

Abstract
Aging is accompanied by increased neuroinflammation, synaptic dysfunction and cognitive deficits both in rodents and humans, yet the onset and progression of these deficits throughout the life span remain unknown. These aging-related deficits affect the quality of life and present challenges to our aging society. Here, we defined age-dependent and progressive impairments of synaptic and cognitive functions and showed that reducing astrocyte-related neuroinflammation through anti-inflammatory drug treatment in aged mice reverses these events. By comparing young (3 months), middle-aged (18 months), aged (24 months) and advanced-aged wild-type mice (30 months), we found that the levels of an astrocytic marker, GFAP, progressively increased after 18 months of age, which preceded the decreases of the synaptic marker PSD-95. Hippocampal long-term potentiation (LTP) was also suppressed in an age-dependent manner, where significant deficits were observed after 24 months of age. Fear conditioning tests demonstrated that associative memory in the context and cued conditions was decreased starting at the ages of 18 and 30 months, respectively. When the mice were tested on hidden platform water maze, spatial learning memory was significantly impaired after 24 months of age. Importantly, subacute treatment with the anti-inflammatory drug ibuprofen suppressed astrocyte activation, and restored synaptic plasticity and memory function in advanced-aged mice. These results support the critical contribution of aging-related inflammatory responses to hippocampal-dependent cognitive function and synaptic plasticity, in particular during advanced aging. Our findings provide strong evidence that suppression of neuroinflammation could be a promising treatment strategy to preserve cognition during aging.
Keywords: Aging, ibuprofen, anti-inflammatory responses, cognitive function, synaptic plasticity
1. Introduction
By 2050, the aged population who are older than 85 years are expected to reach 21 million in the United States (Vincent and Velkof, 2010). Increasing evidence demonstrates that aging accelerates the risk for neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (Mattson and Magnus, 2006). More importantly, aging drives the vulnerability to cognitive impairments even in the absence of neurodegenerative diseases (Hedden and Gabrieli, 2004, Mattson and Magnus, 2006, Morrison and Baxter, 2012). Thus, it is critical to understand how aging affects neuronal systems and cognitive function in both normal and pathological aging. In humans, cognitive aging usually starts at middle age with an increase in forgetting, which may be an early sign of impaired synaptic transmission and plasticity (Christensen, et al., 1999, Colsher and Wallace, 1991, Schonknecht, et al., 2005). The aging-related cognitive decline is progressive, where spatial memory, working memory, executive function as well as processing speed are gradually impaired (Kukolja, et al., 2009, Plancher, et al., 2010, Uttl and Graf, 1993).
Memory deficits are related to impaired hippocampal function; progressive decline of memory is often associated with a decrease in hippocampal volume (Kramer, et al., 2007, Mueller, et al., 2007, Mungas, et al., 2005, Reuter-Lorenz and Park, 2010). Hippocampal-mediated cognitive processes are most vulnerable to aging. Studies in humans and animal models suggest that aging-related cognitive decline is caused by disturbances of synaptic integrity in the hippocampus (Morrison and Baxter, 2012). These synaptic alternations result in increased slow after hyperpolarizations (sAHPs) (Disterhoft, et al., 1996, Landfield and Pitler, 1984, Moyer, et al., 1992), deficits in long-term potentiation (LTP) (Barnes and Kidd, 1979, Barnes, 1994, Shankar, et al., 1998), and long-term depression (LTD) (Norris, et al., 1998, Norris, et al., 1996). The processes that underlie aging-related cognitive decline are vastly complex. Thus, studies that investigate brain aging, the molecular mechanisms involved, neuronal plasticity and possible therapeutic strategies to combat aging-related cognitive deficits are urgent needs for our aging society.
Aging is associated with increased neuroinflammation, diminished motor function and cognitive decline in both humans and mice. In fact, non-steroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen, have been associated with a reduction in the incidence of AD (in t’ Veld, et al., 2001) and increased longevity in simple organisms (He, et al., 2014). Therefore, in this study, we used young (3 month-old), middle-aged (18 month-old), aged (24 month-old) and advanced-aged (30 month-old) mice to address the age-dependent effects on synaptic plasticity, cognitive function and neuroinflammation throughout the life span of mice. The ages of 3, 18, 24 and 30 months in mice are estimated to correspond to 20, 56, 69 and 81 years of age in humans, respectively (Fox, 2007). Furthermore, the 30-month-old mice were subacutely treated with ibuprofen to determine if synaptic plasticity and cognitive deficits are reversible at an advanced age. We report here the aging-dependent synaptic deficits and cognition decline, and show that anti-inflammatory drug ibuprofen is beneficial in rescuing these detrimental effects.
2. Methods
2.1. Animals
Male mice with different ages (3, 18, 24 and 30 months) were obtained from the National Institute on Aging. All mice were housed in standard 12 hr light-dark cycle and fed normal chow ad libitum. In some experiments, mice (30 months-old) were administrated with 0.14 mg/ml ibuprofen in drinking water according to previous methods (Kofidis, et al., 2006) and allowed to drink ad libitum. All animal procedures were approved by the Animal Study Committee at Mayo Clinic and in accordance with the regulations of the American Association for the Accreditation of Laboratory Animal Care.
2.2. Western Blot and ELISA
Samples were homogenized and incubated in PBS containing 1% Triton X-100, supplemented with protease inhibitor mix and PhosSTOP (Roche). Equal amounts of protein (by Bradford assay) were resolved by SDS-PAGE and transferred to PVDF membranes. After the membranes were blocked, proteins were detected with primary antibody for overnight at 4°C. Membrane was probed with LI-COR IRDye secondary antibodies and detected using the Odyssey infrared imaging system (LI-COR). The following antibodies were used in this study: anti-PSD-95 (Cell Signaling), anti-synaptophysin (Millipore), anti-GFAP (Millipore) and anti-β-actin (Sigma) antibodies. Levels of PSD-95, and GFAP were also determined by enzyme-linked immunosorbent assay (ELISA) as previously described (Shinohara, et al., 2013).
2.3. Extracellular Recordings
After at least one week following behavioral testing, mice were sacrificed and hippocampi were dissected out for electrophysiological experimental paradigms in ice-cold cutting solution containing 110 mM sucrose, 60 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 28 mM NaHCO3, 0.6 mM sodium ascorbate, 5 mM glucose, 7 mM MgCl2 and 0.5 mM CaCl2 as previously described (Rogers, et al., 2011, Rogers, et al., 2013). Field excitatory post-synaptic potentials (fEPSPs) were obtained from area CA1 stratum radiatum with the use of a glass microelectrode (2–4 mΩ) filled with artificial cerebrospinal fluid (aCSF) containing 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 25 mM glucose, 1 mM MgCl2 and 2 mM CaCl2. fEPSPs were evoked through stimulation of the Schaffer collaterals using a 0.1 msec biphasic pulse delivered every 20 sec. After a consistent response to a voltage stimulus was established, threshold voltage for evoking fEPSPs was determined and the voltage was increased incrementally every 0.5 mV until the maximum amplitude of the fEPSP was reached (I/O curve). All other stimulation paradigms were induced at the same voltage, defined as 50% of the stimulus voltage used to produce the maximum fEPSP amplitude, for each individual slice. Paired-pulse facilitation (PPF) was induced with two paired-pulses given with an initial delay of 20 msec and the time to the second pulse incrementally increased 20 msec until a final delay of 300 msec was reached. A fEPSP baseline response was then recorded for 20 min. The tetanus used to evoke LTP was a theta-burst stimulation (tbs) protocol consisting of five trains of four pulse bursts at 200 Hz separated by 200 msec, repeated six times with an inter-train interval of 10 sec. Following tbs, fEPSPs were recorded for 60 min. Potentiation was measured as the increase of the mean fEPSP descending slope following tbs normalized to the mean fEPSP descending slope of baseline recordings.
