

Abstract
Globally, gastric cancer is the second leading cause of cancer deaths because of the lack of effective treatments for patients with advanced tumors when curative surgery is not possible. Thus, there is an urgent need to identify molecular targets in gastric cancer that can be used for developing novel therapies and prolonging patient survival. Checkpoint kinase 1 (Chk1) is a crucial regulator of cell cycle transition in DNA damage response (DDR). In our study, we report that Chk1 plays an important role in promoting gastric cancer cell survival and growth, which serves as an effective therapeutic target in gastric cancer. First, Chk1 ablation by small interfering RNA could significantly inhibit cell proliferation and sensitize the effects of ionizing radiation (IR) treatment in both p53 wild type gastric cancer cell line AGS, and p53 mutant cell line MKN1. Secondly, we tested the anticancer effects of Chk1 chemical inhibitor LY2606368, which is a novel Chk1/2 targeted drug undergoing clinical trials in many malignant diseases. We found that LY2606368 can induce DNA damage, and remarkably suppress cancer proliferation and induce apoptosis in AGS and MKN1 cells. Moreover, we identified that LY2606368 can significantly inhibit homologous recombination (HR) mediated DNA repair and thus showed marked synergistic anticancer effect in combination with poly (ADP-ribose) polymerase 1 (PARP1) inhibitor BMN673 in both in vitro studies and in vivo experiments using a gastric cancer PDx model. The synergy between LY2606368 and PARP1 was likely caused by impaired the G2M checkpoint due to LY2606368 treatment, which forced mitotic entry and cell death in the presence of BMN673. In conclusion, we propose that Chk1 is a valued target for gastric cancer treatment, especially Chk1 inhibitor combined with PARP inhibitor may be a more effective therapeutic strategy in gastric cancer.
Keywords: Chk1, LY2606368, DNA damage response, BMN673, gastric cancer
Introduction
Despite the development of early diagnosis and new therapeutic strategy, the clinical outcome of gastric cancer has only been modestly improved [1]. It still ranks as the fourth cause of cancer and the second cause of cancer-related mortality worldwide [2]. Conventionally, surgery is the most effective treatment method for the limited number of patient identified without metastasis to other organs. However, for gastric cancer patients with distant metastasis, chemotherapy and radiotherapy are the major approaches but are relatively ineffective [3]. Due to poor response rate to chemotherapy and radiation, the prognosis of advanced gastric cancer patient remains abysmal. To overcome this clinical obstacle, novel chemotherapy approaches and deeper molecular mechanistic understanding required to implement effective gastric cancer treatment strategies [4,5].
In the past decades, emerging targeted therapy against cell cycle checkpoints have provided the opportunity to improve therapeutic outcome of cancer patient [6,7]. Normally, DNA damage activates the cell cycle checkpoint that causes arrest injury at different cell cycle phases, protecting cells from apoptosis and unscheduled death until DNA damage is repaired. Two major signaling pathways are involved in the DNA damage response; one is the ataxia telangiectasia mutated serine/threonine kinase (ATM)/checkpoint kinase 2 (Chk2) pathway [8], and another is the ATM and Rad3-related serine/threonine kinase (ATR)/checkpoint kinase 1 (Chk1) cascade [9]. In contrast to frequent mutations and/or loss of ATM/Chk2 signaling components, the components involved in ATR/Chk1 pathway are often overexpressed in malignant cells and are considered to promote the tumor tumorigenesis [10]. Given that p53 is the major checkpoint regulator in the G1 phase, and due to the common mutations or loss of p53 function, cancer cells largely depend on the G2M checkpoint to repair endogenous and exogenous DNA damage, thus providing an attractive opportunity for cancer treatment by targeting key regulators of the G2M checkpoint especially in p53 mutant cancer [11,12]. Chk1 is a highly conserved serine-threonine kinase, which is encoded by the CHK1 gene located at chromosome 11q22-23, with its C-terminal SQ/TQ domain regulating translocation between the nuclear compartment and cytoplasm, which is critical for its activity [13,14]. Independent of p53 status, Chk1 can be activated by most DNA damage types including single-stranded DNA breaks and stalled forks causing replication stress, which may lead to the degradation of Cdc25A due to its phosphorylation, and blocks the cells from the entry of mitosis with unrepaired DNA damage. Chk1 is not only involved in G2M arrest but also play a role in G1-S, intra S, and mitotic checkpoints [15]. Thus Chk1 may be an ideal target for cancer therapy [16].
Chk1 overexpression is observed in many cancer types and is associated with poor response to chemotherapy and radiotherapy, including lung cancer and breast cancer and leukemia [17-19]. To date many inhibitors against Chk1 have been designed are in clinical trials. For example, LY2606368 can selectively and potentially inhibit the activity of Chk1. Preclinical data suggests that LY2606368 treatment induces DNA damage and apoptosis, and in phase I clinical trials LY2606368 showed potential anticancer effects in solid tumors [20,21]. However, there is no report on the therapeutic value of targeting Chk1 in gastric cancer what both p53 and ATM aberrations are common. Our study is designed to demonstrate the role of Chk1 in regulating cell proliferation and the response to DNA damage in gastric cancer. Moreover, we also investigated the therapeutic effect of Chk1 inhibitor LY2606368 in gastric cancer cells, and examined the rational combinations with LY2606368 that may be more effective for gastric cancer treatment.
