

Abstract
AIM
To explore the exact interaction between Notch and transforming growth factor (TGF)-β signaling in liver fibrosis.
METHODS
We established a rat model of liver fibrosis induced by concanavalin A. Peripheral blood mononuclear cells (PBMCs) were isolated from the modeled rats, and cultured with γ-secretase inhibitor DAPT and TGF-β inhibitor for 24 h. The mRNA levels of Notch and TGF-β signaling were detected by quantitative real-time polymerase chain reaction. Expression of Notch and TGF-β proteins was analyzed by western blotting.
RESULTS
Compared to control rats, Notch and TGF-β signaling was activated in PBMCs of model rats. Administration of DAPT and TGF-β inhibitor suppressed Notch and TGF-β signal transducer in PBMCs of model rats. DAPT reduced mRNA and protein expression of TGF-β signaling, such as TGF-β1 and Smad3. TGF-β inhibitor also downregulated Notch1, Hes1 and Hes5, and mRNA and protein expression of the Notch signaling pathway.
CONCLUSION
Notch and TGF-β signaling play a role in liver fibrosis. TGF-β signaling upregulates Notch signaling, which promotes TGF-β signaling.
Keywords: Notch, Peripheral blood mononuclear cells, Concanavalin A, Transforming growth factor-β, Liver fibrosis
Core tip: Notch and transforming growth factor (TGF)-β activation plays an important role in liver fibrosis induced by concanavalin A. It has been shown that TGF-β facilitates liver fibrosis. However, the mechanism of action of Notch in liver fibrosis is not fully understood. In this study, we found that excessive activation of TGF-β regulated Notch in liver fibrosis in rats, and that inhibition of TGF-β signaling blocked Notch signaling and vice versa. This may be a complementary mechanism of liver fibrosis, which provides a potential immunotherapeutic strategy.
INTRODUCTION
Liver fibrosis occurs as a result of chronic liver disease and is associated with severe morbidity and mortality. It is a reversible wound-healing response characterized by the accumulation of extracellular matrix (ECM) in liver injury[1]. In the mechanisms of liver fibrosis, activation of resident hepatic stellate cells (HSCs) after liver injury remains a dominant theme[2]. The quiescent HSCs can be induced by transforming growth factor (TGF)-β1 to transdifferentiate into myofibroblasts that secrete ECM[3]. Accumulating evidence indicates that multiple signaling pathways, such as the Notch and TGF-β pathways, are involved in epithelial-to-mesenchymal transition and fibroblast activation, resulting in the development of renal fibrosis[4,5]. Inhibition of Notch signaling can ameliorate renal fibrosis through inhibition of Notch-mediated TGF-β signaling activation[6]. However, whether similar regulation occurs in liver fibrosis, and what happens between the two signaling pathways during the recovery stage of liver fibrosis, needs further clarification.
Notch signaling is highly conserved among many animals. There are four Notch receptors (1-4) and five Notch ligands (delta-like 1, 3 and 4, and Jagged 1 and 2) in mammals[7]. After ligand binding, the Notch receptors experience a series of cleavages catalyzed by the γ-secretase combination, leading to the release of the Notch intracellular domain (NICD), which can be blocked by the γ-secretase inhibitor[8]. The NICD then enters into the nucleus, where it directly induces transcription of its target genes, such as Hes1 and Hes5. Ligand-induced Notch signaling directs a key role in organ formation through its effects on cellular differentiation, proliferation, survival and apoptosis[9]. Recent studies have demonstrated that Notch signaling is also involved in various types of tissue fibroses, including idiopathic pulmonary fibrosis, kidney fibrosis, and cardiac fibrosis[5,10,11]. However, the exact cellular mechanisms are not completely clear.
In this study, we researched the role of Notch signaling in liver fibrosis development and whether inhibition of Notch activation by γ-secretase inhibitor (DAPT) could suppress the TGF-β/Smad signaling pathway in a rat model of concanavalin (Con)A-induced liver fibrosis. We found that Notch signaling is involved in liver fibrosis via activation of the TGF-β/Smad pathway. Administration of DAPT markedly attenuated the TGF-β/Smad signaling pathway in ConA-induced liver fibrosis in rats.
