

Abstract
Hepatocarcinogenesis is a complex process that includes pronounced necroinflammation, unregulated hepatocyte damage, subsequent extensive fibrosis, and carcinogenesis. GPR110 was an adhesion G protein-coupled receptor. Analysis of the expression pattern of Gpr110 in mice displayed that Gpr110 was expressed highly in liver, implicating the tissue compartments where Gpr110 could execute its functions, the role of Gpr110 in the physiological and pathological state of liver remains unclear. Based on a Gpr110 knockout mouse model, we evaluated the role of Gpr110 in hepatocarcinogenesis by using a carbon tetrachloride (CCl4)-induced liver injury and fibrosis model, as well as diethylnitrosamine (DEN) plus CCl4-induced liver cancer model. In this study, we found subdued chronic liver injury, reduced compensatory proliferation, lower liver fibrosis, but enhanced inflammation occurred in Gpr110-/- mice during CCl4 challenge. In addition, Gpr110-/- mice were resistant to liver tumorigenesis induced by DEN plus CCl4 injection. Molecular mechanisms underlying these differences correlated with augmented activation of the IL-6/STAT3 pathway, which exerted hepatoprotective effects during liver damage, fibrosis, and oncogenesis in Gpr110-/- mice. Furthermore, pharmacological inhibition of the activation of the IL-6/STAT3 pathway enhanced hepatic fibrosis and promoted DEN plus CCl4-induced carcinogenesis in Gpr110-/- mice. In summary, absence of Gpr110 decelerates liver fibrosis/cirrhosis progressing into tumorigenesis, due to strengthening activation of the IL-6/STAT3 pathway, leading to a weaker liver injury and fibrosis microenvironment. It is indicated that targeting Gpr110 and activating the IL-6/STAT3 pathway may be considered to be preventive methods for some cirrhosis transition.
Keywords: Gpr110, CCl4, hepatocarcinogenesis, knockout mouse model, fibrosis
Introduction
Hepatocellular carcinoma (HCC) is the typical of chronic tissue injury-associated cancer [1]. Chronic liver injury, which triggers perpetual hepatocellular damage, hepatocyte regeneration, and inflammation, is thought to be the established background that promotes neoplasia in these pathophysiologically distinct diseases [2]. HCC has clearly defined etiological factors, including viral hepatitis, nonalcoholic steatohepatitis (NASH), alcoholic liver disease, andcirrhosis [3]. Cirrhosis, which develops from long-term chronic liver disease, is considered to be the primary risk factor leading to HCC [4]. However, the molecular mechanisms responsible for this malignant transformation are still not understood.
Adhesion GPCRs, which have more than 60 receptors in vertebrate genomes, including 31 members in mice and 33 members in humans [5], are characterized by extremely long N-terminal regions and a GPCR-like seven-pass transmembrane domain [6,7]. In recent years, since aberrant expressions of several adhesion-GPCR molecules have been identified in various human cancers, they have been closely associated with cancer development. Forexample, BAI1 was downregulated in glioblastomas [8,9], colorectal cancer [10] and gastric cancers [11]; GPR124 was identified as a tumor endothelial marker [12], promoting tumor angiogenesis; GPR56 was reported to be highly expressed in human gliomas [13]. With the gradual identification of adhesion GPCRs in various tumors, it is advisable to pay more attention to study the role of these interesting molecules in tumorigenesis and to explore more possibilities for their use in drugs [14].
GPR110, an orphan receptor that is highly conserved among species, belongs to members of the adhesion G protein-coupled receptor family. To date, its physiological function remains unknown. Lum et al. identified GPR110 as a potential oncogene in murine T cell lymphomas during a large-scale retroviral insertion mutagenesis screening [15]. Subsequently, they detected overexpression of the GPR110 transcript and protein in the majority of lung (74%) and prostate (59%) adenocarcinomas, as well as related cell lines. Therefore, it can be assumed that GPR110 may participate in tumor development. However, the relationship between Gpr110 and hepatocarcinogenesis in vivo is as yet unknown.
Here we used Gpr110 knockout mice to study the role of Gpr110 in hepatocarcinogenesis. We found that deficiency of Gpr110 impeded DEN plus CCl4-induced HCC formation. Moreover, the absence of Gpr110 alleviated CCl4-mediated liver injury and hepaticfibrosis, and ultimately delayed the development of cirrhosis and progression into HCC. Mechanistically, disruption of Gpr110 triggered increased activation of the IL-6/STAT3 pathway, which decelerated chronic liver injury-driven hepatocarcinogenesis.
Materials and methods
DEN plus CCl4-induced liver cancer model
Mice with a homozygous deletion of Gpr110 were maintained on a 129SvJ background (Supplementary Figure 2). The procedures described were approved by the Animal Care and Use Committee of Shanghai Jiao Tong University School of Medicine. The liver tumor model induced by DEN plus CCl4 was generated according to the previous reports [16,17]. Briefly, 15-day-old male Gpr110-/- mice and their male wild-type littermates were intraperitoneally injected with a single dose of DEN (25 mg/kg), and then with 5 ml/kg body weight of 10% CCl4 (dissolved in olive oil) twice a week from the age of 4 weeks. The mice were sacrificed at 4, 5, and 6 months post-CCl4, and liver tissues were excised for experiments.
CCl4-induced liver injury
Four- to six-week-old male Gpr110-/- mice and their male wild-type littermates were intraperitoneally injected with a single dose of CCl4 (4 ml/kg, 10% dissolved in olive oil). The mice were sacrificed at 0, 24, 48, and 72 hours post-CCl4, and liver tissues were excised for experiments.
CCl4-induced liver fibrosis
Four- to six-week-old male Gpr110-/- mice and their male wild-type littermates were intraperitoneally injected with CCl4 (4 ml/kg, 10% dissolved in olive oil) three times a week [17,18]. The mice were sacrificed at 0, 2, 4, and 8 weeks post-CCl4, and liver tissues were excised for experiments.
Inhibitor treatment
Specific p-Stat3 inhibitor, Stattic (Selleck Chemicals, USA), was injected at 3.75 mg/kg every other day, i.p., for four weeks, or at 3.75 mg/kg twice a week for four months.