Material and methods
Cell culture
Gastric cancer cell lines AGS (p53 wild type) was obtained from American Type Culture Collection(ATCC, USA), MKN1 was a gift from Dr Sanghoon Lee(MD Anderson cancer center, USA), cells were grown in RMPI 1640 supplemented with 10% fetal bovine serum (Gibco, USA) and 0.5% penicillin/streptomycin (Gibco, USA). Cells were cultured in a humidified cell culture incubator containing 5% carbon dioxide at 37°C, routinely changed the cell culture medium every 3 days.
Drugs and irradiation preparation
PARP inhibitor BMN673 was purchased from Selleckchem (USA), Chk1 inhibitor LY2606368 was bought from Eli Lily and Company (USA), both reagents were dissolved in DMSO for vitro experiment. Irradiation was generated by X-ray irradiator (Radsource, USA) at 250 kv.
Small interfering RNA
AGS and MKN1 were seeded to 6cm cell culture dish with 30% density overnight, and transfected with Chk1 or non-target siRNA (Sigma Aldrich, USA) using oligofectamine transfection reagent (Thermo Fisher Scientific, USA) according to the protocol provided by the manufacture. The following is siRNA sequences of Chk1 and non-target control: Chk1: GCAACAGTATTTCGGTATAAT; non-target control: UAAGGCUAUGAAGAGAUAC.
MTS assays
Proliferation inhibition effect of Chk1 ablation, IR sensitivity, anticancer effect of BMN673 and LY2606368 were detected by MTS Cell Proliferation Colorimetric Assay Kit (Biovision, USA). Cells were seeded into 96 wells cell culture plate, then treated with indicated experiment conditions, then added 20 uL MTS reagent to each well subsequently, after incubated for 2 hours, cell viability of each well was detected on microplate reader (BioTek, USA) at a wavelength of 490 nM.
Colony formation assays
AGS and MKN1 were seeded to 6 well cell culture plate, then treated with indicated conditions, cultured for 10-14 days, washed with PBS twice, then fixed in 4% polymerised formaldehyde, discarded the medium and washed with PBS, stained the cells with crystal violet, colony formation in all groups were calculated.
Apoptosis assay
Annexin V/PI apoptosis kit (Invitrogen, USA) was applied to detect the apoptosis. Cells were treated with indicated reagents, gently trypsinized the cells, then washed the cells with cold PBS twice, resuspended the cells in 500 uL Annexin V binding buffer, added 5 uL Annexin V and 5 uL propidium iodide (PI) into each sample, incubated in the dark room for 5 minutes at room temperature, analyzed the Annexin-V and PI binding using fluorescence-activated cell sorting (FACS) (Beckman Coulter, USA).
Cell cycle analysis
AGS and MKN1 cells were treated with LY2606368, BMN673 or combination of these two drugs for 24 hours, control groups treated with DSMO were settled. After treatment, cells were collected and fixed in 70% ethanol at 4°C overnight, then washed with PBS twice, suspended the cell and stained with PI Staining solution (50 ug/ml PI and RNase in PBS), cell cycle analysis were measured using FACS (Beckman Coulter, USA).
HR repair analysis
As previous reported [22], U2OS cells containing HR repair reporter with direct repeat GFP (DR-GFP) was generated for HR repair capacity analysis. GFP-expressing plasmid was used as a control of transfection efficiency. pCBASecI plasmid was transfected to detect the HR repair capacity. After overexpression for 24 hours, cells were treated with 20 nM LY2606368 for another 24 hours, then GFP positive cells were detected by FACS. For hydroxyurea (HU)-synchronized HR repair assay [22], the same procedure was performed as HR repair analysis assay, then treated with 2 mM Hu for another 16 hours to synchronize the cell cycle, GFP positive cells were detected by FACS.
Western blot
Cells were harvested and lysed in urine buffer supplemented with 1% protease and 1% phosphorylation inhibitors (GeneDepot, USA), Bradford protein assay kit was used to determine the protein concentration for each sample. Total 30 μg protein for each sample was loaded to gradient SDS-PAGE gel, after finishing gel electrophoresis, protein was transferred from gel to PVDF membranes, then blocked in 5% skim milk (Sigma Aldrich, USA) for 1 hours, washed by 0.1% Tween-20 with PBS (PBST) for three times (10 minutes/time), then incubated with primary antibodies overnight at 4°C, washed the membranes by PBST for 10 minutes in the second day, repeated for three times, incubated with anti-mouse or rabbit-HRP conjugated secondary antibodies (Invitrogen, USA, 1:2500 in PBST) for 1 hour, after washing with PBST for another 3 times, the expression of the targeted proteins were detected using ECL solution (BioRad, USA), beta-actin (Sigma Aldrich, USA) was chosen as loading control. The following were the information of the target primary antibodies: anti-Chk1, anti-phospho Chk1(Ser345), anti-γ-H2AX, anti-cleaved capased3, anti-phospho-H3 (S10), anti-Cyclin-B1 were purchased from Cellsignaling Techonology (USA).