2.4. Fear conditioning test
This test was conducted in a sound attenuated chamber with a grid floor capable of delivering an electric shock and freezing was measured with an overhead camera and FreezeFrame software (Actimetrics, Wilmette, IL). Mice were initially placed into the chamber and undisturbed for 2 minutes, during which time baseline freezing behavior was recorded. An 80-dB white noise served as the conditioned stimulus (CS) and was presented for 30 sec. During the final 2 sec of this noise, mice received a mild foot shock (0.5mA), which served as the unconditioned stimulus (US). After 1 minute, another CS-US pair was presented. The mouse was removed 30 sec after the second CS-US pair and returned to its home cage. Twenty-four hours later, each mouse was returned to the test chamber and freezing behavior was recorded for 5 minutes (context test). Mice were returned to their home cage and placed in a different room than previously tested in reduced lighting conditions for a period of no less than one hour. For the auditory CS test, environmental and contextual cues were changed by: wiping testing boxes with 30% isopropyl alcohol instead of 30% ethanol; replacing white house lights with red house lights; placing a colored plastic triangular insert in the chamber to alter its shape and spatial cues; covering the wire grid floor with opaque plastic; and altering the smell in the chamber with vanilla extract. The animals were placed in the apparatus for 3 min and then the auditory CS was presented and freezing was recorded for another 3 min (cued test). Baseline freezing behavior obtained during training was subtracted from the context or cued tests to control for animal variability.
2.5. Hidden platform water maze (HPWM)
Mice were trained to locate an escape platform hidden just beneath the water surface. The training trials lasted for 4 days, 4 trials per day with an inter-trail interval of 1 min. Goal latencies were obtained using automated video tracking software (ANY-maze). On day 5, the platform was removed and mice were allowed to free swim for 60 sec (probe) and time spent in quadrants and number of target platform entries calculated.
2.6. Rotarod test
This test was performed on an accelerating rotarod apparatus (Rotamex-5 Columbus instruments) with a 3cm diameter rod starting at an initial rotation of 4 RPM accelerating to 40 RPM over 5 minutes. Mice were tested for the time spent on the rod during each of four trials per day, for two consecutive days. Testing is completed when the mouse falls off the rod (distance of 12 cm) onto a cushioned platform.
2.7. Open field activity (OFA) and Elevated plus maze (EPM)
General locomotor activity and anxiety were measured with an open field task. Mice were placed in the center of an open-field arena (40 × 40 × 30 cm, W × L × H), and allowed to roam freely for 15 minutes. An overhead camera was used to track movement with AnyMaze software (Stoelting Co., Wood Dale, IL). Mice were analyzed for total locomotor activity by measuring the total distance traveled in the apparatus. Mice were analyzed for anxiety-like behavior by measuring the distance traveled in an imaginary “center” zone (20 × 20 cm) normalized to the total distance traveled (center : total distance ratio). General anxiety levels were measured with the use of the elevated plus maze (EPM). The EPM consists of two well-lit open arms (30 × 5 cm) facing each other and two enclosed arms (30 × 5 × 15 cm) which faced each other. Each arm is attached to a common center open-square platform (4.5 cm) and elevated 40 cm off the floor. Mice were placed in the central open square platform facing the closed arms and allowed to explore for a 5 min period. The total time spent in the open arms and closed arms were recorded and analyzed with ANY-Maze animal activity system (Stoelting Co, Wood Dale, Illinois). Anxiety levels were assessed as the ratio of time spent in the open arms vs. closed arms.
2.8. Statistical analysis
For all data, a one-way ANOVA was used to determine significance (p<0.05). A post-hoc Tukey’s multiple comparison test was used to assess significance between groups and are reported as p-values (*, p<0.05; **p<0.01; ***, p<0.001).
3. Results
Aging is associated with exacerbated astrogliosis and compromised synapses
GFAP expression is increased in activated astrocytes and is an indicator of the astrocyte-related inflammatory state of the central nervous system (CNS) (Haley, et al., 2010). To determine whether aging results in increased activation of astrocytes, we examined the protein levels of GFAP by both Western blotting and ELISA in the brains of wild-type C57BL/6 mice at different ages (3, 18, 24 and 30 months). GFAP levels were significantly elevated in 30 month-old mice when compared to either 3 month-old or 18 month-old mice by Western blot analysis in both the cortex (Fig. 1A) and hippocampus (Fig. 1B). Consistent with the Western blotting results, ELISAs also showed age-dependent activation of astrocytes, where GFAP levels were significantly elevated in 18 month-, 24 month- and 30 month-old mice in the cortex (Fig. 1C) and hippocampus (Fig. 1D). Furthermore, 30 month-old mice also had significantly higher GFAP levels compared to 18 month- and 24 month-old mice in both the cortex (Fig. 1C) and hippocampus (Fig. 1D). Taken together, these findings indicate that aging is significantly accompanied by astrogliosis in mouse brains.
Fig. 1.
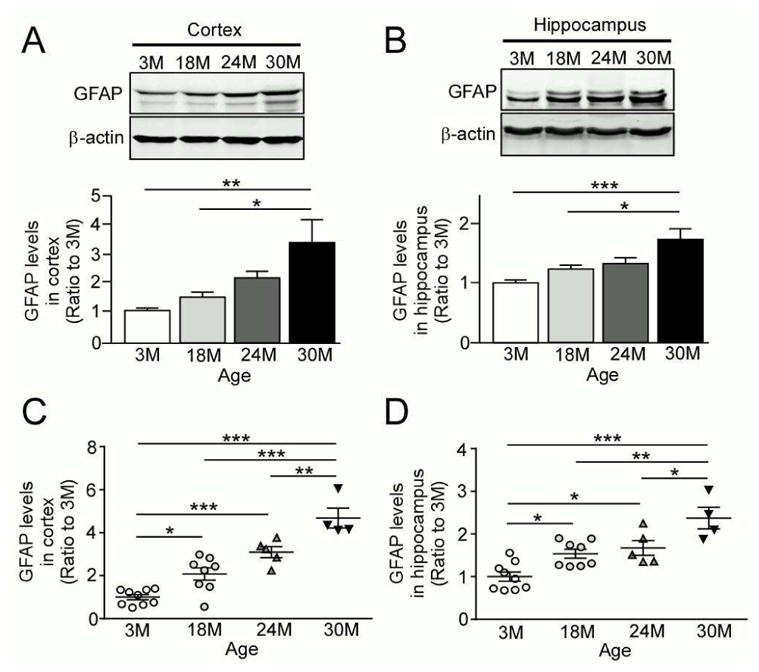
Levels of the astrocyte marker GFAP are increased in an age-dependent manner. (A and B) GFAP levels were analyzed in the cortex (A) and hippocampus (B) from wild-type C57BL/6 mice by Western blotting at the ages of 3, 18, 24 and 30 months (n=4/group). (C and D) GFAP levels were analyzed in the cortex (C) and hippocampus (D) from the above described mice by ELISA (n=4–9/group). Data are plotted as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001.