Histology of mouse liver tissues
Formalin-fixed liver samples were paraffin-embedded, and paraffin sections (5 μm) were stained with hematoxylin and eosin (H&E) for analysis of morphologic changes. Liver fibrosis was determined by Sirius red staining. Immunohistochemistry and Sirius red staining were performed according to routine protocol. The primary antibodies were as follows: anti-Gpr110 (sc-292423, Santa Cruz, CA, USA,1:50), anti-α-SMA (A5228, Sigma, CA, USA, 1:200), anti-F4/80 (ab6604, Abcam, MA, USA, 1:100), anti-AFP (A0200, Abclonal, MA, USA, 1:200), anti-Collagen1 (sc-59772, Santa Cruz, CA, USA, 1:50), and anti-Tgf-β (GTX110630, GeneTex, CA, USA, 100). Apoptosis was assessed by TUNEL staining of paraffin-embedded slides (11684795910, Roche, Mannheim, Germany). Proliferation was assessed by immunostaining for BrdU (#MS-1058-B0, Thermo Fisher Scientific Inc. Rockford, CA, USA, 1:200) staining.
Blood chemistry
Mice were sacrificed at different time points after CCl4 treatment. Blood was drawn via cardiac puncture into heparinized tubes and centrifuged (5,000 rpm, 15 min, 4°C). Plasma was collected and stored at -80°C for further analysis. Serum AST, ALT and other serum biochemical indicators were measured using a Fuji DRICHEM 55500 V (sysmex System, Tokyo, Japan) according to the manufacturer’s instructions.
TUNNEL assay
Apoptotic cells on sections were detected using an In Situ Cell Death Detection Kit (Roche) according to the manufacturer’s protocol. Images were taken with a Nikon fluorescent microscope.
BrdU staining
Cell proliferation in livers were measured by the incorporation of BrdU, which was administered i.p. to mice at a dosage of 50 μg/g body weight 2 h before killing. BrdU incorporation was detected on sections by immunohistochemistry.
Cell culture
The SMMC-7721 HCC cell line was obtained from the Shanghai Cell Bank (Shanghai, China). The Huh7 cell line was obtained from the American Type Culture Collection (Manassas, VA). Cells were routinely cultured in DMEM medium (Hyclone) supplemented with 10% fetal bovine serum (FBS, Gibco) within a humidified incubator containing 5% CO2 at 37°C.
Western blotting
Whole mouse liver tissues were homogenized in Triton lysis buffer (20 mM Tris, pH 7.4, 137 mM NaCl, 10% glycerol, 1% Triton X+100, 2 mM EDTA, 1 mM PMSF, 10 mM NaF, 5 mg/ml aprotinin, 20 mM leupeptin, and 1 mM sodium orthovanadate) and centrifuged at 13,000 rpm at 4°C for 15 min. Protein extracts were separated by 8-16% SDS-PAGE and analyzed using the following primary antibodies: anti-CYP2E1 (A2160, Abclonal, MA, USA, 1:500), anti-a-SMA (A5228, Sigma, CA, USA, 1:200), anti-Collagen1 (sc-59772, Santa Cruz, CA, USA, 1:500), and anti-Tgf-β (GTX110630, GeneTex, CA, USA, 1000), anti-IL-6 (GTX31178, GeneTex, CA, USA, 1:500), anti-p-Stat3 (#9145, Cell Signaling Technology, MA, USA, 1:1000), anti-stat3 (#4904, Cell Signaling Technology, MA, USA, 1:1000), anti-p-ERK (#9101, Cell Signaling Technology, MA, USA, 1:1000), anti-p-P38 (#9211, Cell Signaling Technology, MA, USA, 1:1000), and anti-p50/105 (ADI-KAP-TF112, ENZO, NY, USA, 1:1000). Secondary antibodies were labeled with IRDye 700 (Rockland Immunochemicals). Protein levels were detected using the Odyssey system (LiCor, Lincoln, NE).
Cytokine measurement in serum
Mice were sacrificed at different time points after CCl4 treatment. Blood was drawn via cardiac puncture into heparinized tubes and centrifuged (5,000 rpm, 15 min, 4°C). Serum was collected and stored at -80°C for further analysis. Levels of IL-6, IL-1β, TNF-α and MIP2 were detected with a commercial ELISA kit following the instructions of the manufacturer (Dakewei, Shenzhen, China) (Synergy H1Hybrid Reader, BioTek, USA).
Measurement of IL-6 in liver tissue homogenates
Mice were sacrificed at different time points after CCl4 treatment. For the measurement of IL-6, equal weights of liver tissues (100 mg) from various groups were sonicated in 1 ml of phosphate buffer saline containing protease inhibitor cocktail. Homogenates were cleared by centrifuging at 10,000 rpm at 4°C for 15 min. The supernatants were transferred to another new tube, liver IL-6 levels were normalized to the total protein in the homogenates, which was detected with a commercial ELISA kit following the instructions of the manufacturer (Dakewei, Shenzhen, China) (Synergy H1Hybrid Reader, BioTek, USA).
Quantitative real-time PCR analysis
Total RNA was prepared from mouse liver tissues using Trizol Reagent (Invitrogen) according to the manufacturer’s instructions. Real-time PCR analysis was performed using a SYBR Premix Ex Taq kit (Takara, Dalian, China) on an Eppendorf Mastercycler system according to the manufacturer’s instructions. The expression of specific transcript was normalized against β-actin, and the experiments were repeated in triplicate. The primers used are listed in Supplementary Table 1.
Statistical analysis
Data were presented as means ± SD, while quantitative variables were analyzed using t-test. All statistical tests were two-sided, and P<0.05 was regarded as statistically significant.