Gastric cancer PDX mouse model
All animal work was approved by MD Anderson Cancer Center Institutional Animal Care and Use Committee. The primary tumor was obtained from the surgical specimen (F0) of gastric cancer patient, then transplanted to six weeks old male Balb/c-nude mice, after two generations culture to establish stable PDX tumor, the PDX tumor was transplanted to the leg flank of male nude mice. After the PDX tumor volume reached 50 mm3, then randomly divided to 4 groups (N=5 per group): 1; untreated group as control, 2; BMN673 (0.33 mg/kg orally, one time daily), 3; LY2606368 (2 mg/kg, subcutaneous injection, 3 times/week), 4; Combination group. Tumor volume was measured and recorded once every two days, after nearly 3 weeks treatment, mice were sacrificed, and tumor weight was measured. Tumor volume was calculated by the following formula: tumor volume = length * width2/2.
Statistical analysis
All data in this study were presented with mean value ± standard deviation (SD). The student t test was used to determine the P value between different groups, if the P value was less than 0.05, the result was considered to be valued.
Results
Chk1 ablation can significantly suppress the cell proliferation and sensitize the IR treatment in gastric cancer cells
To evaluate the role of Chk1 on the survival and proliferation of gastric cancer cell lines, AGS and MKN1 were transfected with Chk1 siRNA and control groups with non-target siRNA. As shown in Figure 1A, 1B, Chk1 knockdown in AGS and MKN1 cell lines significantly inhibited cell proliferation. We then assessed the effect of Chk1 knockdown on the responses to IR treatment. We found that Chk1 ablation significantly enhanced the anticancer effects of IR treatment both in AGS and MKN1 cells (Figure 1C). Western blot analysis showed that the siRNA could significant inhibit expression of Chk1 (Figure 1D). Together, these data suggest that targeting Chk1 might be a good choice for gastric cancer treatment.
Figure 1.
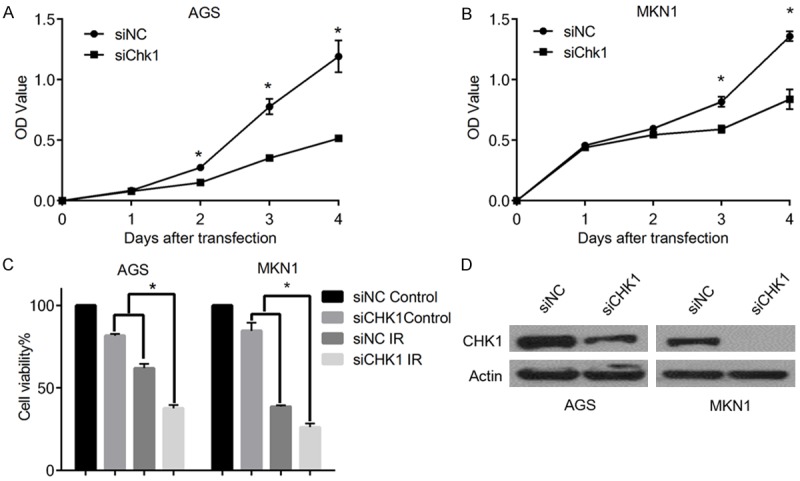
Chk1 ablation can significantly suppress the cell proliferation and sensitize the IR treatment in gastric cancer cells. A, B. Graphical presentation of cell viability at different days examined by MTS assay after Chk1 knockdown. OD values were measured and plotted with respect to time (*P<0.05). C. Graphical presentation of relative (%) cell viability of AGS and MKN1 cells exposed with IR (2 Gy), and with IR (4 Gy) respectively, (*P<0.05). D. Western blot analyses using anti-Chk1 antibodies showing Chk1 siRNA efficiently inhibited the expression of Chk1 in AGS and MKN1 cells. Actin was used as a loading control.
Chk1 inhibitor LY2606368 can induce DNA damage and apoptosis, and can suppress cell proliferation in gastric cancer cells
AGS and MKN1 cells were treated with different concentrations of LY2606368 for 3 days, and cell viability was then evaluated. Our results showed that the viability of cells treated with LY2606368 was significantly reduced in a dose dependent manner for both the cell lines (Figure 2A). Further, clonogenic assay also showed remarkable inhibition of proliferation by LY2606368 in AGS and MKN1 cells (Figure 2B). Moreover, increased levels of cleaved-caspase3 indicated significant apoptosis induction after exposure to LY2606368 (Figure 2C, 2D). Furthermore, Western blot analyses of AGS and MKN1 cells treated with LY2606368 showed substantial reduction in the level of endogenous Chk1, whereas the phosphorylation of Chk1 (Ser345) and the expression of γ-H2AX was significantly increased, indicating the persistence of double-strand break (DSB) in the treatment groups (Figure 2E).
Figure 2.