PSD-95 is a scaffolding protein in the post-synaptic density that organizes postsynaptic proteins and glutamate receptors. It plays a vital role in synaptic plasticity; PSD-95 knockout mice have deficits in hippocampal LTP (Carlisle, et al., 2008, Yao, et al., 2004). Synaptophysin is a presynaptic protein that also plays a critical role in synaptic function and related cognition (Schmitt, et al., 2009). To determine if aging is associated with compromised synaptic integrity, we examined the protein levels of PSD-95 and synaptophysin in our aging cohorts. Western blotting showed that PSD-95 levels were decreased in 30 month-old mice compared to 3 month-old mice in both cortex (Fig. 2A, B) and hippocampus (Fig. 2D, E). Although there was a modest reduction, PSD-95 levels were not significantly reduced in 18 month- or 24 month-old mice when compared to 3 month-old mice (Fig. 2A–E). However, there were no significant differences in synaptophysin levels between cohorts in either the cortex (Fig. 2A, C) or hippocampus (Fig. 2D, F). Reduced PSD-95 levels in 30 month-old advanced-aged mice in cortex (Fig. 2G) and hippocampus (Fig. 2H) were also confirmed by ELISA. Taken together, these findings indicate that significant decline of postsynaptic proteins become evident only after 30 months of age, although presynaptic proteins appear to be relatively preserved during aging.
Fig. 2.

Postsynaptic marker PSD-95 is selectively decreased in the brains of advanced-aged mice. (A–C) Levels of a postsynaptic marker PSD-95 and a presynaptic marker synaptophysin (Syn) were analyzed in the cortex from wild-type C57BL/6 mice by Western blotting at the ages of 3, 18, 24 and 30 months (n=4/group). (D–F) Levels of PSD-95 and synaptophysin were analyzed in the hippocampus from the above described mice by Western blotting (n=4/group). (G and H) PSD-95 levels were analyzed in the cortex (G) and hippocampus (H) from the above described mice by ELISA (n=4–9/group). Data are plotted as mean ± SEM. *p<0.05, **p<0.01.
Aging disturbs hippocampal synaptic plasticity and cognition
With the use of extracellular recording techniques, we determined how aging influences synaptic transmission and plasticity in hippocampi of our aging cohort. We found that theta burst stimulation-induced LTP between Schaffer collateral-CA1 region was diminished in an age-dependent manner (Fig. 3A–C); 24 month- and 30 month-old mice had significant suppression of LTP compared to 3 month-old mice, while 18 month-old mice had a trend for the reduction of LTP compared to 3 month-old mice. Also, there was a significant difference in LTP between 18 month- and 30 month-old mice (Fig. 3D). In addition, we did not observe any significant changes in short-term plasticity evaluated by PPF testing between experimental groups (Fig. 3E), which indicates preserved presynaptic-dependent function. Taken together, these data suggest that LTP is gradually, but progressively, disturbed during aging in the mouse hippocampus likely due to postsynaptic alterations.
Fig. 3.

Mouse hippocampal LTP is suppressed in an aging-dependent manner. (A–C) LTP was induced with theta burst stimulation (5 bursts of 200 Hz separated by 200 msec, repeated 6 times with 10 sec between the 6 trains) after 20 min of baseline recording and changes in fEPSP slope are expressed as a percentage of baseline. LTP expression of 3 month (A–C, n=13), 18 month (A, n=20), 24 month (B, n=14) and 30 month (C, n=14) old wild-type C57BL/6 are shown. (D) The last 5 min of fEPSPs slope recordings were averaged for LTP analysis. (E) Paired-pulse facilitation (PPF) was induced with the use of paired-pulses given with an initial delay of 20 msec and the time to the second pulse was increased 20 msec incrementally until a final delay of 300 msec was reached. Data are plotted as mean ± SEM. *p<0.05, **p<0.01.
To assess aging effects on mouse cognition, we first examined associative fear conditioned learning and memory in our aging cohort. Mice were trained with a standard two-shock protocol as previously described (Cook, et al., 2014, Cook, et al., 2015, Rogers, et al., 2011). To assess if aging diminishes contextual memory, which is hippocampal dependent, the mice were tested for freezing to the context at 24 hours after training. We found that context-dependent freezing was diminished as early as at 18 months of age without progression (Fig. 4A). Changes in fear memory to the cue, which is both amygdala and hippocampal dependent, were significantly diminished only in 30 month-old cohort compared to 3 month-old mice 24 hours after training (Fig. 4B). These results suggest that hippocampal dependent associative learning and memory, rather than amygdala dependent, are vulnerable during aging.
Fig. 4.

Aging compromises cognitive performance in mice. (A and B) Associative memory was assessed in wild-type C57BL/6 mice with the use of fear conditioning with a standard 2-shock protocol at the ages of 3 months (n=14), 18 months (n=14), 24 months (n=14) and 30 months (n=13). Freezing to the context (A) or the cue (B) was assessed for 3 min at 24 hr post-training. (C–F) Hippocampal-dependent spatial memory was tested by a hidden platform water maze (HPWM) paradigm. Mice were trained in the HPWM for 4 days with 4 trials per day. On day 5, the platform was removed and a 60 sec probe trial was conducted. Travel distances were plotted against the training days (C). Frequency of platform crossings (D) and target quadrant entries (E) during the probe trail, and Swim speed (F) were also assessed. Data are plotted as mean ± SEM. *p<0.05, **p<0.01.
To determine if aging affects spatial learning and memory, the mice were tested in the HPWM task. Mice were trained using an experimental paradigm of four trials per day with an inter-trial interval of 1 min. There were no significant differences in distance to find the hidden platform during training days between the cohorts with different ages (Fig. 4C). However, 24 month- and 30 month-old mice had significantly fewer platform crossings (Fig. 4D) and target quadrant entries (Fig. 4E) compared to young mice during the probe trial conducted on day 5, indicating diminished memory retention of platform location. There was a modest decline of target quadrant entries and platform crossing in 18 month-old mice, but this effect was not significant (Fig. 4D, E). These data demonstrate that aging diminishes spatial learning in wild-type mice beginning at 24 months of age.