MATERIALS AND METHODS
Animals
Male Wistar rats (weighing 210-230 g) were supplied by the Experimental Animal and Animal Experiment Center of Qingdao, Shandong, China. They were housed in the animal facility in a 12-h light-dark cycle, and the temperature was maintained at 22-23 °C and relative humidity at 60%. The animals were randomly distributed into two groups: normal group (n = 12), which received a weekly intravenous injection of 300 μL phosphate-buffered saline (PBS) for 8 wk, and the model group (n = 36), which received a weekly intravenous injection of ConA (17.5 mg/kg, in 300 μL PBS) for 8 wk[12,13]. During all experiments, rats were maintained in individually ventilated cages under specific pathogen-free conditions. At 8 wk later, we collected blood via cardiac puncture. Rats were handled and treated in accordance with the strict guiding principles of the National Institutes of Health for Experimental Care and Use of Animals and approved by the Institutional Animal Care and Use Committee of Qingdao University Medical College, Qingdao, China.
Cell culture and treatment
Peripheral blood mononuclear cells (PBMCs) were isolated from model rats by Ficoll-Hypaque density gradient centrifugation (TBD Science, Tianjin, China). The cells were washed three times with sterile PBS, suspended in a concentration of 2 × 106 cells/mL in RPMI 1640 (HyClone, Logan, UT, United States) supplemented with 10% heat-inactivated fetal calf serum and 1% penicillin-streptomycin (HyClone), and cultured at 37 °C in a CO2 incubator. These cells were treated with 0.25 μmol/L DAPT (HY-13027; MedChem Express, Monmouth Junction, NJ, United States) and 0.25 mmol/L TGF-β inhibitor (LY-364947; MedChem Express), with 0.1% dimethyl sulfoxide (DMSO) (Sigma, St. Louis, MO, United States) as a control. After treatment for 24 h, all cells were collected for total RNA and protein extraction.
Liver histopathological observation
After rats were killed, their blood vessels were perfused with PBS, followed by 4% paraformaldehyde. The livers were removed and fixed in 4% paraformaldehyde overnight and embedded in paraffin. Sections (4 μm) were cut, deparaffinized and stained with hematoxylin and eosin. Slides were evaluated using light microscopy by a pathologist on a blinded basis. Representative sections were presented.
Liver function
The serum levels of alanine aminotransferase, aspartate aminotransferase and albumin were measured with a biochemical autoanalyzer (P800; Roche, Basel, Switzerland).
Quantitative reverse transcription polymerase chain reaction
Total RNA was purified from cultured cells using the TRIzol Reagent (Takara, Otsu, Japan). Total RNA was reverse transcribed using the PrimeScript RT Reagent Kit (Takara, Otsu, Japan). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed by mixing cDNA and gene-specific primers with iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, United States), and reactions were carried out in an iCycler Thermal Cycler (Bio-Rad). Results were normalized to rat β-actin based on the threshold cycle (Ct) and relative fold-change calculated by the 2−ΔΔCt method. The primer sequences for mouse Notch 1-4, Jagged1, Hes1, Hes5, TGF-β1, Smad2 and Smad3 are described in Table 1.
Table 1.