Results
Deletion of Gpr110 alleviated CCl4-mediated acute hepatic injury and compensatory proliferation
To determine the expression pattern of Gpr110 in mice, we performed semi-quantitative and real-time reverse transcription (RT)-PCR analyses on various mouse tissues. As shown in Supplementary Figure 1, the highest expression level of Gpr110 mRNA was found in the prostate, and the second highest was found in the kidney and liver, implicating the tissue compartments where Gpr110 could execute its physiological functions in mice. To assess the biological functional role of Gpr110 in vivo, we generated a mouse model with global-targeted deletion of Gpr110. As expected, Gpr110 mRNA and proteins were disrupted in Gpr110-/- mice (Supplementary Figure 2). However, after a global-targeted deletion of Gpr110 mice were born alive and appeared grossly normal (Supplementary Figure 3), suggesting that Gpr110 is not indispensable for normal development. Interestingly, pathological analysis showed a smaller necrosis area existed in Gpr110-/- mice compared with their wild-type littermates after CCl4 challenge (Figure 1A, 1B). Meanwhile, serum alanine transaminase (ALT) and aspartate transaminase (AST) levels were significantly lower in Gpr110-/- mice compared to control mice, resulting in subdued hepatocellular damage in Gpr110-/- mice (Figure 1C, 1D). In addition, TUNEL assay results revealed that the number of apoptotic hepatocytes was significantly lower in Gpr110-/- mice than in wild-type mice post CCl4 injection (Figure 1E, 1F). As we have known, p450 CYP2E1-mediated CCl4 metabolism is required for CCl4-induced liver injury [19]. Therefore, we examined whether the reduction of liver injury in Gpr110-/- mice was due to alterations of CCl4 metabolism. As shown in Supplementary Figure 4, Expression of CYP2E1 was similar in liver tissues from control and Gpr110-/- mice, suggesting no difference between the two groups on CCl4 metabolism. Owing to the high regenerative capacity of the liver, even when some fractions of hepatocytes undergo cell death in response to CCl4, the remaining surviving hepatocytes should undergo a compensatory proliferative response. BrdU-labeling of proliferating cells showed that the number of compensatory proliferating hepatocytes was significantly lower in Gpr110-/- mice than in wild-type mice after CCl4 challenge (Figure 1G, 1H), which indicated that deletion of Gpr110 further impeded hepatic compensatory proliferation after CCl4 treatment. Usually, acute liver diseases are accompanied by inflammation, and inflammation is considered to be an important factor in hepatocellular damage [20,21]. However, we found that Gpr110-/- mice were more susceptible to CCl4-induced liver acute inflammation by detecting of several hepatic inflammatory cytokines and inflammatory cell markers (Supplementary Figure 5). Moreover, clinical data have also shown that inflammation does not always kill hepatocytes during liver damage in some patients [22,23]. In conclusion, these results suggest that inflammation may do not correlate with hepatocellular injury induced by CCl4. Taken together, these observations demonstrate that deficiency of Gpr110 weakened hepatocellular damage, but enhanced inflammation induced by CCl4.
Figure 1.

Disruption of Gpr110 weakened CCl4-induced hepatic acute injury and hepatocyte compensatory proliferation. (A, B) H&E staining of sacrificed Gpr110-/- mice and wild-type littermate liver sections (100×), mice were treated CCl4 for 0-, 24- and 48 hours (A). Scalebar, 100 μm. Percentage of necrosis area were quantified with the ImageJ software and statistical analyzed (B). Values are presented as mean ± SD. *P<0.05; **P<0.01. (C, D) Serum ALT, AST levels were determined at 0, 24 and 48 hours after the CCl4 injection (n=4) and statistical analyzed. Values are presented as mean ± SD. *P<0.05; **P<0.01. (E, F) TUNEL staining of liver sections from 0-, 24- and 48 hours after the CCl4 injection-sacrificed Gpr110-/- mice and wild-type littermate mice liver (200×) (E). Scalebar, 50 μm. Quantified by statistical analyzing percentage of positive cells in 20 high-power fields (F). Values are presented as mean ± SD. *P<0.05; **P<0.01. (G, H) Representative immunohistochemical staining of BrdU, sections from 0- and 48 hours after the CCl4 injection-sacrificed Gpr110-/- mice and wild-type littermate mice liver (200×) (G). Scalebar, 50 μm. Quantified by statistical analyzing percentage of positive cells in 20 high-power fields (H). Values are presented as mean ± SD. *P<0.05; **P<0.01.
Gpr110-/- mice displayed a lower level of hepatic fibrosis compared with wild-type mice after CCl4 administration
To assess whether deletion of Gpr110 also mitigated liver fibrosis, a model of long-term CCl4 administration was used. After two, four, and eight weeks of CCl4 treatment, fibrotic changes were also noticeable, Gpr110-/- mice displayed subdued hepatic fibrosis (Figure 2A, 2B). Western blot and immunohistochemical analysis also confirmed that lower expressions of liver fibrosis-related molecular Collagen-1 and TGF-β in the liver were found in Gpr110-/- mice compared to their wild-type littermates (Figure 2C-F, 2I). Given that hepatic stellate cells (HSCs) have a critical role in inducing fibrosis, and that the α-smooth muscle actin (α-SMA) was considered as the marker of activated HSCs, we detected the expression levels of α-SMA. As shown in Figure 2F-I, decreased α-SMA was found in the Gpr110-/- group after CCl4 treatment. Since chronic inflammation is known to play an important role in fibrosis, we assessed inflammatory markers and infiltrate. As shown in Supplementary Figure 6, the difference did not appear to be significant. Thus, these results demonstrated the importance of Gpr110 in CCl4-mediated chronic liver fibrosis. Collectively, these data suggest that deletion of Gpr110 abated CCl4-mediated hepatocyte injury, which possibly led to subsequent fibrotic changes and tumor formation.
Figure 2.

Deletion of Gpr110 caused less evident liver fibrosis and HSC activation upon chronic CCl4 challenges. (A, B) Mice were treated with CCl4 for 0, 4 and 8 weeks (n=6). Siriusred staining sections from 0-, 4- and 8 weeks after the CCl4 injection-sacrificed Gpr110-/- mice and wild-type littermate mice liver (100×) (A). Scalebar, 100 μm. Percentage of Sirius red positive area were quantified with the ImageJ software and statistical analyzed (B). Values are presented as mean ± SD. *P<0.05; **P<0.01. (C, D) Immunostaining of Collagen1, sections from 0-, 4- and 8 weeks after the CCl4 injection -sacrificed Gpr110-/- mice and wild-type littermate mice liver (100×) (C). Scalebar, 100 μm. Percentage of collagen1 positive area were quantified with the ImageJ software and statistical analyzed (D). Values are presented as mean ± SD. *P<0.05; **P<0.01. (E, F) Immunostaining of Tgf-β, sections from 0-, 4- and 8 weeks after the CCl4 injection-sacrificed Gpr110-/- mice and wild-type littermate mice liver (100×) (E). Scalebar, 100 μm. Percentage of Tgf-β positive area were quantified with the ImageJ software and statistical analyzed (F). Values are presented as mean ± SD. *P<0.05; **P<0.01. (G, H) Immunostaining of α-SMA, sections from 0-, 4- and 8 weeks after the CCl4 injection-sacrificed mice liver (100×) (G). Scalebar, 100 μm. Percentage of Tgf-β positive area were quantified with the ImageJ software and statistical analyzed (H). Values are presented as mean ± SD. *P<0.05; **P<0.01. (I) Western blot analysis of Collagen1, Tgf-β and α-SMA in 0-week, 4-week and 8-week CCl4-treated Gpr110-/- mice and wild-type littermate mice liver.
