

Abstract
The Ten-eleven translocation (TET) family of 5-methylcytosine (5mC) dioxygenases catalyze the conversion of 5mC into 5-hydroxymethylcytosine (5hmC) and further oxidized species to promote active DNA demethylation. Here we engineered a split-TET2 enzyme to enable temporal control of 5mC oxidation and subsequent remodeling of epigenetic states in mammalian cells. We further demonstrate the use of this chemically inducible system to dissect the correlation between DNA hydroxymethylation and chromatin accessibility in the mammalian genome. This chemical-inducible epigenome remodeling tool will find broad use in interrogating cellular systems without altering the genetic code, as well as in probing the epigenotype–phenotype relations in various biological systems.
The addition of a methyl group to the carbon 5 position of cytosine to form 5-methylcytosine (5mC), a process known as DNA methylation, is catalyzed by DNA methyltransferases (DNMTs). 5mC acts as a major epigenetic mark in the mammalian genome that often signals for transcriptional repression, X-chromosome inactivation and transposon silencing.1 Tet-eleven translocation (TET) family of methylcytosine dioxygenases, which catalyzes the successive oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylmethylcytosine (5fC) and 5-carboxymethylcytosine (5caC), has added an additional layer of previously underappreciated epigenetic control over the mammalian genome.2−4 The discovery of TET has sparked intense interest in the epigenetic field to unveil the biological functions of TET proteins and their major catalytic product 5hmC. 5hmC is regarded to serves as an intermediate during TET-mediated active DNA demethylation,2−4 as well as a stable epigenetic mark.5−8 Although it has been widely observed that DNA hydroxymethylation is highly correlated with gene expression and some human disorders,9−11 the causal relations between epigenetic modifications on DNA and the phenotypes often remain challenging to be established, largely owing to the lack of reliable tools to add or remove accurately DNA modifications in the genome at defined temporal and spatial resolution.
To tackle this challenge, we set out to design a chemical-inducible epigenome remodeling tool (CiDER; Scheme 1) to overcome the hurdle facing studies of causal relationships between DNA hydroxymethylation and gene transcription. We chose the catalytic domain of human TET2 (TET2CD, Figure 1), rather than TET1 or TET3, as our target for engineering a split epigenomic modifier because of the following major considerations. First, TET2 is among the most frequently mutated genes in hematological malignancies.10 Exome sequencing in cancer patients has revealed a large panel of disease-associated mutations,12,13 thereby providing abundant information with regard to sensitive spots to be avoided during our selection of split sites. Second, the crystal structures of the catalytic domain of TET2 (TET2CD) in complex with 5mC or 5hmC have been recently determined,14,15 and thus enabled us to prioritize the selection and validation of split sites in a more rationalized manner. Third, the low complexity region (residues 1481–1843) of TET2CD can be replaced by a flexible GS linker without significantly compromising its catalytic activity,15 clearly speaking for the structural malleability of TET2 and the high flexibility to accommodate the insertion of foreign polypeptide sequences. Omitting this large fragment of low complexity region (∼1.2 kb) further allows us to generate constructs with minimized sizes. We therefore set out to test the idea that TET2CD can be split into two inactive fragments and that its enzymatic function can be restored by taking a chemically inducible dimerization approach.
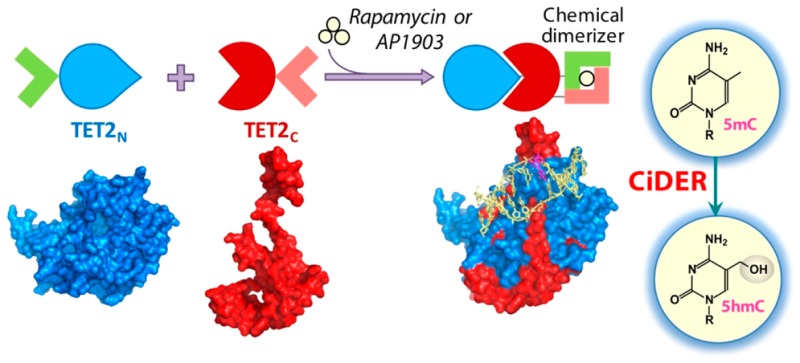
Scheme 1. Design of a Chemical-Inducible Epigenome Remodeling (CiDER) Tool Based on a Split TET2 Enzyme.