Sequences of primers for quantitative real-time reverse transcription polymerase chain reaction
| Gene | Forward | Reverse |
| Notch1 | 5’-TCGTGCTCCTGTTCTTTGTG-3’ | 5’-TTCTCTCCGCTTCTTCTTGC-3’ |
| Notch2 | 5’-TGATGAGCAGGAACAGGAGA-3’ | 5’-ATGAGAAGCCAGGAGAGCAG-3’ |
| Notch3 | 5’-ATACCCACTACGGGATGTGC-3’ | 5’-AGGACGAAGATGACCAGCAG-3’ |
| Notch4 | 5’-GGATGAATGTCGGAGTGACC-3’ | 5’-GGCTACACAAGGGAACCTCA-3’ |
| Jagged1 | 5’-CAGTGGCTTGGGTCTGTTG-3’ | 5’-CATTGTTGGTGGTGTTGTCC-3’ |
| Hes1 | 5’-GTGGGTCCTAACGCAGTGTC-3’ | 5’-GTCAGAAGAGAGAGGTGGGCTA-3’ |
| Hes5 | 5’-ATGCTCAGTCCCAAGGAGAA-3’ | 5’-CTCCAGCAGCAGTTTCAGC-3’ |
| TGF-β1 | 5’-ATTCCTGGCGTTACCTTGG-3’ | 5’-AGCCCTGTATTCCGTTCTCT-3’ |
| Smad2 | 5’-CGGCTGAACTGTCTCCTACC-3’ | 5’-AGGTCTCTCCAACCCTCTGG-3’ |
| Smad3 | 5’-GGTAAAGGATTGCCACCAAA-3’ | 5’-GAACAGCCAGGAAAGGGACT-3’ |
| β-actin | 5’-CACCCGCGAGTACAACCTTC-3’ | 5’-CCCATACCCACCATCACACC-3’ |
Western blot analysis
Western blot analysis was performed using standard procedures with enhanced chemiluminescence using an ECL reagent and visualized by Imager (UVP 810; Biospectrum Upland, CA, United States). PBMCs were homogenized in RIPA lysis buffer (Thermo Scientific, Grand Island, NY, United States), and extracts were centrifuged for 10 min at 14000 × g at 4 °C. Total protein samples were analyzed by 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes by a wet blotting procedure (100 V, 2 h, 4 °C). Blocking buffer (5%) incubation was followed by incubation with primary antibodies at 4 °C overnight using the following concentrations: Notch1 (1:1000, Cat. No. 4380; Cell Signaling Technology, Danvers, MA, United States); Hes1 (1:1000, Cat. No. ab108937; Abcam, Cambridge, MA, United States); Hes5 (1:1000, Cat. No. ab194111; Abcam); TGF-β (1:1000, Cat. No. 3711; Cell Signaling Technology); Smad3 (1:1000, Cat. No. ab40854; Abcam); and β-actin (1:5000, Cat. No. TA-09; Zhongshan Jinqiao Biotechnology, Beijing, China), which acted as the internal control for normalization of protein expression. The quantification of densitometry, which was calculated by a multimedia color image analysis system (Image-Pro Plus 6.0), was measured as a relative objective index.
Statistical analysis
Quantitative data are expressed as mean ± SD. Statistical comparisons among multiple groups were performed by Tukey’s post hoc test and one-way ANOVA. Student’s t-test was used to compare the difference between two groups. P ≤ 0.05 was considered significant.
RESULTS
Notch signaling and TGF-β signaling are activated in liver fibrosis
To determine whether the Notch signaling and TGF-β signaling pathways were involved in liver fibrosis, we tested the expression of Notch- and TGF-β-related genes in liver fibrosis in rats. At 8 wk after intravenous injection of ConA, hematoxylin-eosin staining and liver function tests demonstrated modeling success compared with normal rats (Figure 1A and Table 2). qRT-PCR analysis revealed that the mRNA levels of Notch1, Hes1, Hes5, TGF-β1 and Smad3 were significantly increased in PBMCs of model rats as compared with controls. Notch2, Notch3, Jagged1 and Smad2 were detectable, but in contrast to Notch1, their levels did not differ between models and controls. Notch4 and HeyL were not detectable (aP < 0.05, eP < 0.001; Figure 1B). Through western blot analysis, we observed that the protein levels of Notch 1, Hes1, Hes5, TGF-β1 and Smad3 were increased after liver fibrosis (eP < 0.001; Figure 1C and D). These results suggest that Notch and TGF-β signaling is activated in liver fibrosis in rats.
Figure 1.