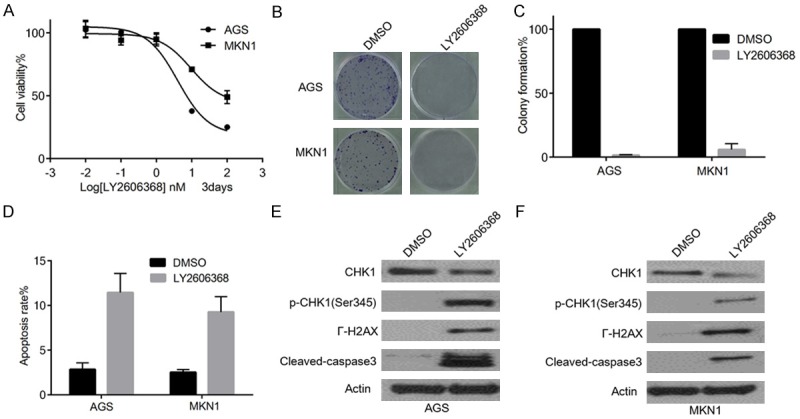
Chk1 inhibitor LY2606368 can induce DNA damage and apoptosis, and can suppress cell proliferation in gastric cancer cells. A. Graphical presentation of % cell viability of AGS and MKN1 cells measured 3 days after treatment with LY2606368. B. Clonogenic assay in AGS and MKN1 cells. Cells were treated with LY2606368 for 3 days. Cell viability in AGS and MKN1 cells were significantly inhibited in a dosage-dependent manner. C. Graphical presentation of relative (%) colony formation of AGS and MKN1 cells in clonogenic assay as described in B after exposure to 25 nM LY2606368 for 24 hours. D. Graphical presentation of apoptosis in AGS and MKN1 cells, measured using Annexin V/PI after exposure to 25 nM LY2606368 for 24 hours. Significant apoptosis was observed in AGS and MKN1 cells staining. E, F. Western blot analyses of AGS and MKN1 cells treated with LY2606368 for 24 hours. Endogenous Chk1, γ-H2AX, cleaved caspase3, and p-Chk1 (Ser345) were detected using their respective antibodies as shown left to each panel.
Chk1 inhibitor LY2606368 reduces HR repair
As proper cell cycle regulation is essential for efficient DNA repair, we further investigate whether LY2606368 may impair HR repair, which is a predominant repair mechanism utilized by cells in S and G2/M phases. We utilized a HR repair reporter assay, where in the pCBASecI plasmid was transfected to induce DSB. The ability of cells to repair DSBs through HR can be examined by the ratio of GFP positive cells to negative cells. As showed in Figure 3A, LY2606368 significantly impaired HR repair capacity. We also found that LY2606368 also caused S phase arrest (Figure 3B). Thus we used HU to synchronized the cell cycle to test whether Chk1 inhibition may regulate HR repair in addition to its effect on controlling cell cycle transition. We found that HR repair was decreased in LY2606368 treatment group even after HU exposure (Figure 3C, 3D). Together, our data demonstrated that LY2606368 could repress HR repair independent of its effect on cell cycle progression.
Figure 3.
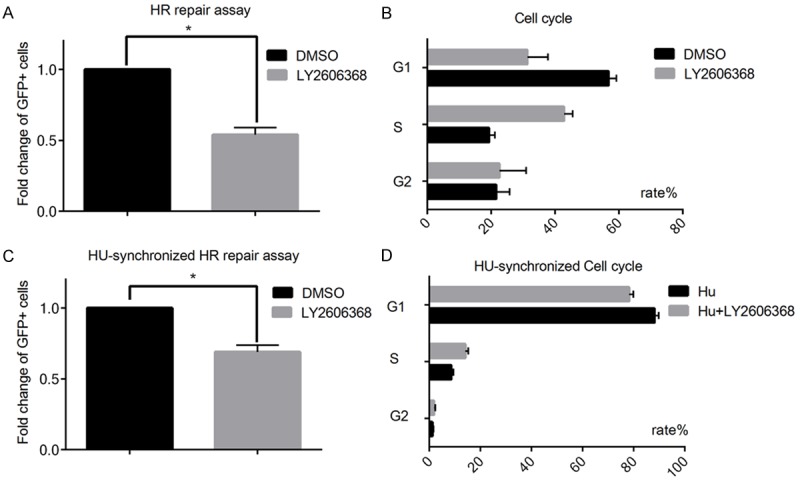
Chk1 inhibitor LY2606368 can suppress HR repair capacity. DR-GFP cells were used to detect the effect of LY2606368 on HR repair. A. LY2606368 treatment (20 nM) for 24 hours suppresses the HR repair capacity (P<0.05, LY20606368 VS DMSO). Each value was presented with the percentage of the GFP positive cells in pCBASecI transfected cells. The fold change was calculated, and data was showed as Mean ± SD. B. Cell cycle in HR repair assay was analyzed. LY2606368 induces S phase arrest in DR-GFP cells. C. HU was used to synchronize the cell cycle distribution for 16 hours before HR repair analysis, fold change was showed as mean ± SD. D. Cell cycle analysis for HU-synchronized HR repair assays showed there was no significant cell cycle alternation between DMSO control and LY2606368 treated groups after HU synchronization.
LY2606368 has synergistic anticancer effects with the PARP inhibitor BMN673 in gastric cancer cells
Given that, Chk1 inhibits HR repair, and PARP inhibitors are more effect in HR repair deficient cells, it is likely that Chk1 inhibition could enhance the effect of PARP inhibitor by decreasing HR repair capacity. To address this possibility, we examined whether the PARP inhibitor BMN673 has synergistic anticancer effect with LY2606368 in gastric cancer cells. Chou-Talalay method was applied to calculate the combination index (CI) of BMN673 and LY2606368 in AGS and MKN1 cells [23]. As shown in Figure 4A, 4B, significant synergy was observed between BMN673 and LY2606369. Moreover, the combination treatment was associated with decreased colony formation and more apoptosis in AGS and MKN1 cells (Figure 4C-E). Furthermore, increased expression levels of cleaved caspase3 in combination groups also supported the synergistic effect.