FKBP12/FRB heterodimerization or FKBP-F36 V homodimerization modules are fused with two inactive fragments of a split TET2CD. Upon the addition of rapamycin or AP1903, split TET2CD fragments reassemble into a functional methylcytosine dioxygenase to catalyze the conversion of 5mC into 5hmC and further oxidized species, thus promoting DNA demethylation to remodel the epigenetic landscapes in mammalian cells.
Figure 1.
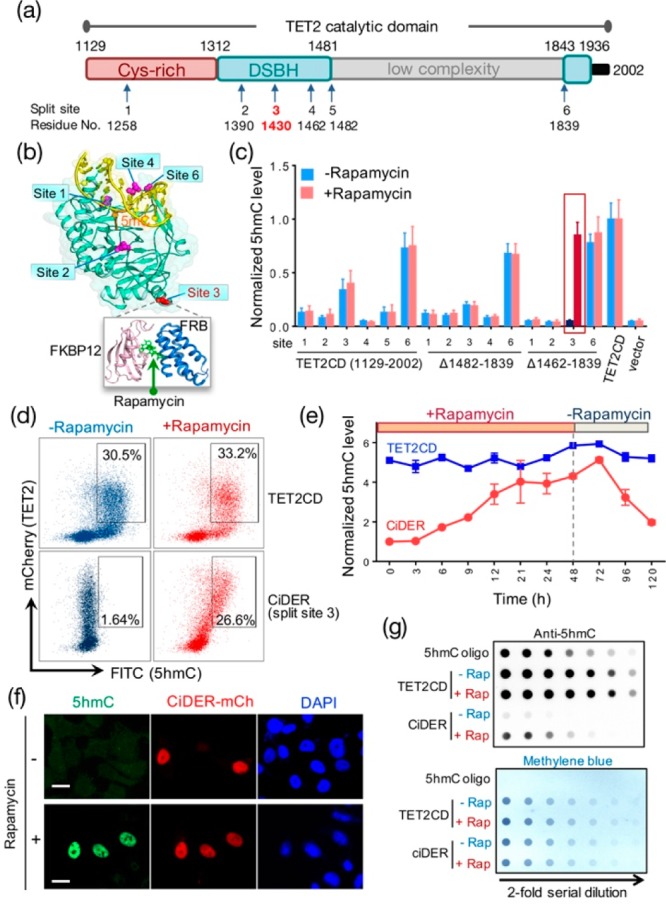
An engineered split-TET2 enzyme for inducible DNA hydroxymethylation in mammalian cells. (a) Domain architecture of the catalytic domain of TET2 (TET2CD; aa 1129–2002) and positions of selected split sites. DSBH, double stranded beta helix. (b) Split sites mapped to the 3D structure of TET2CD (PDB entry: 4NM6). A rapamycin-inducible heterodimerization module composed of FKBP12 and FRB was inserted individually into the selected split sites. (c) Screening and optimization of split-TET2CD constructs to achieve chemical-inducible 5hmC generation in HEK293T cells. The construct with insertion of FKBP12-T2A-FRB at split site 3 and deletion of the low complexity region (Δ1462–1839) stood out as the best candidate (termed “CiDER”). AP1903-incucible homodimerization of a mutant FKBP12 (F36 V) can also be engineered into this position to restore the catalytic activity of split-TET2CD (Figure S2). (d) Quantification of CiDER-mediated 5hmC production by flow cytometry. HEK293T cells transfected with mCherry (mCh)-tagged CiDER or mCh-TET2CD (positive control) were immunostained with an anti-5hmC primary antibody and an FITC-labeled secondary antibody. (e) Time course of rapamycin (200 nM)-induced production of 5hmC in HEK293T cells expressing CiDER or TET2CD (as positive control). Rapamycin was washed away 48 h after incubation with cells. (f) Representative fluorescent images of 5hmC (green), CiDER-mCh (red), and nuclear staining with DAPI (blue) in HEK293T cells before and after rapamycin (200 nM) treatment. (g) Dot-blot assay to quantify rapamycin (200 nM)-induced changes of 5hmC levels in genomic DNA purified from HEK293T cells expressing CiDER or TET2CD. A synthetic oligonucleotide with a known amount of 5hmC was used as a positive control. The loading control was shown in the bottom panel by methylene blue staining of total amounts of input DNA. See Supporting Information (Figures S1,2) for more results and sequences. n = 5. Scale bar = 10 μm.