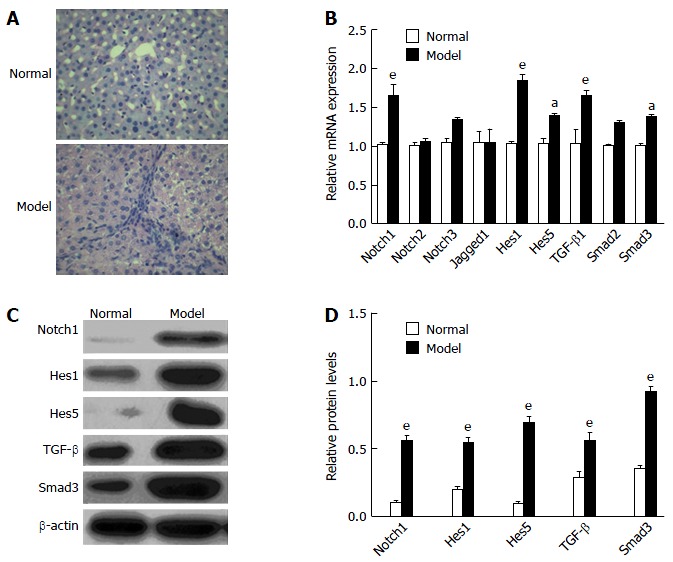
Notch signaling and transforming growth factor-β signaling pathways were activated in liver fibrosis rats. A: Representative hematoxylin-eosin staining in the livers from normal or model group rats (× 200); B: qRT-PCR analyzed mRNA expression of Notch- and TGF-β-related genes in the peripheral blood mononuclear cells (PBMCs, 2 × 106) from model rats and control group; C and D: Protein expression and quantification of Notch1, Hes1, Hes5, TGF-β and Smad3 were measured by western blot analysis of PBMCs (2 × 106) from model rats and control group. aP < 0.05, eP < 0.001 vs normal group, n = 12 per group. TGF-β: Transforming growth factor-β.
Table 2.
Levels of serum biochemical markers in rats
| Group | ALT (U/L) | AST (U/L) | Albumin (g/L) |
| Control | 47.32 ± 5.21 | 167.68 ± 14.31 | 34.23 ± 2.62 |
| Model | 75.13 ± 9.68a | 252.06 ± 22.83a | 25.60 ± 1.90a |
Data are presented as mean ± SD.
P < 0.05 vs control, n = 12 per group. ALT: Alanine aminotransferase; AST: Aspartate aminotransferase.
DAPT inhibits the Notch and TGF-β signaling pathway in vitro
Because TGF-β signaling was involved in the development of liver fibrosis[14], we tested whether DAPT affected this pathway in PBMCs of liver fibrosis rats. TGF-β1 and Smad3 expression was significantly downregulated in the PBMCs of liver fibrosis rats treated with DAPT, compared with the DMSO groups (Figure 2). The levels of Notch1, Hes1 and Hes5 genes were significantly attenuated in the DAPT-treated group at the same time. Protein was extracted from the PBMCs of DAPT- or DMSO-treated rats for western blotting. We observed a significant reduction in the expression of TGF-β1 and Smad3 protein, and a decrease in Notch1, Hes1 and Hes5 protein (Figure 2B and C). These changes indicated that inhibiting Notch signaling suppresses the activation of TGF-β signaling in PBMCs of liver fibrosis model rats.
Figure 2.