Figure 4.
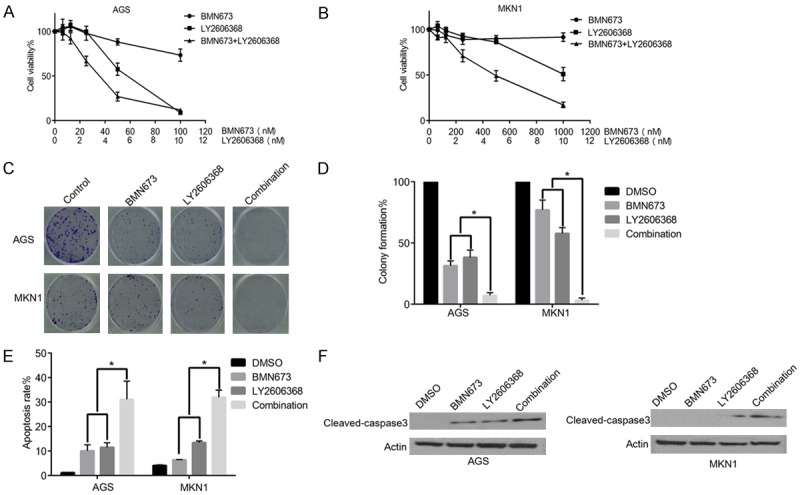
LY2606368 can sensitize the anticancer effect of PARP inhibitor BMN673 in gastric cancer cells. A, B. Graphical presentation of relative (%) cell viability of AGS and MKN1 cells after treatment with indicated drugs for 5 days. Cell viability was detected by MTS assays, significant synergistic anticancer effect between LY2606368 and BMN673 was observed in AGS and MKN1 cell, CI value of EC50 is 0.76 in AGS, and 0.41 in MKN1 (CI<1 indicates synergy). C. Clonogenic assay showed that LY2606368 (5 nM) and BMN673 (10 nM) combination inhibits cell survival of AGS and MKN1 cells. D. Graphical presentation of relative (%) colony formation in clonogenic assay as described in C (*P<0.05). E. Graphical presentation of (%) rate of apoptosis in AGS and MKN1 cells after treatment with 5 nM LY2606368, 1 µM BMN673, or both (AGS for 72 hours, MKN1 for 48 h) (*P<0.05). F. Western blot analyses of AGS and MKN1 cells treated with LY2606368, BMN673, or both. Combination of BMN673 and LY2606368 can induce more expression of apoptosis marker cleaved caspase3.
LY2606368 could force mitotic entry of G2M phase cells induced by BMN673 in gastric cancer cells
To detect potential mechanism of underlying synergistic effect of the BMN673 and LY2606368 combination in gastric cancer cells, cell cycle analyses were performed. As showed in Figure 5A, BMN673 induced a G2M arrest in AGS and MKN1 cells. Chk1 inhibition by LY2606368 impaired the G2M checkpoint, which resulted in decreased G2M arrest in combination groups (Figure 5B). Western blot analyses of AGS and MKN1 cells showed that BMN673 treatment increased the level of the G2M phase marker Cyclin-B1, and in contrast LY2606368 inhibited Cyclin B1 induction by BMN673. The expression level of mitotic marker p-H3 was increased in combination groups suggesting that the blockade was in fact in M phase (Figure 5C, 5D). Together these results suggest that BMN673 and LY2606368 combination could force mitotic entry of G2M phase cells with unrepaired DNA damage, which may lead to the synergistic anticancer effect of BMN673 and LY2606368.
Figure 5.
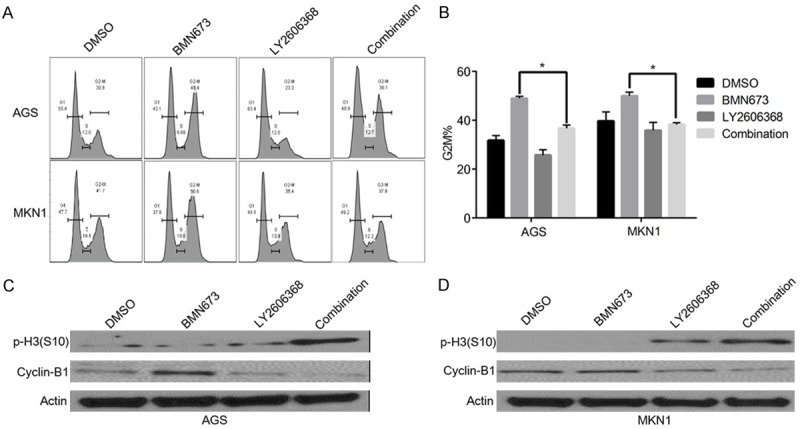
LY2606368 could force mitotic entry of G2M phase cells induced by BMN673 in gastric cancer cells. A, B. Cell cycle analysis of AGS and MKN1 cells after treatment with BMN673 (1 µM), LY2606368 (10 nM), or combination for 24 hours. C, D. Western blot analyses of AGS and MKN1 cells respectively after treatment with BMN673 (1 µM), LY2606368 (10 nM), or combination for 24 hours using antibodies as indicated left to each panel.
