholar]
- 35.Rohde D, Schon C, Boerries M, Didrihsone I, Ritterhoff J, Kubatzky KF, Volkers M, Herzog N, Mahler M, Tsoporis JN, Parker TG, Linke B, Giannitsis E, Gao E, Peppel K, Katus HA, Most P. S100A1 is released from ischemic cardiomyocytes and signals myocardial damage via Toll-like receptor 4. EMBO Mol Med. 2014;6:778–794. doi: 10.15252/emmm.201303498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serebruany VL, Solomon SR, Herzog WR, Gurbel PA. Plasma fibronectin during myocardial ischemia-reperfusion: effects of magnesium, diltiazem, and a novel Mac-1 inhibitor. Am J Hematol. 1998;57:309–314. doi: 10.1002/(sici)1096-8652(199804)57:4<309::aid-ajh7>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 37.Shang L, Ananthakrishnan R, Li Q, Quadri N, Abdillahi M, Zhu Z, Qu W, Rosario R, Toure F, Yan SF, Schmidt AM, Ramasamy R. RAGE modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK-3beta signaling pathways. PloS one. 2010;5:e10092. doi: 10.1371/journal.pone.0010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma AK, LaPar DJ, Stone ML, Zhao Y, Kron IL, Laubach VE. Receptor for advanced glycation end products (RAGE) on iNKT cells mediates lung ischemia-reperfusion injury. Am J Transplant. 2013;13:2255–2267. doi: 10.1111/ajt.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tian Y, French BA, Kron IL, Yang Z. Splenic leukocytes mediate the hyperglycemic exacerbation of myocardial infarct size in mice. Basic Res Cardiol. 2015;110:39. doi: 10.1007/s00395-015-0496-3. [DOI] [PubMed] [Google Scholar]
- 41.Tian Y, Gigliotti JC, Wu D, Klibanov AL, Kron IL, Yang Z. Abstract 12972: Ultrasound of the Spleen Reduces Myocardial Ischemia-Reperfusion Injury by Stimulating a Splenic Anti-inflammatory Pathway. Circulation. 2015;132:A12972–A12972. [Google Scholar]
- 42.Tian Y, Zhang W, Xia D, Modi P, Liang D, Wei M. Postconditioning inhibits myocardial apoptosis during prolonged reperfusion via a JAK2-STAT3-Bcl-2 pathway. J Biomed Sci. 2011;18:53. doi: 10.1186/1423-0127-18-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Laan AM, Ter Horst EN, Delewi R, Begieneman MP, Krijnen PA, Hirsch A, Lavaei M, Nahrendorf M, Horrevoets AJ, Niessen HW, Piek JJ. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur Heart J. 2014;35:376–385. doi: 10.1093/eurheartj/eht331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 45.Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, Chadban SJ. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol. 2010;21:1878–1890. doi: 10.1681/ASN.2009101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wystrychowski W, Filipczyk L, Cierpka L, Obuchowicz E, Wiecek A, Wystrychowski A. Splenectomy attenuates the course of kidney ischemia-reperfusion injury in rats. Transplant Proc. 2014;46:2558–2561. doi: 10.1016/j.transproceed.2014.09.056. [DOI] [PubMed] [Google Scholar]
- 47.Xiao L, Zhang Y, Berr SS, Chordia MD, Pramoonjago P, Pu L, Pan D. A novel near-infrared fluorescence imaging probe for in vivo neutrophil tracking. Mol Imaging. 2012;11:372–382. [PubMed] [Google Scholar]
- 48.Xu H, Yao Y, Su Z, Yang Y, Kao R, Martin CM, Rui T. Endogenous HMGB1 contributes to ischemia-reperfusion-induced myocardial apoptosis by potentiating the effect of TNF-α/JNK. Am J Physiol Heart Circ Physiol. 2011;300:H913–921. doi: 10.1152/ajpheart.00703.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu Y, Huo Y, Toufektsian MC, Ramos SI, Ma Y, Tejani AD, French BA, Yang Z. Activated platelets contribute importantly to myocardial reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;290:H692–699. doi: 10.1152/ajpheart.00634.2005. [DOI] [PubMed] [Google Scholar]
- 50.Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93:865–873. doi: 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang XM, Cui L, White J, Kuck J, Ruchko MV, Wilson GL, Alexeyev M, Gillespie MN, Downey JM, Cohen MV. Mitochondrially targeted Endonuclease III has a powerful anti-infarct effect in an in vivo rat model of myocardial ischemia/reperfusion. Basic Res Cardiol. 2015;110:3. doi: 10.1007/s00395-014-0459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Z, Day YJ, Toufektsian MC, Ramos SI, Marshall M, Wang XQ, French BA, Linden J. Infarct-sparing effect of A2A-adenosine receptor activation is due primarily to its action on lymphocytes. Circulation. 2005;111:2190–2197. doi: 10.1161/01.CIR.0000163586.62253.A5. [DOI] [PubMed] [Google Scholar]
- 53.Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, French BA, Linden J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation. 2006;114:2056–2064. doi: 10.1161/CIRCULATIONAHA.106.649244. [DOI] [PubMed] [Google Scholar]
- 54.Yang Z, Linden J, Berr SS, Kron IL, Beller GA, French BA. Timing of adenosine 2A receptor stimulation relative to reperfusion has differential effects on infarct size and cardiac function as assessed in mice by MRI. Am J Physiol Heart Circ Physiol. 2008;295:H2328–2335. doi: 10.1152/ajpheart.00091.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Z, Tian Y, Liu Y, Hennessy S, Kron IL, French BA. Acute hyperglycemia abolishes ischemic preconditioning by inhibiting Akt phosphorylation: normalizing blood glucose before ischemia restores ischemic preconditioning. Oxid Med Cell Longev. 2013;2013:329183. doi: 10.1155/2013/329183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang BJ, Men XJ, Lu ZQ, Li HY, Qiu W, Hu XQ. Splenectomy protects experimental rats from cerebral damage after stroke due to anti-inflammatory effects. Chin Med J (Engl) 2013;126:2354–2360. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.