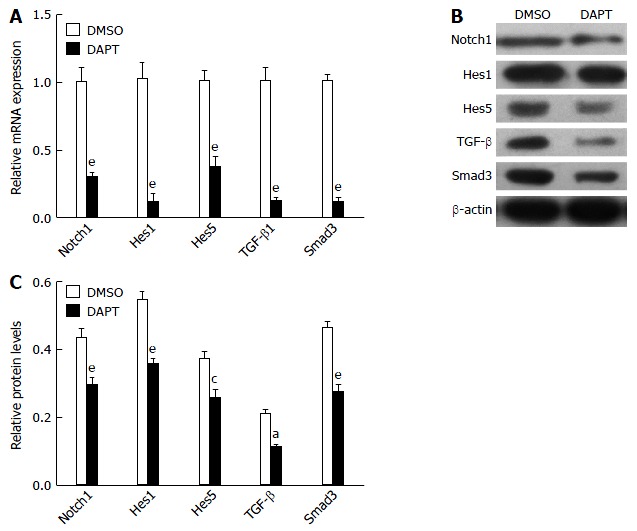
Notch γ-secretase inhibitor attenuates notch and transforming growth factor-β signaling pathways in peripheral blood mononuclear cells of liver fibrosis rats. A: mRNA expression for TGF-β1 Smad3 and Notch1, and Notch target genes Hes1 and Hes5 in peripheral blood mononuclear cells (PBMCs) (2 × 106) from rats treated with DAPT or DMSO for 24 h, as detected by qRT-PCR; B and C: Western blot analysis of protein expression of Notch1, Hes1, Hes5, TGF-β and Smad3 and quantification in PBMCs (2 × 106) from rats treated with DAPT or DMSO for 24 h. aP < 0.05, cP < 0.01, eP < 0.001 vs DMSO group, n = 12 per group. TGF-β: Transforming growth factor-β.
TGF-β inhibitor blocks the Notch and TGF-β signaling pathways in vitro
To further examine possible crosstalk between the Notch and TGF-β signaling pathways, the PBMCs from liver fibrosis rats were treated with TGF-β inhibitor. TGF-β inhibitor caused suppression of Notch1, Hes1, Hes5, TGF-β and Smad3 genes (Figure 3A), demonstrating that canonical TGF-β signaling pathway activity promotes the expression of Notch signaling pathway genes. Protein expression of Notch1, Hes1, Hes5, TGF-β and Smad3 in PBMCs cultured for 24 h with TGF-β inhibitor or DMSO was detected by western blot analysis (Figure 3B and C). These results showed that treatment with TGF-β inhibitor effectively blocked the Notch signaling pathway in PBMCs from liver fibrosis rats.
Figure 3.
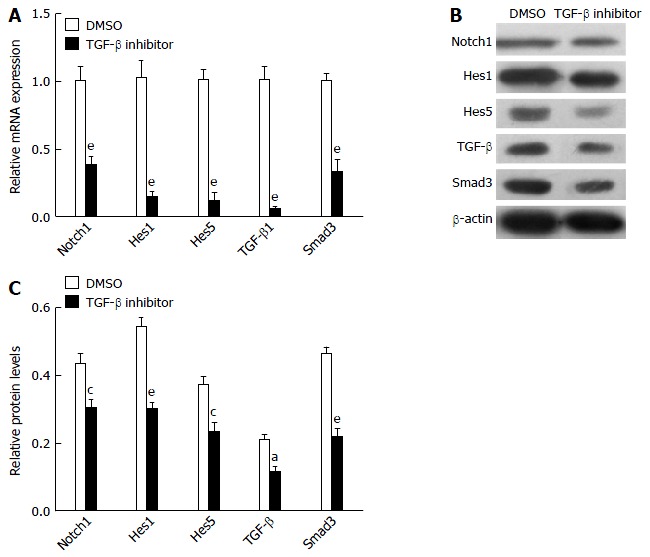
Transforming growth factor-β inhibitor blocks Notch and transforming growth factor-β signaling pathways in peripheral blood mononuclear cells of liver fibrosis rats. A: qRT-PCR analyzed mRNA expression of TGF-β1 Smad3 and Notch1, and Notch target genes Hes1 and Hes5 in peripheral blood mononuclear cells (PBMCs) (2 × 106) from rats treated with DAPT or DMSO for 24 h; B and C: Western blot analysis of Notch1, Hes1, Hes5, TGF-β and Smad3 and quantification in PBMCs (2 × 106) from rats treated with DAPT or DMSO for 24 h. aP < 0.05, cP < 0.01, eP < 0.001 vs DMSO group, n = 12 per group. TGF-β: Transforming growth factor-β.