LY2606368 and BMN673 combination has synergistic anticancer effect in gastric cancer PDX model
In order to determine the anticancer effect of the LY2606368 and BMN673 combination in vivo, we developed a gastric cancer patient derived tumor graft model (PDX). In this model, we found that BMN673 and LY2606368 combination showed a significant inhibition of tumor growth compared to single drug treatment using BMN673 or LY2606368 (Figure 6A, 6B). Twenty days after treatment, compared to the single drug treatment group, the tumor volume and weight in the combination group was significantly decreased (Figure 6C, 6D). In summary, results from a PDX model support the rationale to assess the combination of LY2606368 and PARP inhibitor as a therapeutic strategy in gastric cancer.
Figure 6.
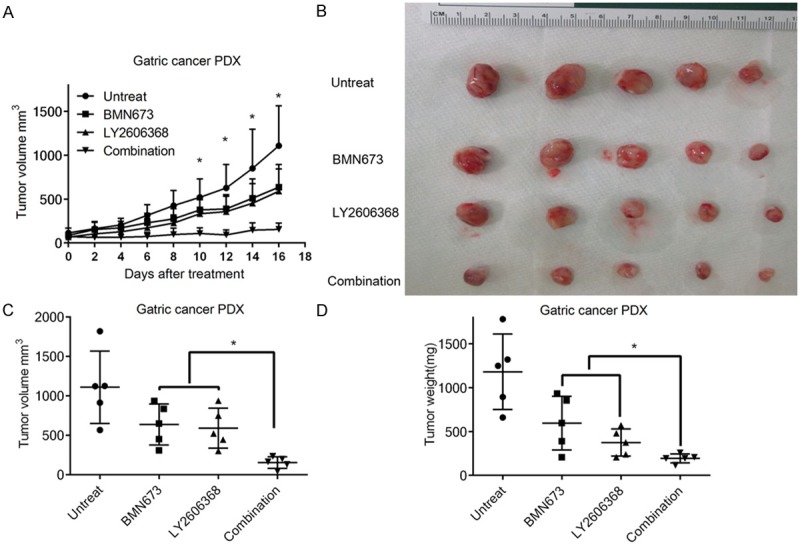
LY2606368 and BMN673 combination has synergistic anticancer effect in gastric cancer PDX model. Six weeks old nude mice with subcutaneous implanted gastric cancer PDX tumor were treated with vehicle control, BMN673 (0.33 mg/kg), LY2606368 (2 mg/kg) or combination of these two drugs for 16 days. Statistical significance differences between groups were calculated using unpaired t test. Significant differences were indicated by asterisk (P<0.05) A. Tumor volume of each group was measured in indicated days of treatment. Data were showed as mean ± SD. B. Images of gastric cancer PDX tumor of each group were presented at the same time of study ending. C, D. Tumor volume and tumor weight in each group were measured at the end of the treatment and presented.
Discussion
Although chemotherapy and radiotherapy only modestly improves clinical outcome of advanced gastric cancer, this benefit is compromised by toxicity and side effects. Thus it is important to identify key factors involved in chemoradiotherapy response, and this may provide chance to develop safer and more effective therapeutic strategies [24]. Given the important role of Chk1 in the response to DNA damage induced chemotherapy drugs and radiotherapy [25], and that activation of ATR/Chk1 pathway may allow cancer cells to escape from the toxicity induced by DNA damage by preventing the entry of the damaged cells into mitosis [26], we propose Chk1 as a validated therapeutic target in gastric cancer. In our studies, first we evaluated the role of Chk1 in cell proliferation in gastric cancer cells. We found that Chk1 ablation is sufficient to significantly suppress cell proliferation. Importantly, proliferation inhibition by Chk1 ablation can be observed both in p53 mutant and wild type gastric cancer cells. However, given that Chk1 is involved in nearly all cell cycle checkpoints, Chk1 inhibition may prove toxic to normal cells.
Because Chk1 regulates activation cell cycle checkpoint in response to DNA damage, Chk1 is an important marker that can predict the response to radiotherapy [27,28]. Further Chk1 expression levels correlate with and radiotherapy response in p53 wild type gastric cancer cells [29]. However, it remains unknown whether the ability of Chk1 inhibitors to sensitize gastric cancer cells to radiation is dependent on the context of genetic alterations in gastric cancer. Loss of p53 function is frequently identified in gastric cancer patients, which plays a critical role in regulating cellular response to radiation through its functions in controlling gene transcription, cell cycle transition and apoptosis. We found that Chk1 knockdown can both enhance the anticancer effect of IR treatment in both p53 wild type and mutant cells, an important observation in gastric cancer.