To develop a split-TET2CD system, we selected six sites in TET2CD, composed of a Cys-rich region and a double-stranded β-helix (DSBH) fold, on the basis of a crystal structure of TET2-DNA complex that lacks a low complexity region15 (Figure 1a,b). A synthetic gene encoding FK506 binding protein 12 (FKBP12) and FKBP rapamycin binding domain (FRB) of the mammalian target of rapamycin,16,17 separated by a self-cleaving T2A polypeptide sequence18,19 in the middle, was individually inserted into the selected split sites within TET2CD or TET2CD that lacks the low complexity region (Figure S1a). We transfected each mCherry-tagged construct into human embryonic kidney 293T (HEK293T) or HeLa cells, both of which have extremely low basal 5hmC levels. Once expressed in HEK293T cells, the fusion protein was self-cleaved into two fragments (Figure S1b). The two fragments could reconstitute a functional enzyme in vitro, as reflected by inducible production of 5hmC after incubating the enriched fragments with a synthetic 5mC-containing dsDNA oligo in the presence of rapamycin (Figure S1c). By using an antibody that specifically recognizes 5hmC, we compared the global 5hmC levels in transfected cells before and after rapamycin treatment by flow cytometry (Figure 1c,d). After screening over 15 constructs, we identified a split-TET2CD variant (designated as CiDER for chemical-inducible epigenome remodeling tool; see Supporting Information for the sequences) that exhibited rapamycin-inducible restoration of enzymatic activity. We found that almost no background activity of CiDER was detected prior to the addition of rapamycin (Figure 1c,d). Following the addition of rapamycin, the total 5hmC amount was restored to a level comparable to cells expressing intact TET2CD proteins (split site 3; Figure 1c,d).
We further monitored chemical-induced 5hmC production at different time points and noticed that the functional restoration reached a maximum approximately 48 h after rapamycin treatment. Notably, approximately 3 days after withdrawal of rapamycin, the 5hmC level returned to its basal level, presumably owing to the constant dilution of 5hmC after multiple rounds of cell division (Figure 1e). By contrast, the 5hmC level in the control group expressing an intact TET2CD remained largely unaltered under the same experimental conditions. Next, we sought further confirmation of rapamycin-inducible generation of 5hmC by two additional methods: immunostaining with an antibody against 5hmC in individual cells (Figures 1f and S1d) and a more quantitative dot-blot assay that measures the total amounts of 5hmC in the whole cell population (Figure 1g). Both assays clearly demonstrated that rapamycin elicited robust production of 5hmC in CiDER-expressing HEK293T cells with negligible background activity.
To examine systematically the CiDER-mediated changes in the epigenetic landscapes between untreated and rapamycin-treated mammalian cells (Figure 2a), we performed genome-wide 5hmC mapping in HEK293T cells by immunoprecipitation (IP) of bisulfite-treated DNA with a highly specific homemade antibody against cytosine-5-methylenesulfonate (CMS).20,21 Compared with the untreated control group (0 h), we observed a large number (n = 13 125) of differentially hydroxymethylated genomic regions (DHMRs) in cells treated with rapamycin (48 h), with the majority (n = 11 808) showing significant gain of 5hmC (Figure 2b, Table S1).
Figure 2.