Aging is associated with decreased motor function and motor memory. In fact, young mice had faster swim speed compared to the aged and advanced-aged mice in HPWM task (Fig. 4F). Thus, we next tested these mice on the rotarod spinning wheel task. Mice were tested for the time spent on the rod during each of the four trials per day, for two consecutive days. Although all groups spent significantly more time on the rod during the last trial of training compared to the first trial, 3 month-old mice spent significantly more time on the rod compared to 18, 24 and 30 month-old cohorts (Fig. 5A). These findings suggest that middle-age, aged and advanced-aged mice have motor function deficits compared to young mice, but maintain motor memory function at least up to 30 months of age.
Fig. 5.
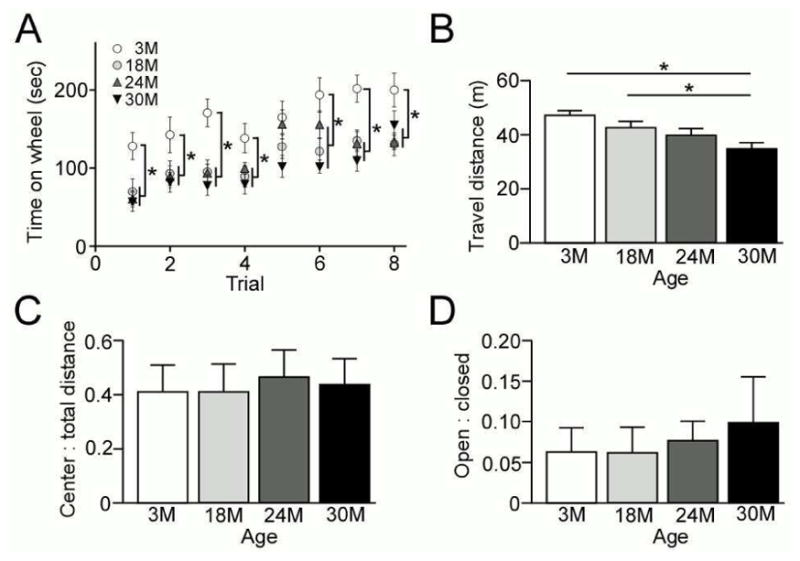
Aged mice exhibit locomotor deficits. (A) Wild-type C57BL/6 mice were tested on a standard rotarod apparatus for two days with 4 trials per day at the ages of 3 months (n=14), 18 months (n=14), 24 months (n=14) and 30 months (n=13). (B and C) The mice were tested in the open field assay and distance traveled and center:total distance ratio were analyzed. Note, advanced aged mice (30 month old mice, Black; n=13) travel less total distance compared to 3 month old (white; n=14) and 18 month old (red; n=14) mice (*, p<0.05). Data represent the distance traveled (B) and the center:total distance ratio (C). (D) Mice underwent elevated plus maze testing for anxiety-like behavior. Data represent the ratio of time spent in the open arms vs. the closed arms of the elevated maze. Data are plotted as mean ± SEM. *p<0.05.
Furthermore, the mice were tested in the open field and elevated plus maze (EPM) to determine if aging alters somatosensory input and anxiety levels. We found that 30 month-old mice traveled significantly less than the 3 month- and 18 month-old mice in the open field (Fig. 5B), indicating a general decrease in locomotor activity. However, we found that aging had no effect on the center:total distance ratio in the open field among the groups, suggesting that anxiety-like behavior was not impacted by aging (Fig. 5C). Consistent with this, the EPM test also did not detect age-dependent differences in the ratio of time spent in the open arms to the closed arms of the elevated maze (Fig. 5D). Thus, aging has no significant effect on anxiety levels in these mice, and therefore the aging effects observed in spatial or associative learning are not attributable to alterations in anxiety, but rather cognitive function.
Aging-related LTP and cognitive deficits are rescued with subacute ibuprofen treatment
To determine if the aging-associated neuroinflammation and synaptic deficits are reversible with pharmacological approaches, we subacutely treated 29 month-old mice with or without ibuprofen administered in drinking water for 6 weeks. Following the treatment, levels of GFAP, PSD-95 and synaptophysin were analyzed in the hippocampus by Western blotting (Fig. 6A–C). While GFAP levels were elevated in the advanced-aged 30 month-old mice compared to young 3 month-old mice, the mice treated with ibuprofen had significantly reduced levels of GFAP (Fig. 6B), comparable to those of young mice. However, there were no ibuprofen-related alterations in the levels of PSD-95 (Fig. 6C). Taken together, these findings suggest that the chronic and progressive increase of neuroinflammation is reversible, even at an advanced age, through pharmacological approaches.
Fig. 6.
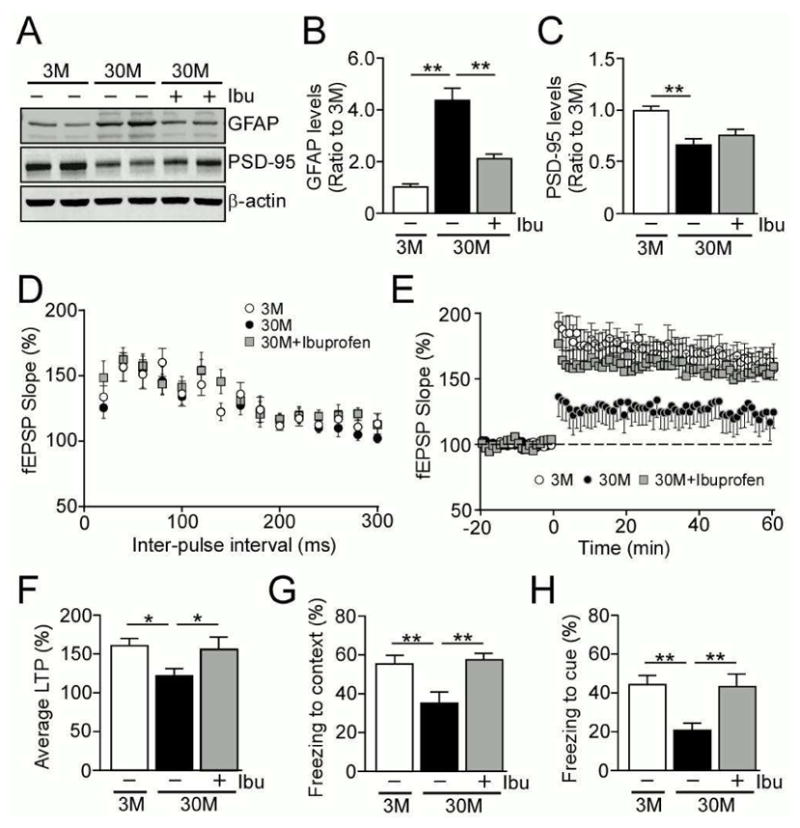
Subacute ibuprofen treatment rescues the aging-related LTP deficit and cognitive decline. (A–C) Levels of GFAP and PSD-95 were analyzed by Western blotting in the hippocampus of 3 month and 30 month old wild-type C57BL/6 with or without ibuprofen treatment (n=4/group). (D–F) LTP of 3 month (n=11) and 30 month (n=12) old wild-type C57BL/6 without ibuprofen treatment, and 30 month old mice (n=12) with ibuprofen treatment were measured. PPF was induced with the use of paired-pulses given with an initial delay of 20 msec and the time to the second pulse was increased 20 msec incrementally until a final delay of 300 msec was reached (D). The LTP expressions of the mice are shown (E). The last 5 min of fEPSPs slope recordings were averaged for LTP analysis (F). (G and H) The mice were analyzed by fear conditioning test with a standard 2-shock protocol at the ages of 3 months (n=20) and 30 months (n=15) without ibuprofen treatment, and 30 months (n=15) with ibuprofen treatment. Freezing to the context (G) or the cue (H) was assessed for 3 min at 24 hr post-training. Data are plotted as mean ± SEM. *p<0.05, **p<0.01.