DISCUSSION
We showed that there is an interaction between the Notch and TGF-β signaling pathways in ConA-induced liver fibrosis rats. We also showed that Notch signaling and TGF-β signaling are activated in liver fibrosis rats, with prominent expression of the Notch1 receptor, TGF-β1 accumulation and increased transcription of target genes such as Hes1, Hes5 and Smad3 in PBMCs of model rats. Furthermore, we found that treatment with the γ-secretase inhibitor DAPT significantly inhibited Notch and TGF-β signaling in PBMCs of liver fibrosis rats, similar to the TGF-β inhibitor.
In the earliest stages, all forms of fibrosis are inflammatory-immunological reactions, and elements of both the innate and adaptive immune systems are involved in subsequent profibrotic processes[15]. Growing evidence shows that Notch signaling plays a crucial role in the pathogenesis of multiple inflammatory diseases[16,17]. Our findings of increased activation of Notch signaling are consistent with ConA-induced liver fibrosis. We have previously shown an imbalance between T helper 17 and T regulatory cells in liver fibrosis model rats. In addition, TGF-β plays an important part in both normal and diseased conditions in the liver and other organs[14]. The direct targets in the TGF-β1 pathway, Smads (Smad2, and especially Smad3), are critical mediators of fibrogenesis[18]. In a recent study, TGF-β was shown to induce transport, Golgi organization and unfolded protein response, thereby facilitating fibrogenesis in murine hepatic fibrosis[19]. Furthermore, the Notch and TGF-β signaling pathways cooperatively regulate forkhead box P3 expression and regulatory T cell maintenance in vitro and in vivo, and integration of the TGF-β and Notch1 pathways may be an important mechanism for the maintenance of immune homeostasis in the periphery[20,21]. Our study also showed that TGF-β signaling was markedly increased in PBMCs of liver fibrosis rats. However, the cellular and molecular mechanisms by which Notch and TGF-β activation facilitates liver fibrosis have not been previously explored. Our study demonstrated that Notch signaling is mediated by the TGF-β/Smad signaling in liver fibrosis.
Evidence for the important role of Notch signaling is growing, such as the crosstalk between Notch and Hedgehog in liver injury, as well as the Notch inhibitor restriction of the TGF-β/Smad2/3 signaling pathway in kidney fibrosis[6,22] and even TGF-b/Smad crosstalk with Notch in many diseases[23]. However, the relation between Notch and TGF-β signaling pathway in liver fibrosis remains unclear. Previously, it has been shown that Notch3 and Hes1 are critically involved in CCl4-induced liver fibrosis in rats[24]. Hes1 and Hes5, as the targets of Notch affect chondrogenesis and chondrocyte hypertrophy in cartilage maintenance and osteoarthritis[25,26]. In our study, we found that Notch1 was markedly increased in PBMCs during liver fibrogenesis. This may reveal the distinctive fibrotic mechanism that exists between liver and peripheral tissues. Studies analyzing the therapeutic potential of inhibitors of the γ-secretase complex that is required for release of the active NICD support this method as effective for targeting Notch[27]. It should be noted that our study examined only the interaction between the Notch and TGF-β signaling pathways in vitro, and we now need to explore the interaction in vivo in model rats and patients with liver fibrosis.
In summary, we have demonstrated that the Notch pathway is activated in liver fibrosis rats and that inhibition of Notch and TGF-β signaling reduces the mRNA and protein expression in immune-induced rat liver fibrosis. Thus, we conclude that Notch signaling exerts fibrogenesis mediated by the TGF-β/Smad signaling pathway, although additional research is needed to clarify the nuances of this process.
COMMENTS
Background
Liver fibrosis is distributed widely and a serious threat to human health. It is difficult to cure the disease because the pathogenesis is not completely understood. The current study suggests that liver damage is related to immune factors, and Notch and transforming growth factor (TGF)-β signaling play an important role in it. Blocking the two pathways may alleviate liver fibrosis.