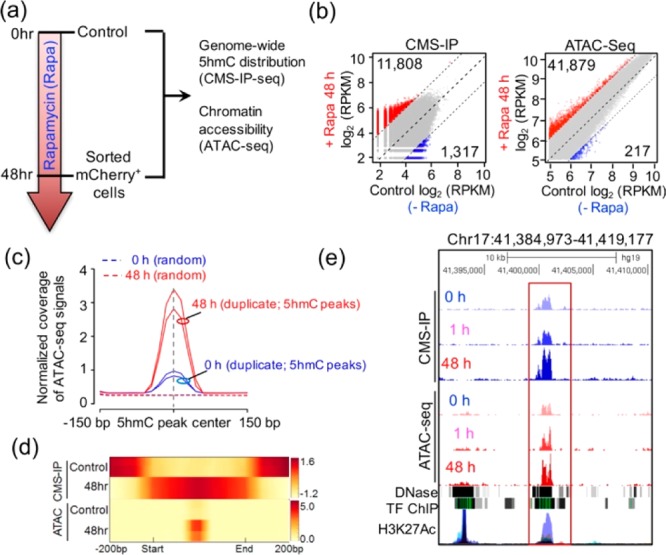
CiDER-mediated chemical inducible remodeling of epigenetic states to increase chromatin accessibility in mammalian cells. (a) Schematic of the experimental setup 0 or 48 h after 200 nM rapamycin treatment, genomic DNA samples from HEK293T cells were subjected to genome-wide 5hmC profiling and ATAC-seq to monitor chromatin accessible regions in the genome. (b) Scatter plots of differential 5hmC peaks and ATAC peaks between 0 and 48 h treatment groups. Red and blue dots represent significantly up- or down-regulated ATAC/5hmC peaks at 48 h, respectively. The gray dots represent peaks without significant changes at 48 h. (c) Normalized coverage of ATAC-seq signals (0 h and 48h; with biological duplicates) were plotted 150 bp up- and downstream of the centers of 5hmC-peaks at 0 h (control). (d) Distribution of averaged 5hmC enrichment (2 upper panels) and ATAC-seq peak enrichment (2 lower panels) at control ATAC/5hmC peaks regions in CiDER-expressing HEK293T cells at time 0 (control) or 48 h following rapamycin treatment. (e) Representative genome browser view of one HMDR in the LINC00854 locus on chromosome 17 that showed rapamycin-induced gain of 5hmC and ATAC-seq peaks, which was overlaid with traces representing DNase I hypersensitive sites, transcriptional factor binding (TF ChIP) and H3K27Ac enrichment.
Very recently, it has been shown that TET loss-of-function with the resultant reduction of genomic 5hmC is correlated with reduced numbers of chromatin accessible regions, thus suggesting a potential role of TET and/or 5hmC in the maintenance of chromatin accessibility in mammals.6,22 This led us to hypothesize that 5hmC-enriched regions might mark more accessible chromatin regions. Because increased chromatin accessibility is often indirectly indicated by a lower DAPI staining of the nuclear euchromatin,23 we examined if the intensity of 5hmC staining is correlated with DAPI staining levels in two types of cells, the mouse embryonic stem cells that are rich in 5hmC (Figure S3a) and rapamycin-treated HEK293T cells that expressed chemical-inducible TET2CD (Figure 2a and Figure S3b,c). In each individual cell, high 5-hmC areas in the same nucleus were generally correlated with pronouncedly decreased nuclear staining by DAPI, and vice versa. In both cell types, we observed a general inverse relationship between intensities of 5hmC and DAPI staining (Figure S3), a clear indication of the involvement of 5hmC in the regulation of chromatin accessibility.
The causal relationship between 5hmC and chromatin accessibility, nonetheless, remains unresolved, primarily owing to the lack of tools to control temporally the hydroxymethylation of 5mC in living cells. To clarify this, we tested if CiDER-mediated chemical-inducible deposition of 5hmC in the genome would cause changes in the chromatin architecture. We unbiasedly profiled the genome-wide chromatin accessibility by ATAC-seq (assay for transposase-accessible chromatin using sequencing24) before and after rapamycin treatment in the same batch of cells (Figure S4; Table S1). We observed that 41 879 regions were more accessible in rapamycin-treated cells compared to untreated cells (Figure 2b, right and Figure 2c). By taking a genome-wide approach to compare the 5hmC enrichment and ATAC-Seq peaks, we observed a substantial enrichment of ATAC-seq peaks within the 5hmC enriched regions (Figure 2c,d). In parallel, we performed the principal component analysis (PCA) of ATAC-seq and 5hmC enriched regions. Both parameters (ATAC-Seq and 5hmC peaks) displayed similar distribution patterns on the plots before (0 h) and after (48 h) rapamycin treatment (Figure S5), clearly indicating that CiDER-mediated chemical inducible generation of 5hmC was strongly correlated with the increase of chromatin accessible regions. The observed overlapping enrichment of 5hmC in chromatin accessible regions (Figure 2c,d) further imply that CiDER, with its enzymatic function restored by rapamycin, could actively mark these chromatin regions with 5hmC to maintain their accessibility in mammalian cells. To demonstrate this further, a genome browser view of one DHMR located in the LINC00854 locus is shown in Figure 2e. The distal region of LINC00854 contains moderate levels of both 5hmC and chromatin accessible regions prior to the addition of rapamycin (0 h), and showed a substantial gain of 5hmC and ATAC-Seq peaks after rapamycin-induced restoration of TET2 enzymatic activity (48 h). This region might also function as distal-regulatory regions (e.g., enhancers) as it is enriched with an enhancer mark (Histone H3 lysine 27 acetylation, H3K27ac) as well as DNase I hypersensitivity regions identified from the ENCODE database. Taken together, by controlling the formation of 5hmC at real time in living cells, we demonstrated that TET-mediated 5mC oxidation constitutes one important epigenetic regulatory mechanism to modulate chromatin accessibility without altering the genetic code.