Absence of Gpr110 restrained carcinogen-induced hepatocarcinogenesis, weakened fibrosis, and prolonged survival time
It has been well established that most patients with HCC developed as a result of chronic injury, fibrosis, and hepatocarcinogenesis. Meanwhile, using the Cancer Genome Atlas (TCGA) database, we found the gene copy number of GPR110 in hepatocellular carcinoma was significantly different from the level in normal samples (Supplementary Figure 7). To elucidate whether Gpr110 regulated hepatocarcinogenesis in vivo, we used 15-day-old Gpr110-/- mice. Gpr110-/- mice and their wild-type littermates were subjected to DEN one time at 15-day age, and they were consistently given hepatotoxin CCl4 injections starting at four weeks of age (Figure 3A). This model revealed representative features of human HCC development, reflected the multiple stages of hepatocarcinogenesis, and faithfully recapitulated the natural history of HCC, including chronic injury, inflammation, and fibrosis. Gpr110-/- mice showed an obvious diminution of tumor number and tumor load after DEN plus CCl4 injections (Figure 3B-D). As expected, H&E staining and immunostaining of anti-alpha fetoprotein (AFP) antibody definitely confirmed that the tumors were hepatocellular carcinoma in the Gpr110-/- mice and wild-type mice (Figure 3E). The survival curves revealed that the survival of Gpr110-/- mice was apparently longer than that of wild-type mice (P<0.05, Figure 3F). As illustrated in Figure 3G, after four months of CCl4 treatment only a 20% occurrence rate was observed in the surface of livers in the Gpr110-/- mice, whereas nearly 100% of Gpr110 wild-type mice had developed single- or multiple-surface nodular tumors. Moreover, deletion of Gpr110 in tumor tissue masses significantly reduced tumor growth in the nude mice tumor xenograft model (Supplementary Figure 8). Tumor tissue masses from Gpr110-/- mice and wild-type mice were subcutaneously inoculated into nude mice, a month later, we found that tumor tissue masses from the wild-type mice were completely developed, while tumor tissue masses from Gpr110-/- mice had all disappeared. These tumors were well delineated and exhibited enlargement of hepatocytic plates, disruption of the reticulin network, and absence of portal tracts. Sirius red staining showed less severe liver fibrosis in Gpr110-/- mice (Figure 3H, 3I). However, disruption of Gpr110 did not affect the proliferation, apoptosis, vascularization and inflammation of tumors in vivo (Supplementary Figures 9 and 10). Furthermore, overexpression of GPR110 did not affect the proliferation, migration, or invasion of the human hepatocellular carcinoma cell lines SMMC-7721 or Huh7 in vitro (Supplementary Figure 11). The above findings indicate that Gpr110 may not regulate the growth of liver tumor cells, but that it accelerates injury-driven liver tumor initiation. They also suggest that Gpr110-related hepatocarcinogenesis is associated with chronic liver injury and fibrosis. Absence of Gpr110 decelerates liver fibrosis/cirrhosis from progressing into tumorigenesis.
Figure 3.

Deletion of Gpr110 decelerated hepatocarcinogenesis in chronically injured livers. (A) Schematic diagram of the progression of DEN plus CCl4-induced mouse HCC model. (B) Macroscopic liver appearance of Gpr110-/- mice and wild-type littermates injected with DEN plus CCl4 at different time points (n=6). (C, D) Statistical analysis of Tumor number and Tumor load about Gpr110-/- mice and wild-type littermates injected with DEN plus CCl4 at different time points (n=6) Values are presented as mean ± SD. *P<0.05; **P<0.01. (E) H&E staining and immunohistochemical staining of AFP in tumor area about liver sections from Gpr110-/- mice and wild-type littermates injected with DEN plus CCl4 after 5 months. Scalebar, 50 μm. (F) The survival curves of Gpr110-/- mice (n=10) and control littermates (n=7) (P<0.05) after DEN+CCl4 treated. (G) After 4 months CCl4 treated Gpr110-/- mice (n=6) and control littermates (n=6), Tumor incidence was determined and statistical analyzed (P<0.05). (H, I) Liver sections from (b) were collected for Sirius red staining (100×) (H). Scalebar, 100 μm. Quantified and statistical analyzed (I). Values are presented as mean ± SD. *P<0.05; **P<0.01.
Gpr110 deficiency resulted in increased IL-6/STAT3 pathway activation in mice liver tumor
To explore the possible mechanism for the decreased susceptibility of Gpr110-/- mice to carcinogen-induced carcinogenesis, we detected several typical pathways involved in carcinogenesis as well as belonging to the GPCR pathway. Although CCl4 induced activation of MEK, ERK, NF-kB, JNK, and p38/MAPK, there were no differences to be distinguished between the Gpr110-/- mice and control littermates (Supplementary Figure 12). Interestingly, we detected obvious enhanced activation of STAT3 and increased IL-6 secretion in the liver tumors from DEN plus CCl4-treated Gpr110-/- mice (Figure 4A-C). It has been reported that activation of STAT3 in hepatocytes plays a protective role in preventing liver tumorigenesis induced by chronic CCl4 challenge [24]. Meanwhile, transient overexpression of GPR110 in the transfected human hepatocellular carcinoma cell lines SMMC-7721 and Huh-7 inhibited IL-6 expression and STAT3 activation (Figure 4D-F). Thus, these results suggest that Gpr110 deficiency leads to increased IL-6/STAT3 pathway activation in mice liver tumor and enhanced IL-6/STAT3 pathway activation in Gpr110-/- mice liver most likely contributes to decelerate tumorigenesis after DEN plus CCl4 exposure.
Figure 4.