Research frontiers
TGF-β is one of the key factors that activates quiescent hepatic stellate cells, which transform into myofibroblasts, and it also promotes collagen production. Notch crosstalk with TGF-β is involved in fibrosis of many organs, including the liver, and overexpression of Notch intracellular domain and Smad3 activates their target gene transcription.
Innovations and breakthroughs
The present study showed that Notch signaling and TGF-β signaling regulate each other in liver fibrosis, which provides a new insight into the immune mechanism of liver fibrosis.
Applications
The results of this study may have a positive impact on patients with liver fibrosis and its outcome.
Terminology
Notch is a highly conserved signaling pathway among all animal species, and plays a key role in regulating cellular differentiation, proliferation, survival and apoptosis.
Peer-review
This article provides a particular answer that is only associated with the white blood cells being present in the circulation of the hepatic fibrosis-induced rats, presuming that the peripheral blood mononuclear cells (PBMCs) fraction is characteristic and indicative of the fibrotic state, and that the PBMC fraction may have a role in hepatic fibrogenesis.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: The study was reviewed and approved by the Qingdao University Medical College Institutional Review Board.
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Qingdao University Medical College (No. SCXK20090007).
Conflict-of-interest statement: We declare that there are no conflicts of interest to disclose.
Data sharing statement: No additional data are available.
Peer-review started: December 23, 2016
First decision: January 10, 2017
Article in press: February 17, 2017
P- Reviewer: Abdelgawad IA, Ji G, Lendvai G S- Editor: Qi Y L- Editor: Filipodia E- Editor: Zhang FF
References
- 1.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 2.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25:195–206. doi: 10.1016/j.bpg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schnaper HW, Jandeska S, Runyan CE, Hubchak SC, Basu RK, Curley JF, Smith RD, Hayashida T. TGF-beta signal transduction in chronic kidney disease. Front Biosci (Landmark Ed) 2009;14:2448–2465. doi: 10.2741/3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S, Kato H, Pullman J, Gessler M, Haase VH, et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest. 2010;120:4040–4054. doi: 10.1172/JCI43025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao Z, Zhang J, Peng X, Dong Y, Jia L, Li H, Du J. The Notch γ-secretase inhibitor ameliorates kidney fibrosis via inhibition of TGF-β/Smad2/3 signaling pathway activation. Int J Biochem Cell Biol. 2014;55:65–71. doi: 10.1016/j.biocel.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 7.Maillard I, Adler SH, Pear WS. Notch and the immune system. Immunity. 2003;19:781–791. doi: 10.1016/s1074-7613(03)00325-x. [DOI] [PubMed] [Google Scholar]
- 8.Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, Jacobs RT, Zacco A, Greenberg B, Ciaccio PJ. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82:341–358. doi: 10.1093/toxsci/kfh254. [DOI] [PubMed] [Google Scholar]
- 9.Fiúza UM, Arias AM. Cell and molecular biology of Notch. J Endocrinol. 2007;194:459–474. doi: 10.1677/JOE-07-0242. [DOI] [PubMed] [Google Scholar]
- 10.Plantier L, Crestani B, Wert SE, Dehoux M, Zweytick B, Guenther A, Whitsett JA. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax. 2011;66:651–657. doi: 10.1136/thx.2010.151555. [DOI] [PubMed] [Google Scholar]