Importantly, we found that the Chk1 inhibitor LY2606368 sensitizes gastric cancer cells to the effect of the PARP inhibitor BMN673. This combination decreased cell viability and induced significant apoptosis both in p53 wild and mutant gastric cancer cells. Moreover, the dosage used in the combination is low, consistent with the potential for decreased toxicity to normal cells. It has been shown that PARP inhibitors could selectively target BRCA1 and BRCA2 mutant cancer cells through synthetic lethal effect in HRD [30,31]. Although PARP inhibitors showed profound anticancer effect as single agent, PARP inhibition also showed considerable effect when combined with other therapeutic reagents, such as EGFR inhibitors [32], and PI3K/mTOR inhibitors [33]. In our study, we showed that Chk1 could be considered as a potential target for combinations with PARP inhibitors in gastric cancer. Most tumor cells largely rely on the G2M checkpoint for DNA damage response due to lack of G1M checkpoint function, due to aberrant p53 function. When gastric cancer cells were exposed to the PARP inhibitor, the G2M checkpoint was activated and cells were arrested in the G2M phase until the DNA damage was repaired. We propose that the combination of a PARP inhibitor with Chk1 inhibitor largely inhibits the function of the G2M checkpoint, and speeds up the cell cycle, forcing the cell into mitosis with unrepaired DNA damage, and eventually leading to apoptosis and cell death.
Taken together, we demonstrate that Chk1 is credentialed target for gastric cancer therapy, and targeting Chk1 could sensitize cells to the anticancer effect of the PARP inhibitor BMN673. This provides a potential therapeutic strategy for advanced gastric cancer patient.
Acknowledgements
This research was supported by National Natural Science Foundation of China (No. 81572413), Natural Science Foundation of Hubei Province (No. 2015CFB378), Research Fund of Public welfare in Health, Industry Health and Family Plan Committee of China (No. 201402015), and the Scientific Research Training Program for Young Talents of Wuhan Union Hospital.
Disclosure of conflict of interest
None.
References
- 1.Tsai SH, Liu CA, Huang KH, Lan YT, Chen MH, Chao Y, Lo SS, Li AF, Wu CW, Chiou SH, Yang MH, Shyr YM, Fang WL. Advances in laparoscopic and robotic gastrectomy for gastric cancer. Pathol Oncol Res. 2017;23:13–17. doi: 10.1007/s12253-016-0131-0. [DOI] [PubMed] [Google Scholar]
- 2.Venerito M, Linkw A, Rokkas T, Malfertheiner P. Gastric cancer-clinical and epidemiological aspects. Helicobacter. 2016;21(Suppl 1):39–44. doi: 10.1111/hel.12339. [DOI] [PubMed] [Google Scholar]
- 3.Ajani JA, D’Amico TA, Almhanna K, Bentrem DJ, Chao J, Das P, Denlinger CS, Fanta P, Farjah F, Fuchs CS, Gerdes H, Gibson M, Glasgow RE, Hayman JA, Hochwald S, Hofstetter WL, Ilson DH, Jaroszewski D, Johung KL, Keswani RN, Kleinberg LR, Korn WM, Leong S, Linn C, Lockhart AC, Ly QP, Mulcahy MF, Orringer MB, Perry KA, Poultsides GA, Scott WJ, Strong VE, Washington MK, Weksler B, Willett CG, Wright CD, Zelman D, McMillian N, Sundar H. Gastric cancer, version 3.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2016;14:1286–1312. doi: 10.6004/jnccn.2016.0137. [DOI] [PubMed] [Google Scholar]
- 4.Joo MK, Park JJ, Chun HJ. Recent updates of precision therapy for gastric cancer: towards optimal tailored management. World J Gastroenterol. 2016;22:4638–50. doi: 10.3748/wjg.v22.i19.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulte N, Ebert MP, Hartel N. Gastric cancer: new drugs-new strategies. Gastrointest Tumors. 2014;1:180–94. doi: 10.1159/000380786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manic G, Obrist F, Sistigu A, Vitale I. Trial Watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol Cell Oncol. 2015;2:e1012976. doi: 10.1080/23723556.2015.1012976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen T, Stephens PA, Middleton FK, Curtin NJ. Targeting the S and G2 checkpoint to treat cancer. Drug Discov Today. 2012;17:194–202. doi: 10.1016/j.drudis.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 8.Awasthi P, Foiani M, Kumar A. ATM and ATR signaling at a glance. J Cell Sci. 2015;128:4255–62. doi: 10.1242/jcs.169730. [DOI] [PubMed] [Google Scholar]
- 9.Yazinski SA, Zou L. Functions, regulation, and therapeutic implications of the ATR checkpoint pathway. Annu Rev Genet. 2016;50:155–173. doi: 10.1146/annurev-genet-121415-121658. [DOI] [PubMed] [Google Scholar]
- 10.Sarmento LM, Póvoa V, Nascimento R, Real G, Antunes I, Martins LR, Moita C, Alves PM, Abecasis M, Moita LF, Parkhouse RM, Meijerink JP, Barata JT. CHK1 overexpression in T-cell acute lymphoblastic leukemia is essential for proliferation and survival by preventing excessive replication stress. Oncogene. 2015;34:2978–90. doi: 10.1038/onc.2014.248. [DOI] [PubMed] [Google Scholar]