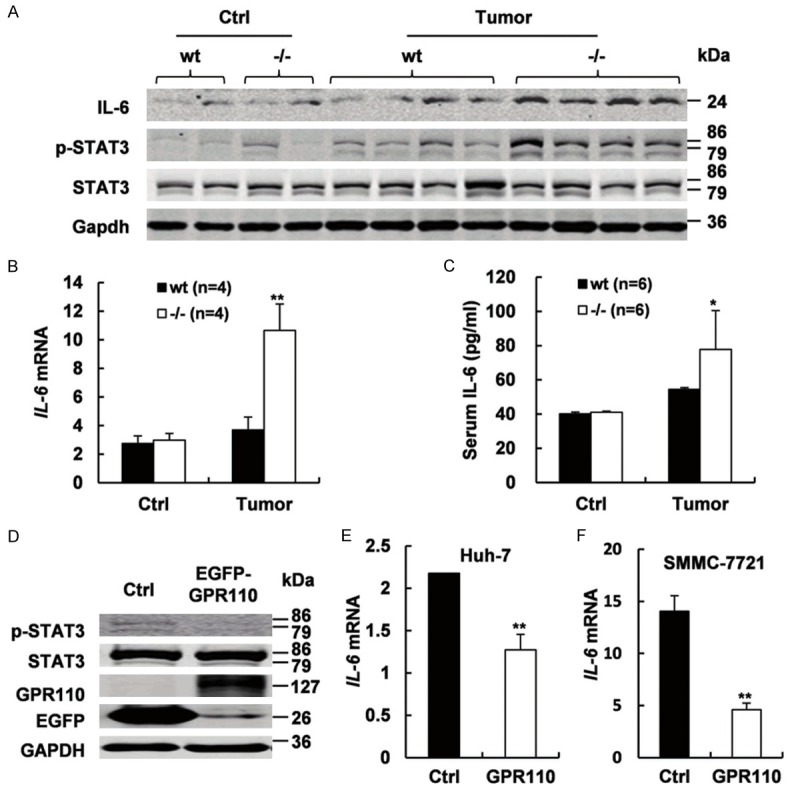
Gpr110-/- mice exhibited sustained increased IL-6/STAT3 pathway activation in liver tumors. A. Immunoblot staining analysis of IL-6, p-STAT3, and total STAT3 in olive oil and DEN plus CCl4-treated 5 month Gpr110-/- mice and wild-type littermate mice liver (n=6). B. qRT-PCR analysis of IL-6 expression in olive oil and DEN plus CCl4-treated 5 months Gpr110-/- mice and wild-type littermate mice livers (n=4). Values are presented as mean ± SD from three independent experiments. *P<0.05; **P<0.01. C. ELISA analysis of serum levels of IL-6 in olive oil and DEN plus CCl4-treated 5 months Gpr110-/- mice and wild-type littermate mice livers (n=4). Values are presented as mean ± SD. *P<0.05; **P<0.01. D. Western blot analysis of GFP, p-STAT3, and total STAT3 in Huh-7 and SMMC-7721 cells after instantaneous overexpression of GPR110. E, F. qRT-PCR analysis of IL-6 expression in Huh-7 and SMMC-7721 cells after instantaneous overexpression of GPR110. Values are presented as mean ± SD from three independent experiments. *P<0.05; **P<0.01.
Increased IL-6/STAT3 pathway activation was detected during liver injury and fibrosis in Gpr110-/- mice
Subsequently, we investigated whether IL-6/STAT3 pathway persistently activated in the early stage of CCl4 induced liver injury and fibrosis in Gpr110-/- mice. Interleukin-6 (IL-6), an important pro-inflammatory cytokine, was found to be higher in Gpr110-/- mice during liver injury and fibrosis (Figure 5A-F). Constantly increased STAT3 activation was also detected in the hepatocytes of the Gpr110-/- group after CCl4 treatment (Figure 5G, 5H). As well as, we assessed a few of classical STAT3 target genes, Mcl-1, Socs, FOXO3A, FOXO1, Pim-1 and c-Myc by real-time PCR, except for Pim-1 and c-Myc, the others were all higher expressed in Gpr110-/- mice after 24 h CCl4 treatment (Figure 5I). Interleukin-6 (IL-6) has been shown to be a hepatoprotective cytokine in this model [25], as well as in several other liver injury models, including ischemia/reperfusion, partial hepatectomy, alcoholic and nonalcoholic fatty liver, and Con A-induced T cell hepatitis model [26,27]. Numerous studies have demonstrated the hepatoprotective role of IL-6 against liver injury, despite its pro-inflammatory effect [28,29]. The hepatoprotective effects of IL-6 are mediated mainly via activation of signal transducers and activators of transcription 3 (STAT3) in the hepatocytes [26]. The IL-6/STAT3 pathway has been shown to exert hepatoprotective effects during liver injury, fibrosis and hepatocarcinogenesis by regulating the expression of a wide array of genes, including anti-apoptotic and anti-oxidative genes that protect against hepatocyte damage [24,30]. In summary, these results suggest that a persistently enhanced IL-6/STAT3 pathway activation due to deletion of Gpr110 is most likely to alleviate hepatocellular damage and fibrosis and delay the development of cirrhosis and progression into HCC.
Figure 5.

Enhanced IL-6/STAT3 pathway activation was detected in Gpr110-/- mice during liver injury and fibrosis. (A-C) qRT-PCR, ELISA analysis of serum levels of IL-6 and ELISA analysis of liver levels of IL-6 expression in Gpr110-/- mice and wild-type littermates treated with CCl4 for 0, 12, 24, and 48 hours (n=4). Values are presented as mean ± SD from three independent experiments. *P<0.05; **P<0.01. (D-F) qRT-PCR, ELISA analysis of serum levels of IL-6 and ELISA analysis of liver levels of IL-6 expression in Gpr110-/- mice and wild-type littermates treated with CCl4 for 0, 2, 4, and 8 weeks (n=4). Values are presented as mean ± SD from three independent experiments. *P<0.05; **P<0.01. (G) Immunoblot staining analysis of IL-6, p-STAT3, and total STAT3 in Gpr110-/- mice and wild-type littermate liver treated with CCl4 for 0 h, 24 h and four weeks. (H) Quantification and statistical analysis of STAT3 activation detected by Immunoblot staining in (G). Values are presented as mean ± SD. *P<0.05; **P<0.01. (I) qRT-PCR analysis of classical STAT3 target genes, Mcl-1, Socs, FOXO3A, FOXO1, Pim-1 and c-Myc in Gpr110-/- mice and wild-type littermate liver treated with CCl4 for 24 h. Values are presented as mean ± SD from three independent experiments. *P<0.05; **P<0.01.