To determine effects of ibuprofen treatment on synaptic plasticity in advanced-aged mice, 30 month-old mice were treated with or without ibuprofen containing water for 6 weeks. Following treatment, LTP was measured in the Schaffer collateral-CA1 region and compared with young 3 month-old mice. PPF testing to measure presynaptic function detected no significant changes among the experimental groups (Fig. 6D). While 30 month-old mice had significantly reduced LTP compared to 3 month-old mice, subacute treatment with ibuprofen restored LTP in the advanced-aged mice to the levels that are indistinguishable from young mice (Fig. 6E, F). Furthermore, consistent with the results from LTP, associative memory as tested by fear conditioning was also improved with subacute ibuprofen treatment in the 30 month-old mice both in contextual (Fig. 6G) and cued (Fig. 6H) conditions. These findings suggest that aging-related deficits of LTP and associative memory can be rescued through the subacute treatment with anti-inflammatory drugs such as ibuprofen.
4. Discussion
In spite of the fact that cognitive performance is unequivocally influenced by aging, our knowledge regarding the pathogenesis of aging-related cognitive decline is still limited. Effective therapeutic interventions for the pathogenic conditions have also not been established. Rodent models of aging and cognitive decline are used to mimic human aging in order to identify the underlying molecular mechanisms of cognitive aging. In humans, aging is associated with a decline of working memory, executive function, and processing speed. Notably, impaired episodic memory, including spatial memory is associated with aging (Kukolja, et al., 2009, Plancher, et al., 2010, Uttl and Graf, 1993). The age-related impairment is identified as a mild deficit in acquisition of information and an increase in forgetfulness (Davis, et al., 2003). The decline of cognitive function in humans is progressive, usually starting at middle age, and an increase in forgetting may be an early sign of impaired synaptic transmission and plasticity (Christensen, et al., 1999, Colsher and Wallace, 1991, Mungas, et al., 2005, Schonknecht, et al., 2005).
In rodents, memory deficits are related to impaired hippocampal function. The progressive decline of memory is associated with a decrease in hippocampal volume (Kramer, et al., 2007, Mueller, et al., 2007, Mungas, et al., 2005, Reuter-Lorenz and Park, 2010). The “calcium hypothesis of brain aging” states that aging alters calcium regulation, which results in impairment of neuronal function (Khachaturian, 1984, Landfield and Pitler, 1984). Specifically in rat models of aging, calcium-dependent processes associated with learning and memory are altered in the hippocampus. These changes include increased slow after hyperpolarizations (sAHP) (Disterhoft, et al., 1996, Landfield and Pitler, 1984, Moyer, et al., 1992), deficits in LTP (Barnes and Kidd, 1979, Barnes, 1994, Shankar, et al., 1998), and increased long-term depression (Norris, et al., 1998, Norris, et al., 1996). Furthermore, the decline of these hippocampal processes are correlated with memory deficits in hippocampal-dependent memory tasks (Thibault and Landfield, 1996). However, to our knowledge, a comprehensive study of the effects of aging on cognitive function, synaptic integrity and inflammation have not been conducted in a mouse model of aging. In this study, we have comprehensively assessed the effects of aging on mouse cognitive function and synaptic plasticity by comparing these phenotypes in wild-type mice with increasing ages (3, 18, 24 and 30 months). We found that mice had hippocampal-dependent associative memory deficits at 18 months of age and older as tested in contextual fear conditioning. In fact, associative memory observed as a decrease in recall and recognition is one of the cognitive abilities that are predominantly impaired with aging in humans (Johnson, et al., 2008, Troyer, et al., 2011). On the other hand, spatial cognitive deficits were first observed in older 24 month-old mice as tested by HPWM task in our mouse cohorts. Thus, these variable vulnerabilities across the lifespan suggest that aging has distinctive effects on different types of learning and memory. Synaptic plasticity monitored by LTP generally represents the cellular mechanisms of learning and memory (McNaughton, et al., 1986, Morris, et al., 1986a, Moser, et al., 1998). Consistent with the results from HPWM test, significant LTP deficits were noticeable in 24 month-old mice and this decline was further exacerbated in 30 month-old mice. In rats, impairments in temporal/spatial learning memory (Morris, et al., 2013, Morris, et al., 1982, Morris, et al., 1986b, Moser, et al., 1998, Thibault and Landfield, 1996) and LTP (Rex, et al., 2005, Rex, et al., 2006, Wang, et al., 2012) begin as early as 18 months of age. The differences observed between rats and mice may be explained by species variance and/or methods used for experimentation or analysis. Nonetheless, our studies are in general agreement with what is observed in the aging rat model and validate that mice can be used to model CNS and cognitive aging.
Neuroinflammation is a common feature of brain aging (Ojo, et al., 2015). GFAP expression is increased in activated astrocytes, representing the inflammatory state of the CNS (Haley, et al., 2010). GFAP levels progressively increase with age in humans, rhesus macaques and rodents (Haley, et al., 2010, Middeldorp and Hol, 2011). We also demonstrate that upregulation of GFAP levels had already begun to increase at the age of 18 months in mice, which progressively increase in an age-dependent manner. Of note, the age-dependent astrocyte activation preceded spatial cognitive deficits and LTP suppression in our mouse cohorts. Aging-related changes in LTP have been reported for all regions of the hippocampus, where the neuroinflammatory changes and oxidative stress are likely central events that diminish LTP (Lynch, 1998a, Lynch, 1998b, Lynch, 2010). Indeed, repeated mild traumatic brain injury leads to chronic neuroinflammation in mice, resulting in synaptic plasticity deficits and associative cognitive deficits (Aungst, et al., 2014). This evidence demonstrates the impact of neuroinflammation on synaptic plasticity.