To confer spatial control over the CiDER system, it is our immediate future plan to fuse it with a catalytically inactive Cas9 or its orthologues, which will enable loci-specific targeting and precise control of 5mC oxidation or DNA demethylation in the genome. Ideally, the epigenome remodeling is a powerful strategy for interrogating, perturbing and engineering cellular system without altering the genetic code, and could thus serve as a high entry point for reprogramming cell fate and disease intervention.
Acknowledgments
We thank the financial supports from the National Institutes of Health grant (R01GM112003), the Welch Foundation (BE-1913), the American Cancer Society (RSG-16-215-01 TBE), the Cancer Prevention and Research Institute of Texas (RR140053, RP120348 and RP170002), the American Heart Association (16IRG27250155) and by an allocation from the Texas A&M University Health Science Center Startup Fund.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b01459.
Experimental details and supplementary figures (PDF)
Author Contributions
† These authors contributed equally
The authors declare no competing financial interest.
Supplementary Material
References
- Li E.; Zhang Y. Cold Spring Harbor Perspect. Biol. 2014, 6, a019133. 10.1101/cshperspect.a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M.; Koh K. P.; Shen Y.; Pastor W. A.; Bandukwala H.; Brudno Y.; Agarwal S.; Iyer L. M.; Liu D. R.; Aravind L.; Rao A. Science 2009, 324, 930–5. 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y. F.; Li B. Z.; Li Z.; Liu P.; Wang Y.; Tang Q.; Ding J.; Jia Y.; Chen Z.; Li L.; Sun Y.; Li X.; Dai Q.; Song C. X.; Zhang K.; He C.; Xu G. L. Science 2011, 333, 1303–7. 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S.; Shen L.; Dai Q.; Wu S. C.; Collins L. B.; Swenberg J. A.; He C.; Zhang Y. Science 2011, 333, 1300–3. 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt C. G.; Gnerlich F.; Smits A. H.; Pfaffeneder T.; Jansen P. W.; Bauer C.; Munzel M.; Wagner M.; Muller M.; Khan F.; Eberl H. C.; Mensinga A.; Brinkman A. B.; Lephikov K.; Muller U.; Walter J.; Boelens R.; van Ingen H.; Leonhardt H.; Carell T.; Vermeulen M. Cell 2013, 152, 1146–59. 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Lio C. W.; Zhang J.; Gonzalez-Avalos E.; Hogan P. G.; Chang X.; Rao A.. eLife 2016, 5, DOI: 10.7554/eLife.18290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellinger M. W.; Song C. X.; Chong J.; Lu X. Y.; He C.; Wang D. Nat. Struct. Mol. Biol. 2012, 19, 831–3. 10.1038/nsmb.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M.; Kirchner A.; Stazzoni S.; Muller M.; Wagner M.; Schroder A.; Carell T. Angew. Chem., Int. Ed. 2016, 55, 11797–800. 10.1002/anie.201605994. [DOI] [PubMed] [Google Scholar]