Phospho-STAT3 inhibitor restored the sensitivity of Gpr110-/- mice to CCl4-induced liver fibrosis and carcinogenesis
As Gpr110-/- mice displayed persistently increased IL-6/STAT3 activation during CCl4 treatment, and IL-6/STAT3 exerted a hepatoprotective function during CCl4 challenge, it seemed likely to account for the decreased susceptibility of Gpr110-/- mice to CCl4-induced liver fibrosis and hepatocarcinogenesis. To test this hypothesis, we treated both groups with four weeks of CCl4 with or without a specific phospho-STAT3 inhibitor, Stattic [31]. Correspondingly, activation of STAT3 was inhibited, and liver fibrosis in Gpr110-/- mice were more severe than that in control mice after long-termCCl4 plus Stattic treatment (Figure 6A-C). During the process of DEN plus CCl4-induced hepatocarcinogenesis, simultaneously injected with Stattic, phospho-STAT3 levels in Gpr110-/- group distinctly declined (Figure 6D). As anticipated, liver cancers in Gpr110-/- mice were completely accelerated, and liver cirrhosis was seriously exacerbated (Figure 6E-G). Taken together, this implies that deletion of Gpr110 increases IL-6/STAT3 activation, which protects against CCl4-induced hepatocyte damage, cirrhosis, and decelerates malignant transformation (Figure 6H).
Figure 6.

p-STAT3 inhibitor promoted liver fibrosis and carcinogenesis in Gpr110-/- mice after CCl4 exposure. (A) Immunoblot staining analysis of p-STAT3 and total STAT3 in Gpr110-/- mice and wild-type littermate liver. Mice were treated with CCl4 and CCl4 plus Stattic for four weeks (n=6). (B, C) Sirius red staining of liver sections from (a) (100×) (B). Scalebar, 100 μm. Quantifications and statistical analysis (C). Values are presented as mean ± SD. *P<0.05; **P<0.01. (D) Immunoblot staining analysis of p-STAT3 and total STAT3 in DEN plus CCl4 and DEN plus CCl4 plus Stattic-treated Gpr110-/- mice and wild-type mice livers (n=6). (E) Macroscopic liver appearance of Gpr110-/- mice and wild-type littermates. H&E (200×). Scalebar, 50 μm. Sirius red staining (100×) of mice treated with CCl4 or CCl4 plus Stattic for four months. Scalebar, 100 μm. (F, G) Tumor number and Tumor load from (E) were quantified and statistical analyzed (n=6). Values are presented as mean ± SD. *P<0.05; **P<0.01. (H) Schematic representation of the function and potential mechanism of Gpr110 in liver malignant transformation.
Discussion
HCC carcinogenesis is an intricate process that usually develops from chronic liver injury, hepatocyte regeneration, and cirrhosis. It is triggered by a variety of etiologic factors, dysregulations of related signal pathways, and genetic alterations, eventually leading to malignant transformation and carcinogenesis [32,33]. So far, many scientific studies have focused on the possible mechanisms involved in the progression from premalignant lesions to overt carcinomas. However, the intercessors that are responsible for a high risk of developing HCC in a chronically injured liver remain to be fully explored.
GPR110 has been identified as a proto-oncogene, and has also shown high expression levels in lung and prostate cancer, suggesting that it plays a role in tumor pathology. In our study, we found DEN plus CCl4 treatment resulted in fewer dysplastic nodules in Gpr110-/- mice than in their wild-type littermates. This strategy of induced liver cancer, which recapitulates human HCC development, can reflect the multiple stages of hepatocarcinogenesis, which including continuous DNA damage, fibrogenesis, cirrhosis, and tumor formation. Therefore, the restraint of carcinogen-induced hepatocarcinogenesis by Gpr110 deficency may be of the result of weakening persistent hepatocyte damage and fibrosis in the microenvironment.
The hepatotoxicity of CCl4 is mediated by the direct induction of hepatocellular injury and necrosis. Once CCl4 is injected, the cytochrome p450 (CYP2E1) in hepatocytes metabolizes it into trichloromethyl radicals (CCl3*), which elicit the production of reactive oxygen intermediates and then cause lipid peroxidation and membrane damage [19]. Repeated and prolonged administration of CCl4 for up to two years induces 50% HCC carcinogenesis in mice [34,35]. Moreover, tumors developed using this strategy displayed typical features of human HCC. CCl4 can induce hepatocytic death, and the surviving hepatocytes undergo compensatory proliferation. Reduced proliferation of hepatocytes was found in Gpr110-/- mice compared to wild-type mice after CCl4 stimulation, which was consistent with a smaller necrosis area in Gpr110-/- mice compared to their wild-type littermates. Because compensatory proliferation is critical for tumor development, the most likely interpretation of our results is that disruption of Gpr110 inhibited malignant transformation of liver cirrhosis by decreased compensatory growth in the microenvironment of inflammation and fibrillation.
In general, liver injury is accompanied by inflammation, which is generally believed to play a key role in accelerating the progression of liver damage. However, clinical data revealed that inflammation was not always consistent with hepatocellular damage in some patients. We showed that inflammation may attenuate liver necrosis induced by CCl4 in Gpr110-/- mice. Such a mechanism was associated with a hepatoprotective cytokine, IL-6, and IL-6/STAT3 pathway increased activation. IL-6 has been shown to be a hepatoprotective cytokine in this liver injury model.
The IL-6/STAT3 pathway was observed to be constantly increased in the Gpr110-/- group after CCl4 treatment. IL-6 is a pleiotropic cytokine with beneficial effects for the liver; numerous studies have demonstrated the hepatoprotective role of IL-6 against liver injury despite its pro-inflammatory effect [28,29]. IL-6 knockout mice were reported to be more susceptible to CCl4-induced liver injury and fibrosis [25]. Other studies have demonstrated that liver fibrosis was reduced in IL-6-deficient mice after CCl4 treatment [36,37]. The hepatoprotective effect of hepatic STAT3 has been well documented in various models of liver injury [22,30,38,39]. Our postulation was that disruption of Gpr110 increased activation of the IL-6/STAT3 pathway, which protected against hepatic damage and fibrosis induced by CCl4, consequently suppressing injury-driven liver tumor initiation. In addition, a specific phospho-STAT3 inhibitor significantly blocked disruption of Gpr110-mediated IL-6/STAT3 activation, and promoted hepatic fibrosis and HCC formation. Gpr110 is very likely involved in CCl4-induced IL-6/STAT3 pathway activation.