- 11.Russell JL, Goetsch SC, Gaiano NR, Hill JA, Olson EN, Schneider JW. A dynamic notch injury response activates epicardium and contributes to fibrosis repair. Circ Res. 2011;108:51–59. doi: 10.1161/CIRCRESAHA.110.233262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang J, Zhang B, Shen RW, Liu JB, Gao MH, Li Y, Li YY, Zhang W. Preventive effect of halofuginone on concanavalin A-induced liver fibrosis. PLoS One. 2013;8:e82232. doi: 10.1371/journal.pone.0082232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang J, Zhang B, Shen RW, Liu JB, Gao MH, Geng X, Li Y, Li YY, Zhang W. The effect of antifibrotic drug halofugine on Th17 cells in concanavalin A-induced liver fibrosis. Scand J Immunol. 2014;79:163–172. doi: 10.1111/sji.12144. [DOI] [PubMed] [Google Scholar]
- 14.Inagaki Y, Okazaki I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56:284–292. doi: 10.1136/gut.2005.088690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wick G, Grundtman C, Mayerl C, Wimpissinger TF, Feichtinger J, Zelger B, Sgonc R, Wolfram D. The immunology of fibrosis. Annu Rev Immunol. 2013;31:107–135. doi: 10.1146/annurev-immunol-032712-095937. [DOI] [PubMed] [Google Scholar]
- 16.Cheng YL, Choi Y, Sobey CG, Arumugam TV, Jo DG. Emerging roles of the γ-secretase-notch axis in inflammation. Pharmacol Ther. 2015;147:80–90. doi: 10.1016/j.pharmthera.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Hans CP, Koenig SN, Huang N, Cheng J, Beceiro S, Guggilam A, Kuivaniemi H, Partida-Sánchez S, Garg V. Inhibition of Notch1 signaling reduces abdominal aortic aneurysm in mice by attenuating macrophage-mediated inflammation. Arterioscler Thromb Vasc Biol. 2012;32:3012–3023. doi: 10.1161/ATVBAHA.112.254219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C, Song X, Li Y, Han F, Gao S, Wang X, Xie S, Lv C. Low-dose paclitaxel ameliorates pulmonary fibrosis by suppressing TGF-β1/Smad3 pathway via miR-140 upregulation. PLoS One. 2013;8:e70725. doi: 10.1371/journal.pone.0070725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maiers JL, Kostallari E, Mushref M, deAssuncao TM, Li H, Jalan-Sakrikar N, Huebert RC, Cao S, Malhi H, Shah VH. The unfolded protein response mediates fibrogenesis and collagen I secretion through regulating TANGO1 in mice. Hepatology. 2017;65:983–998. doi: 10.1002/hep.28921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samon JB, Champhekar A, Minter LM, Telfer JC, Miele L, Fauq A, Das P, Golde TE, Osborne BA. Notch1 and TGFbeta1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood. 2008;112:1813–1821. doi: 10.1182/blood-2008-03-144980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ostroukhova M, Qi Z, Oriss TB, Dixon-McCarthy B, Ray P, Ray A. Treg-mediated immunosuppression involves activation of the Notch-HES1 axis by membrane-bound TGF-beta. J Clin Invest. 2006;116:996–1004. doi: 10.1172/JCI26490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie G, Karaca G, Swiderska-Syn M, Michelotti GA, Krüger L, Chen Y, Premont RT, Choi SS, Diehl AM. Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology. 2013;58:1801–1813. doi: 10.1002/hep.26511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb Perspect Biol. 2017;9:pii: a022137. doi: 10.1101/cshperspect.a022137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Zheng S, Qi D, Zheng S, Guo J, Zhang S, Weng Z. Inhibition of Notch signaling by a γ-secretase inhibitor attenuates hepatic fibrosis in rats. PLoS One. 2012;7:e46512. doi: 10.1371/journal.pone.0046512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rutkowski TP, Kohn A, Sharma D, Ren Y, Mirando AJ, Hilton MJ. HES factors regulate specific aspects of chondrogenesis and chondrocyte hypertrophy during cartilage development. J Cell Sci. 2016;129:2145–2155. doi: 10.1242/jcs.181271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z, Chen J, Mirando AJ, Wang C, Zuscik MJ, O’Keefe RJ, Hilton MJ. A dual role for NOTCH signaling in joint cartilage maintenance and osteoarthritis. Sci Signal. 2015;8:ra71. doi: 10.1126/scisignal.aaa3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene. 2008;27:5124–5131. doi: 10.1038/onc.2008.226. [DOI] [PubMed] [Google Scholar]