- Ko M.; Huang Y.; Jankowska A. M.; Pape U. J.; Tahiliani M.; Bandukwala H. S.; An J.; Lamperti E. D.; Koh K. P.; Ganetzky R.; Liu X. S.; Aravind L.; Agarwal S.; Maciejewski J. P.; Rao A. Nature 2010, 468, 839–43. 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Rao A. Trends Genet. 2014, 30, 464–74. 10.1016/j.tig.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Zhao B. S.; He C. Chem. Rev. 2015, 115, 2225–39. 10.1021/cr500470n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhommeau F.; Dupont S.; Della Valle V.; James C.; Trannoy S.; Masse A.; Kosmider O.; Le Couedic J. P.; Robert F.; Alberdi A.; Lecluse Y.; Plo I.; Dreyfus F. J.; Marzac C.; Casadevall N.; Lacombe C.; Romana S. P.; Dessen P.; Soulier J.; Viguie F.; Fontenay M.; Vainchenker W.; Bernard O. A. N. Engl. J. Med. 2009, 360, 2289–2301. 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- Langemeijer S. M.; Kuiper R. P.; Berends M.; Knops R.; Aslanyan M. G.; Massop M.; Stevens-Linders E.; van Hoogen P.; van Kessel A. G.; Raymakers R. A.; Kamping E. J.; Verhoef G. E.; Verburgh E.; Hagemeijer A.; Vandenberghe P.; de Witte T.; van der Reijden B. A.; Jansen J. H. Nat. Genet. 2009, 41, 838–42. 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- Hashimoto H.; Pais J. E.; Zhang X.; Saleh L.; Fu Z. Q.; Dai N.; Correa I. R. Jr.; Zheng Y.; Cheng X. Nature 2014, 506, 391–395. 10.1038/nature12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L.; Li Z.; Cheng J.; Rao Q.; Gong W.; Liu M.; Shi Y. G.; Zhu J.; Wang P.; Xu Y. Cell 2013, 155, 1545–55. 10.1016/j.cell.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Banaszynski L. A.; Liu C. W.; Wandless T. J. J. Am. Chem. Soc. 2005, 127, 4715–4721. 10.1021/ja043277y. [DOI] [PubMed] [Google Scholar]
- Clackson T.; Yang W.; Rozamus L. W.; Hatada M.; Amara J. F.; Rollins C. T.; Stevenson L. F.; Magari S. R.; Wood S. A.; Courage N. L.; Lu X.; Cerasoli F. Jr.; Gilman M.; Holt D. A. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 10437–42. 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahimi A.; Vande Velde G.; Reumers V.; Toelen J.; Thiry I.; Vandeputte C.; Vets S.; Deroose C.; Bormans G.; Baekelandt V.; Debyser Z.; Gijsbers R. Hum. Gene Ther. 2009, 20, 845–860. 10.1089/hum.2008.188. [DOI] [PubMed] [Google Scholar]
- Kim J. H.; Lee S. R.; Li L. H.; Park H. J.; Park J. H.; Lee K. Y.; Kim M. K.; Shin B. A.; Choi S. Y. PLoS One 2011, 6, e18556. 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor W. A.; Pape U. J.; Huang Y.; Henderson H. R.; Lister R.; Ko M.; McLoughlin E. M.; Brudno Y.; Mahapatra S.; Kapranov P.; Tahiliani M.; Daley G. Q.; Liu X. S.; Ecker J. R.; Milos P. M.; Agarwal S.; Rao A. Nature 2011, 473, 394–7. 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Pastor W. A.; Zepeda-Martinez J. A.; Rao A. Nat. Protoc. 2012, 7, 1897–908. 10.1038/nprot.2012.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovic O.; Smits A. H.; de la Calle Mustienes E.; Tena J. J.; Ford E.; Williams R.; Senanayake U.; Schultz M. D.; Hontelez S.; van Kruijsbergen I.; Rayon T.; Gnerlich F.; Carell T.; Veenstra G. J.; Manzanares M.; Sauka-Spengler T.; Ecker J. R.; Vermeulen M.; Gomez-Skarmeta J. L.; Lister R. Nat. Genet. 2016, 48, 417–26. 10.1038/ng.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach K. E.; Li X.; Li Y.; Song C. X.; Han J. W.; Kim S.; Namburi S.; Hermetz K.; Kim J. J.; Rudd M. K.; Yoon Y. S.; Ren B.; He C.; Jin P. PLoS Genet. 2011, 7, e1002154. 10.1371/journal.pgen.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J. D.; Giresi P. G.; Zaba L. C.; Chang H. Y.; Greenleaf W. J. Nat. Methods 2013, 10, 1213–8. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.