There is mounting evidence that adhesion GPCRs can couple to G proteins to activate a variety of different downstream signaling pathways. Therefore, disruption of Gpr110 may lead to the dysregulation of GPCR’s downstream signaling pathways and relevant molecules which are associated with tumorigenesis. Although CCl4 induced ERK, NF-kB, JNK, and p38/MAPK activation, almost no differences between the Gpr110-/- mice and control mice indicated that these pathways were not involved.
It has been well established that almost 90% of HCC cases develop as a result of cirrhosis in clinical practice [3,40]. Now, a number of primary treatment options, such as antiviral therapy and weight loss strategies, are variably effective for the prevention of HCC. Unfortunately, a significant number of patients still progress to end-stage liver disease. Thus, it is urgently necessary for new therapeutic strategies to be developed for the prevention or treatment of hepatic fibrosis, cirrhosis, and HCC. Our results demonstrated the significance of Gpr110 by regulating IL-6/STAT3 to allay cirrhosis and decelerate injury-driven liver tumor initiation, and suggested that targeting GPR110 and IL-6/STAT3 activation has the potential for liver disease therapy. Recently, a new strategy targeting multiple GPCRs was proposed for cancer treatment. In light of our studies, a combination of targeting GPR110 and IL-6/STAT3 activation may profoundly improve liver cirrhosis therapies. Further studies are needed to investigate the way by which Gpr110, as a receptor, regulates the secretion of IL-6, as well as the activation of the IL-6/STAT3 signaling pathway.
Acknowledgements
We thank Shanghai Research Center for Model Organisms for the generation of Gpr110 knockout mice. This work was partially supported by grants from National Natural Science Foundation of China (81430028, 81502383), the Ministry of Science and Technology of China (2011BAI15B02), the grants from the Science and Technology Commission of Shanghai Municipality (13DZ2280600, 15DZ2290800), and the E-Institutes of Shanghai Municipal Education Commission (E03003).
Disclosure of conflict of interest
None.
Abbreviations
- HCC
hepatocellular carcinoma
- ALT
alanine transaminase
- AST
aspartate transaminase
- DEN
diethylnitrosamine
- CCl4
carbon tetrachloride
- TUNEL
terminal-deoxynucleotidyl transferase mediated nick end labeling
- BrdU
5-bromodeoxyuridine
- CCR2
CC chemokine receptor 2
- α-SMA
α-smoothmuscle actin
- AFP
alpha fetoprotein
- IL-6
interleukin-6
- STAT3
signal transducer and activator of transcription
- mRNA
messenger RNA
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
Supporting Information
References
- 1.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 2.Mu X, Espanol-Suner R, Mederacke I, Affo S, Manco R, Sempoux C, Lemaigre FP, Adili A, Yuan D, Weber A, Unger K, Heikenwalder M, Leclercq IA, Schwabe RF. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J Clin Invest. 2015;125:3891–3903. doi: 10.1172/JCI77995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 4.Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–436. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 5.Bjarnadottir TK, Fredriksson R, Hoglund PJ, Gloriam DE, Lagerstrom MC, Schioth HB. The human and mouse repertoire of the adhesion family of G-protein-coupled receptors. Genomics. 2004;84:23–33. doi: 10.1016/j.ygeno.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 7.Bjarnadottir TK, Fredriksson R, Schioth HB. The adhesion GPCRs: a unique family of G protein-coupled receptors with important roles in both central and peripheral tissues. Cell Mol Life Sci. 2007;64:2104–2119. doi: 10.1007/s00018-007-7067-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishimori H, Shiratsuchi T, Urano T, Kimura Y, Kiyono K, Tatsumi K, Yoshida S, Ono M, Kuwano M, Nakamura Y, Tokino T. A novel brain-specific p53-target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene. 1997;15:2145–2150. doi: 10.1038/sj.onc.1201542. [DOI] [PubMed] [Google Scholar]
- 9.Vallon M, Essler M. Proteolytically processed soluble tumor endothelial marker (TEM) 5 mediates endothelial cell survival during angiogenesis by linking integrin alpha(v)beta3 to glycosaminoglycans. J Biol Chem. 2006;281:34179–34188. doi: 10.1074/jbc.M605291200. [DOI] [PubMed] [Google Scholar]
- 10.Hatanaka H, Oshika Y, Abe Y, Yoshida Y, Hashimoto T, Handa A, Kijima H, Yamazaki H, Inoue H, Ueyama Y, Nakamura M. Vascularization is decreased in pulmonary adenocarcinoma expressing brain-specific angiogenesis inhibitor 1 (BAI1) Int J Mol Med. 2000;5:181–183. doi: 10.3892/ijmm.5.2.181. [DOI] [PubMed] [Google Scholar]
- 11.Lee JH, Koh JT, Shin BA, Ahn KY, Roh JH, Kim YJ, Kim KK. Comparative study of angiostatic and anti-invasive gene expressions as prognostic factors in gastric cancer. Int J Oncol. 2001;18:355–361. [PubMed] [Google Scholar]
- 12.Carson-Walter EB, Watkins DN, Nanda A, Vogelstein B, Kinzler KW, St Croix B. Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res. 2001;61:6649–6655. [PubMed] [Google Scholar]
- 13.Shashidhar S, Lorente G, Nagavarapu U, Nelson A, Kuo J, Cummins J, Nikolich K, Urfer R, Foehr ED. GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene. 2005;24:1673–1682. doi: 10.1038/sj.onc.1208395. [DOI] [PubMed] [Google Scholar]
- 14.Lin HH. Adhesion family of G protein-coupled receptors and cancer. Chang Gung Med J. 2012;35:15–27. doi: 10.4103/2319-4170.106170. [DOI] [PubMed] [Google Scholar]