Most importantly, our findings clearly demonstrated that subacute ibuprofen treatment rescues LTP deficits as well as contextual and associative memory deficits in advanced-aged mice, which is accompanied with reduced GFAP levels in the brain. While subacute ibuprofen treatment improves memory and synaptic plasticity in amyloid AD mouse models (Kotilinek, et al., 2008, Van Dam, et al., 2010), to our knowledge, this is a first report showing the protective effects of ibuprofen against cognitive decline in aged wild-type mice. Ibuprofen is one of the classical types of NSAIDs, which suppresses the prostaglandin biogenesis from arachidonic acid by inhibiting functions of cyclooxygenase (COX)-1 and COX-2 (Cunningham and Skelly, 2012). Chronic inflammation occurs in the hippocampus of aged rats. Interleukin-1β (IL1β), tumor necrosis factor α, and prostaglandin E2 increase with age in the rat hippocampus. Importantly, chronic oral treatment with celecoxib, a selective COX-2 inhibitor, rescues the age-dependent increase in hippocampal neuroinflammation in rats (Casolini, et al., 2002). Treatment with sulindac, a non-specific COX inhibitor, also prevents the age-related increase in the pro-inflammatory cytokine IL1β in the hippocampus of aged rats. This rescue is accompanied by reduced age-related deficits in hippocampal-dependent learning and memory as assessed in the radial arm water maze and contextual fear conditioning tasks. Sulindac treatment also attenuated an age-related decrease in the NR1 and NR2B NMDA receptor subunits, which are associated with PSD-95 at the post-synaptic cleft (Mesches, et al., 2004). Since prostaglandins trigger diverse inflammatory cascades in the brain (Cunningham and Skelly, 2012), subacute ibuprofen treatment may also rescue aging-related cognitive declines by preventing the prostaglandin-mediated neuroinflammation via COX-2 inhibition. However, COX-2 is controversially required to mediate the consolidation of hippocampal-dependent memory in rats (Teather, et al., 2002). Therefore, it is possible that the beneficial effects of ibuprofen treatment may be independent of COX-2. Consistent with our results, proteomic analysis of the hippocampus of mice treated with ibuprofen for 6 months also identified the down-regulations of GFAP (Matsuura, et al., 2015), although further studies are needed to determine whether ibuprofen directly influences GFAP levels or indirectly modify them by blocking prostaglandin-mediated neuroinflammation. Thus, astrocytes are predicted to play a critical role in regulating cognitive functions during aging.
In addition, aging-related cognitive deficits are likely associated with reduced postsynaptic density in hippocampal excitatory synapses (Nicholson, et al., 2004). We also show that PSD-95 is significantly decreased in the cortex and hippocampus at 30 months of age, while the presynaptic marker synaptophysin was unaltered. Post-synaptic receptors for neurotransmitters are most highly concentrated in the post-synaptic density (Kennedy, 2000), where PSD-95 serves as a scaffolding protein (de Bartolomeis, et al., 2013). In fact, mutant PSD-95 causes LTP deficits and cognitive declines in a mouse model (Migaud, et al., 1998). However, since PSD-95 was only decreased at 30 months of age in our aging mouse cohorts, deficits of synaptic integrity cannot account for all of the deficits observed in the aging mouse. Furthermore, we found that subacute ibuprofen treatment rescues aging-related cognitive declines without ameliorating the reduction of PSD-95 levels. Thus, the activation of astrocytes and/or immune system may be an initiating event to cause the disturbances of postsynaptic density during aging, while it is possible that they are an independent phenomenon.
In summary, our studies expand the knowledge for aging-related cognitive decline using the aging mouse model at middle-aged, aged and advanced ages. We demonstrate that astrogliosis continues to increase throughout the life span of the mouse, which is associated with decreased synaptic plasticity and hippocampal-dependent cognitive deficits. Importantly, the aging-associated deficits in synaptic plasticity and cognition are rescued with subacute ibuprofen treatment. Therefore, our findings provide strong evidence that astrocyte-related neuroinflammation is a major factor contributing to aging-related cognitive deficits, which is a promising therapeutic target to treat aging-related synaptic and cognitive decline.
Highlights.
Aging disturbs hippocampal synaptic plasticity and cognition
Astrocyte-related neuroinflammation contributes to aging-related cognitive deficits
Aging-related synaptic and cognitive deficits are rescued with ibuprofen treatment
Acknowledgments
This work was supported by NIH grants R01AG035355, R01AG046205, R01AG027924, 1RF1AG051504, P50AG016574, and P01NS074969 (to G.B.), a Cure Alzheimer’s Fund (to G.B.), a fellowship from the BrightFocus (to C.-C.L.), and New Investigator Research Grant from the Alzheimer’s Association (to T.K.).
Footnotes
Disclosure statement
The authors have no conflicts of interest to disclosure in relation to this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aungst SL, Kabadi SV, Thompson SM, Stoica BA, Faden AI. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab. 2014 doi: 10.1038/jcbfm.2014.75.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes IE, Kidd EA. Disappearing Dycal. Br Dent J. 1979;147(5):111. doi: 10.1038/sj.bdj.4804296. [DOI] [PubMed] [Google Scholar]
- Barnes S. After transduction: response shaping and control of transmission by ion channels of the photoreceptor inner segments. Neuroscience. 1994;58(3):447–59. doi: 10.1016/0306-4522(94)90072-8. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Fink AE, Grant SG, O’Dell TJ. Opposing effects of PSD-93 and PSD-95 on long-term potentiation and spike timing-dependent plasticity. J Physiol. 2008;586(Pt 24):5885–900. doi: 10.1113/jphysiol.2008.163469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casolini P, Catalani A, Zuena AR, Angelucci L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J Neurosci Res. 2002;68(3):337–43. doi: 10.1002/jnr.10192. [DOI] [PubMed] [Google Scholar]
- Christensen H, Mackinnon AJ, Korten AE, Jorm AF, Henderson AS, Jacomb P, Rodgers B. An analysis of diversity in the cognitive performance of elderly community dwellers: individual differences in change scores as a function of age. Psychol Aging. 1999;14(3):365–79. doi: 10.1037//0882-7974.14.3.365. [DOI] [PubMed] [Google Scholar]
- Colsher PL, Wallace RB. Longitudinal application of cognitive function measures in a defined population of community-dwelling elders. Ann Epidemiol. 1991;1(3):215–30. doi: 10.1016/1047-2797(91)90001-s. [DOI] [PubMed] [Google Scholar]