- 15.Lum AM, Wang BB, Beck-Engeser GB, Li L, Channa N, Wabl M. Orphan receptor GPR110, an oncogene overexpressed in lung and prostate cancer. BMC Cancer. 2010;10:40. doi: 10.1186/1471-2407-10-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R, Lefkowitch JH, Bower M, Friedman R, Sartor RB, Rabadan R, Schwabe RF. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao X, Fu J, Xu A, Yu L, Zhu J, Dai R, Su B, Luo T, Li N, Qin W, Wang B, Jiang J, Li S, Chen Y, Wang H. Gankyrin drives malignant transformation of chronic liver damage-mediated fibrosis via the Rac1/JNK pathway. Cell Death Dis. 2015;6:e1751. doi: 10.1038/cddis.2015.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun H, Chen L, Zhou W, Hu L, Li L, Tu Q, Chang Y, Liu Q, Sun X, Wu M, Wang H. The protective role of hydrogen-rich saline in experimental liver injury in mice. J Hepatol. 2011;54:471–480. doi: 10.1016/j.jhep.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 19.Basu S. Carbon tetrachloride-induced lipid peroxidation: eicosanoid formation and their regulation by antioxidant nutrients. Toxicology. 2003;189:113–127. doi: 10.1016/s0300-483x(03)00157-4. [DOI] [PubMed] [Google Scholar]
- 20.Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50:1258–1266. doi: 10.1016/j.jhep.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heydtmann M, Adams DH. Chemokines in the immunopathogenesis of hepatitis C infection. Hepatology. 2009;49:676–688. doi: 10.1002/hep.22763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horiguchi N, Lafdil F, Miller AM, Park O, Wang H, Rajesh M, Mukhopadhyay P, Fu XY, Pacher P, Gao B. Dissociation between liver inflammation and hepatocellular damage induced by carbon tetrachloride in myeloid cell-specific signal transducer and activator of transcription 3 gene knockout mice. Hepatology. 2010;51:1724–1734. doi: 10.1002/hep.23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mofrad P, Contos MJ, Haque M, Sargeant C, Fisher RA, Luketic VA, Sterling RK, Shiffman ML, Stravitz RT, Sanyal AJ. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology. 2003;37:1286–1292. doi: 10.1053/jhep.2003.50229. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Lafdil F, Wang L, Park O, Yin S, Niu J, Miller AM, Sun Z, Gao B. Hepatoprotective versus oncogenic functions of STAT3 in liver tumorigenesis. Am J Pathol. 2011;179:714–724. doi: 10.1016/j.ajpath.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovalovich K, DeAngelis RA, Li W, Furth EE, Ciliberto G, Taub R. Increased toxin-induced liver injury and fibrosis in interleukin-6-deficient mice. Hepatology. 2000;31:149–159. doi: 10.1002/hep.510310123. [DOI] [PubMed] [Google Scholar]
- 26.Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol. 2005;2:92–100. [PubMed] [Google Scholar]
- 27.Jin X, Zhang Z, Beer-Stolz D, Zimmers TA, Koniaris LG. Interleukin-6 inhibits oxidative injury and necrosis after extreme liver resection. Hepatology. 2007;46:802–812. doi: 10.1002/hep.21728. [DOI] [PubMed] [Google Scholar]
- 28.Blindenbacher A, Wang X, Langer I, Savino R, Terracciano L, Heim MH. Interleukin 6 is important for survival after partial hepatectomy in mice. Hepatology. 2003;38:674–682. doi: 10.1053/jhep.2003.50378. [DOI] [PubMed] [Google Scholar]
- 29.Wuestefeld T, Klein C, Streetz KL, Betz U, Lauber J, Buer J, Manns MP, Muller W, Trautwein C. Interleukin-6/glycoprotein 130-dependent pathways are protective during liver regeneration. J Biol Chem. 2003;278:11281–11288. doi: 10.1074/jbc.M208470200. [DOI] [PubMed] [Google Scholar]
- 30.Klein C, Wustefeld T, Assmus U, Roskams T, Rose-John S, Muller M, Manns MP, Ernst M, Trautwein C. The IL-6-gp130-STAT3 pathway in hepatocytes triggers liver protection in T cell-mediated liver injury. J Clin Invest. 2005;115:860–869. doi: 10.1172/JCI200523640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scuto A, Kujawski M, Kowolik C, Krymskaya L, Wang L, Weiss LM, Digiusto D, Yu H, Forman S, Jove R. STAT3 inhibition is a therapeutic strategy for ABC-like diffuse large B-cell lymphoma. Cancer Res. 2011;71:3182–3188. doi: 10.1158/0008-5472.CAN-10-2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding J, Wang H. Multiple interactive factors in hepatocarcinogenesis. Cancer Lett. 2014;346:17–23. doi: 10.1016/j.canlet.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 33.Montalto G, Cervello M, Giannitrapani L, Dantona F, Terranova A, Castagnetta LA. Epidemiology, risk factors, and natural history of hepatocellular carcinoma. Ann N Y Acad Sci. 2002;963:13–20. doi: 10.1111/j.1749-6632.2002.tb04090.x. [DOI] [PubMed] [Google Scholar]
- 34.Sun B, Karin M. The therapeutic value of targeting inflammation in gastrointestinal cancers. Trends Pharmacol Sci. 2014;35:349–357. doi: 10.1016/j.tips.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCay PB, Lai EK, Poyer JL, DuBose CM, Janzen EG. Oxygen- and carbon-centered free radical formation during carbon tetrachloride metabolism. Observation of lipid radicals in vivo and in vitro. J Biol Chem. 1984;259:2135–2143. [PubMed] [Google Scholar]
- 36.Sun R, Tian Z, Kulkarni S, Gao B. IL-6 prevents T cell-mediated hepatitis via inhibition of NKT cells in CD4+ T cell- and STAT3-dependent manners. J Immunol. 2004;172:5648–5655. doi: 10.4049/jimmunol.172.9.5648. [DOI] [PubMed] [Google Scholar]
- 37.Rio A, Gassull MA, Aldeguer X, Ojanguren I, Cabre E, Fernandez E. Reduced liver injury in the interleukin-6 knockout mice by chronic carbon tetrachloride administration. Eur J Clin Invest. 2008;38:306–316. doi: 10.1111/j.1365-2362.2008.01939.x. [DOI] [PubMed] [Google Scholar]
- 38.Haga S, Terui K, Zhang HQ, Enosawa S, Ogawa W, Inoue H, Okuyama T, Takeda K, Akira S, Ogino T, Irani K, Ozaki M. Stat3 protects against Fas-induced liver injury by redox-dependent and -independent mechanisms. J Clin Invest. 2003;112:989–998. doi: 10.1172/JCI17970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakamori R, Takehara T, Ohnishi C, Tatsumi T, Ohkawa K, Takeda K, Akira S, Hayashi N. Signal transducer and activator of transcription 3 signaling within hepatocytes attenuates systemic inflammatory response and lethality in septic mice. Hepatology. 2007;46:1564–1573. doi: 10.1002/hep.21837. [DOI] [PubMed] [Google Scholar]
- 40.Aravalli RN, Steer CJ, Cressman EN. Molecular mechanisms of hepatocellular carcinoma. Hepatology. 2008;48:2047–2063. doi: 10.1002/hep.22580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.