- Cook C, Dunmore JH, Murray ME, Scheffel K, Shukoor N, Tong J, Castanedes-Casey M, Phillips V, Rousseau L, Penuliar MS, Kurti A, Dickson DW, Petrucelli L, Fryer JD. Severe amygdala dysfunction in a MAPT transgenic mouse model of frontotemporal dementia. Neurobiol Aging. 2014;35(7):1769–77. doi: 10.1016/j.neurobiolaging.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Kang SS, Carlomagno Y, Lin WL, Yue M, Kurti A, Shinohara M, Jansen-West K, Perkerson E, Castanedes-Casey M, Rousseau L, Phillips V, Bu G, Dickson DW, Petrucelli L, Fryer JD. Tau deposition drives neuropathological, inflammatory and behavioral abnormalities independently of neuronal loss in a novel mouse model. Hum Mol Genet. 2015;24(21):6198–212. doi: 10.1093/hmg/ddv336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Skelly DT. Non-steroidal anti-inflammatory drugs and cognitive function: are prostaglandins at the heart of cognitive impairment in dementia and delirium? J Neuroimmune Pharmacol. 2012;7(1):60–73. doi: 10.1007/s11481-011-9312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis HP, Small SA, Stern Y, Mayeux R, Feldstein SN, Keller FR. Acquisition, recall, and forgetting of verbal information in long-term memory by young, middle-aged, and elderly individuals. Cortex. 2003;39(4–5):1063–91. doi: 10.1016/s0010-9452(08)70878-5. [DOI] [PubMed] [Google Scholar]
- de Bartolomeis A, Sarappa C, Buonaguro EF, Marmo F, Eramo A, Tomasetti C, Iasevoli F. Different effects of the NMDA receptor antagonists ketamine, MK-801, and memantine on postsynaptic density transcripts and their topography: Role of Homer signaling, and implications for novel antipsychotic and pro-cognitive targets in psychosis. Prog Neuropsychopharmacol Biol Psychiatry. 2013;46:1–12. doi: 10.1016/j.pnpbp.2013.06.010. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Thompson LT, Moyer JR, Jr, Mogul DJ. Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus. Life Sci. 1996;59(5–6):413–20. doi: 10.1016/0024-3205(96)00320-7. [DOI] [PubMed] [Google Scholar]
- Fox JG. The mouse in biomedical research. 2. Elsevier; AP, Amsterdam ; Boston: 2007. [Google Scholar]
- Haley GE, Kohama SG, Urbanski HF, Raber J. Age-related decreases in SYN levels associated with increases in MAP-2, apoE, and GFAP levels in the rhesus macaque prefrontal cortex and hippocampus. Age (Dordr) 2010;32(3):283–96. doi: 10.1007/s11357-010-9137-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Tsuchiyama SK, Nguyen QT, Plyusnina EN, Terrill SR, Sahibzada S, Patel B, Faulkner AR, Shaposhnikov MV, Tian R, Tsuchiya M, Kaeberlein M, Moskalev AA, Kennedy BK, Polymenis M. Enhanced longevity by ibuprofen, conserved in multiple species, occurs in yeast through inhibition of tryptophan import. PLoS Genet. 2014;10(12):e1004860. doi: 10.1371/journal.pgen.1004860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedden T, Gabrieli JD. Insights into the ageing mind: a view from cognitive neuroscience. Nat Rev Neurosci. 2004;5(2):87–96. doi: 10.1038/nrn1323. nrn1323 [pii] [DOI] [PubMed] [Google Scholar]
- in t’ Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345(21):1515–21. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Schmitz TW, Asthana S, Gluck MA, Myers C. Associative learning over trials activates the hippocampus in healthy elderly but not mild cognitive impairment. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn. 2008;15(2):129–45. doi: 10.1080/13825580601139444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MB. Signal-processing machines at the postsynaptic density. Science. 2000;290(5492):750–4. doi: 10.1126/science.290.5492.750. 8929 [pii] [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Scientific challenges and opportunities related to Alzheimer’s disease. Clin Pharm. 1984;3(5):522–3. [PubMed] [Google Scholar]
- Kofidis T, Lebl DR, Swijnenburg RJ, Greeve JM, Klima U, Robbins RC. Allopurinol/uricase and ibuprofen enhance engraftment of cardiomyocyte-enriched human embryonic stem cells and improve cardiac function following myocardial injury. Eur J Cardiothorac Surg. 2006;29(1):50–5. doi: 10.1016/j.ejcts.2005.10.015. S1010-7940(05)00773-6 [pii] [DOI] [PubMed] [Google Scholar]
- Kotilinek LA, Westerman MA, Wang Q, Panizzon K, Lim GP, Simonyi A, Lesne S, Falinska A, Younkin LH, Younkin SG, Rowan M, Cleary J, Wallis RA, Sun GY, Cole G, Frautschy S, Anwyl R, Ashe KH. Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain. 2008;131(Pt 3):651–64. doi: 10.1093/brain/awn008. 131/3/651 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer AF, Cassavaugh N, Horrey WJ, Becic E, Mayhugh JL. Influence of age and proximity warning devices on collision avoidance in simulated driving. Hum Factors. 2007;49(5):935–49. doi: 10.1518/001872007X230271. [DOI] [PubMed] [Google Scholar]
- Kukolja J, Thiel CM, Wilms M, Mirzazade S, Fink GR. Ageing-related changes of neural activity associated with spatial contextual memory. Neurobiol Aging. 2009;30(4):630–45. doi: 10.1016/j.neurobiolaging.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Pitler TA. Prolonged Ca2+-dependent after hyperpolarizations in hippocampal neurons of aged rats. Science. 1984;226(4678):1089–92. doi: 10.1126/science.6494926. [DOI] [PubMed] [Google Scholar]
- Lynch MA. Age-related impairment in long-term potentiation in hippocampus: a role for the cytokine, interleukin-1 beta? Prog Neurobiol. 1998a;56(5):571–89. doi: 10.1016/s0301-0082(98)00054-9. [DOI] [PubMed] [Google Scholar]
- Lynch MA. Analysis of the mechanisms underlying the age-related impairment in long-term potentiation in the rat. Rev Neurosci. 1998b;9(3):169–201. doi: 10.1515/revneuro.1998.9.3.169. [DOI] [PubMed] [Google Scholar]
- Lynch MA. Age-related neuroinflammatory changes negatively impact on neuronal function. Frontiers in aging neuroscience. 2010;1:6. doi: 10.3389/neuro.24.006.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura K, Otani M, Takano M, Kadoyama K, Matsuyama S. The influence of chronic ibuprofen treatment on proteins expressed in the mouse hippocampus. Eur J Pharmacol. 2015;752:61–8. doi: 10.1016/j.ejphar.2015.01.047. S0014-2999(15)00080-1 [pii] [DOI] [PubMed] [Google Scholar]
- Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nat Rev Neurosci. 2006;7(4):278–94. doi: 10.1038/nrn1886. nrn1886 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton BL, Barnes CA, Rao G, Baldwin J, Rasmussen M. Long-term enhancement of hippocampal synaptic transmission and the acquisition of spatial information. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1986;6(2):563–71. doi: 10.1523/JNEUROSCI.06-02-00563.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesches MH, Gemma C, Veng LM, Allgeier C, Young DA, Browning MD, Bickford PC. Sulindac improves memory and increases NMDA receptor subunits in aged Fischer 344 rats. Neurobiol Aging. 2004;25(3):315–24. doi: 10.1016/S0197-4580(03)00116-7. [DOI] [PubMed] [Google Scholar]
- Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol. 2011;93(3):421–43. doi: 10.1016/j.pneurobio.2011.01.005. S0301-0082(11)00006-2 [pii] [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O’Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396(6710):433–9. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Morris GP, Clark IA, Zinn R, Vissel B. Microglia: a new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol Learn Mem. 2013;105:40–53. doi: 10.1016/j.nlm.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986a;319(6056):774–6. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O’Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297(5868):681–3. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Morris RG, Hagan JJ, Rawlins JN. Allocentric spatial learning by hippocampectomised rats: a further test of the “spatial mapping” and “working memory” theories of hippocampal function. Q J Exp Psychol B. 1986b;38(4):365–95. [PubMed] [Google Scholar]
- Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13(4):240–50. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser EI, Krobert KA, Moser MB, Morris RG. Impaired spatial learning after saturation of long-term potentiation. Science. 1998;281(5385):2038–42. doi: 10.1126/science.281.5385.2038. [DOI] [PubMed] [Google Scholar]
- Moyer JR, Jr, Thompson LT, Black JP, Disterhoft JF. Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J Neurophysiol. 1992;68(6):2100–9. doi: 10.1152/jn.1992.68.6.2100. [DOI] [PubMed] [Google Scholar]
- Mueller SG, Stables L, Du AT, Schuff N, Truran D, Cashdollar N, Weiner MW. Measurement of hippocampal subfields and age-related changes with high resolution MRI at 4T. Neurobiol Aging. 2007;28(5):719–26. doi: 10.1016/j.neurobiolaging.2006.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungas D, Harvey D, Reed BR, Jagust WJ, DeCarli C, Beckett L, Mack WJ, Kramer JH, Weiner MW, Schuff N, Chui HC. Longitudinal volumetric MRI change and rate of cognitive decline. Neurology. 2005;65(4):565–71. doi: 10.1212/01.wnl.0000172913.88973.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DA, Yoshida R, Berry RW, Gallagher M, Geinisman Y. Reduction in size of perforated postsynaptic densities in hippocampal axospinous synapses and age-related spatial learning impairments. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24(35):7648–53. doi: 10.1523/JNEUROSCI.1725-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Halpain S, Foster TC. Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J Neurosci. 1998;18(9):3171–9. doi: 10.1523/JNEUROSCI.18-09-03171.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Korol DL, Foster TC. Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. J Neurosci. 1996;16(17):5382–92. doi: 10.1523/JNEUROSCI.16-17-05382.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo JO, Rezaie P, Gabbott PL, Stewart MG. Impact of age-related neuroglial cell responses on hippocampal deterioration. Frontiers in aging neuroscience. 2015;7:57. doi: 10.3389/fnagi.2015.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plancher G, Gyselinck V, Nicolas S, Piolino P. Age effect on components of episodic memory and feature binding: A virtual reality study. Neuropsychology. 2010;24(3):379–90. doi: 10.1037/a0018680. [DOI] [PubMed] [Google Scholar]
- Reuter-Lorenz PA, Park DC. Human neuroscience and the aging mind: a new look at old problems. J Gerontol B Psychol Sci Soc Sci. 2010;65(4):405–15. doi: 10.1093/geronb/gbq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Kramar EA, Colgin LL, Lin B, Gall CM, Lynch G. Long-term potentiation is impaired in middle-aged rats: regional specificity and reversal by adenosine receptor antagonists. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25(25):5956–66. doi: 10.1523/JNEUROSCI.0880-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Lauterborn JC, Lin CY, Kramar EA, Rogers GA, Gall CM, Lynch G. Restoration of long-term potentiation in middle-aged hippocampus after induction of brain-derived neurotrophic factor. Journal of neurophysiology. 2006;96(2):677–85. doi: 10.1152/jn.00336.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JT, Rusiana I, Trotter J, Zhao L, Donaldson E, Pak DT, Babus LW, Peters M, Banko JL, Chavis P, Rebeck GW, Hoe HS, Weeber EJ. Reelin supplementation enhances cognitive ability, synaptic plasticity, and dendritic spine density. Learn Mem. 2011;18(9):558–64. doi: 10.1101/lm.2153511. 18/9/558 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JT, Zhao L, Trotter JH, Rusiana I, Peters MM, Li Q, Donaldson E, Banko JL, Keenoy KE, Rebeck GW, Hoe HS, D’Arcangelo G, Weeber EJ. Reelin supplementation recovers sensorimotor gating, synaptic plasticity and associative learning deficits in the heterozygous reeler mouse. J Psychopharmacol. 2013;27(4):386–95. doi: 10.1177/0269881112463468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt U, Tanimoto N, Seeliger M, Schaeffel F, Leube RE. Detection of behavioral alterations and learning deficits in mice lacking synaptophysin. Neuroscience. 2009;162(2):234–43. doi: 10.1016/j.neuroscience.2009.04.046. [DOI] [PubMed] [Google Scholar]
- Schonknecht P, Pantel J, Kruse A, Schroder J. Prevalence and natural course of aging-associated cognitive decline in a population-based sample of young-old subjects. Am J Psychiatry. 2005;162(11):2071–7. doi: 10.1176/appi.ajp.162.11.2071. [DOI] [PubMed] [Google Scholar]
- Shankar S, Teyler TJ, Robbins N. Aging differentially alters forms of long-term potentiation in rat hippocampal area CA1. J Neurophysiol. 1998;79(1):334–41. doi: 10.1152/jn.1998.79.1.334. [DOI] [PubMed] [Google Scholar]
- Shinohara M, Petersen RC, Dickson DW, Bu G. Brain regional correlation of amyloid-beta with synapses and apolipoprotein E in non-demented individuals: potential mechanisms underlying regional vulnerability to amyloid-beta accumulation. Acta Neuropathol. 2013;125(4):535–47. doi: 10.1007/s00401-013-1086-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teather LA, Packard MG, Bazan NG. Post-training cyclooxygenase-2 (COX-2) inhibition impairs memory consolidation. Learn Mem. 2002;9(1):41–7. doi: 10.1101/lm.43602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272(5264):1017–20. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- Troyer AK, D’Souza NA, Vandermorris S, Murphy KJ. Age-related differences in associative memory depend on the types of associations that are formed. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn. 2011;18(3):340–52. doi: 10.1080/13825585.2011.553273. [DOI] [PubMed] [Google Scholar]
- Uttl B, Graf P. Episodic spatial memory in adulthood. Psychol Aging. 1993;8(2):257–73. doi: 10.1037//0882-7974.8.2.257. [DOI] [PubMed] [Google Scholar]
- Van Dam D, Coen K, De Deyn PP. Ibuprofen modifies cognitive disease progression in an Alzheimer’s mouse model. J Psychopharmacol. 2010;24(3):383–8. doi: 10.1177/0269881108097630. 0269881108097630 [pii] [DOI] [PubMed] [Google Scholar]
- Vincent GK, Velkof VA. The next four decades: the older population in the United States: 2010 to 2050. Washington, DC: US Census Bureau; 2010. [Google Scholar]
- Wang BW, Hok V, Della-Chiesa A, Callaghan C, Barlow S, Tsanov M, Bechara R, Irving E, Virley DJ, Upton N, O’Mara SM. Rosiglitazone enhances learning, place cell activity, and synaptic plasticity in middle-aged rats. Neurobiol Aging. 2012;33(4):835e13–30. doi: 10.1016/j.neurobiolaging.2011.08.013. [DOI] [PubMed] [Google Scholar]
- Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, Caron MG. Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron. 2004;41(4):625–38. doi: 10.1016/s0896-6273(04)00048-0. [DOI] [PubMed] [Google Scholar]