- 11.Dillon MT, Good JS, Harrington KJ. Selective targeting of the G2/M cell cycle checkpoint to improve the therapeutic index of radiotherapy. Clin Oncol (R Coll Radiol) 2014;26:257–65. doi: 10.1016/j.clon.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Chen T, Stephens PA, Middleton FK, Curtin NJ. Targeting the S and G2 checkpoint to treat cancer. Drug Discov Today. 2012;17:194–202. doi: 10.1016/j.drudis.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 13.Golsteyn RM. The position of Chk1 in cancer research. Trends Cell Biol. 2001;11:191. doi: 10.1016/s0962-8924(01)01977-8. [DOI] [PubMed] [Google Scholar]
- 14.Walworth NC, Bernards R. rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science. 1996;271:353–6. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- 15.Fofaria NM, Kim SH, Srivastava SK. Piperine causes G1 phase cell cycle arrest and apoptosis in melanoma cells through checkpoint kinase-1 activation. PLoS One. 2014;9:e94298. doi: 10.1371/journal.pone.0094298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manic G, Obrist F, Sistigu A, Vitale I. Trial Watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol Cell Oncol. 2015;2:e1012976. doi: 10.1080/23723556.2015.1012976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grabauskiene S, Bergeron EJ, Chen G, Chang AC, Lin J, Thomas DG, Giordano TJ, Beer DG, Morgan MA, Reddy RM. CHK1 levels correlate with sensitization to pemetrexed by CHK1 inhibitors in non-small cell lung cancer cells. Lung Cancer. 2013;82:477–84. doi: 10.1016/j.lungcan.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarmento LM, Póvoa V, Nascimento R, Real G, Antunes I, Martins LR, Moita C, Alves PM, Abecasis M, Moita LF, Parkhouse RM, Meijerink JP, Barata JT. CHK1 overexpression in T-cell acute lymphoblastic leukemia is essential for proliferation and survival by preventing excessive replication stress. Oncogene. 2015;34:2978–90. doi: 10.1038/onc.2014.248. [DOI] [PubMed] [Google Scholar]
- 19.Al-Kaabi MM, Alshareeda AT, Jerjees DA, Muftah AA, Green AR, Alsubhi NH, Nolan CC, Chan S, Cornford E, Madhusudan S, Ellis IO, Rakha EA. Checkpoint kinase1 (CHK1) is an important biomarker in breast cancer having a role in chemotherapy response. Br J Cancer. 2015;112:901–11. doi: 10.1038/bjc.2014.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, Naing A, Bauer TM, Piha-Paul S, Johnson FM, Kurzrock R, Golden L, Hynes S, Lin J, Lin AB, Bendell J. Phase I study of LY2606368, a checkpoint kinase 1 inhibitor, in patients with advanced cancer. J. Clin. Oncol. 2016;34:1764–71. doi: 10.1200/JCO.2015.64.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, Beckmann R, Barda D, Marshall MS. LY2606368 causes replication catastrophe and antitumor effects through CHK1-dependent mechanisms. Mol Cancer Ther. 2015;14:2004–13. doi: 10.1158/1535-7163.MCT-14-1037. [DOI] [PubMed] [Google Scholar]
- 22.Peng G, Chun-Jen Lin C, Mo W, Dai H, Park YY, Kim SM, Peng Y, Mo Q, Siwko S, Hu R, Lee JS, Hennessy B, Hanash S, Mills GB, Lin SY. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat Commun. 2014;5:3361. doi: 10.1038/ncomms4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 24.Fanta PT, Roeland E, Shen JP, Shimabukuro KA, Hwang M, Ramamoorthy S, Lowy AM. Molecular predictors of treatment response in colon and gastric cancer. J. Clin. Oncol. 2012;30:619. [Google Scholar]
- 25.Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun. 2015;6:6823. doi: 10.1038/ncomms7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yazinski SA, Zou L. Functions, regulation, and therapeutic implications of the ATR checkpoint pathway. Annu Rev Genet. 2016;50:155–173. doi: 10.1146/annurev-genet-121415-121658. [DOI] [PubMed] [Google Scholar]
- 27.Busch CJ, Kröger MS, Jensen J, Kriegs M, Gatzemeier F, Petersen C, Münscher A, Rothkamm K, Rieckmann T. G2-checkpoint targeting and radiosensitization of HPV/p16-positive HNSCC cells through the inhibition of Chk1 and Wee1. Radiother Oncol. 2017;122:260–266. doi: 10.1016/j.radonc.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 28.Gill MR, Harun SN, Halder S, Boghozian RA, Ramadan K, Ahmad H, Vallis KA. A ruthenium polypyridyl intercalator stalls DNA replication forks, radiosensitizes human cancer cells and is enhanced by Chk1 inhibition. Sci Rep. 2016;6:31973. doi: 10.1038/srep31973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bargiela-Iparraguirre J, Prado-Marchal L, Fernandez-Fuente M, Gutierrez-González A, Moreno-Rubio J, Muñoz-Fernandez M, Sereno M, Sanchez-Prieto R, Perona R, Sanchez-Perez I. CHK1 expression in Gastric Cancer is modulated by p53 and RB1/E2F1: implications in chemo/radiotherapy response. Sci Rep. 2016;6:21519. doi: 10.1038/srep21519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tutt A, Robson M, Garber JE, Domchek S, Audeh MW, Weitzel JN, Friedlander M, Carmichael J. Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer. J. Clin. Oncol. 2009;27:CRA501. [Google Scholar]
- 31.Audeh MW, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Carmichael J, Tutt A. Phase II trial of the oral PARP inhibitor olaparib (AZD2281) in BRCA-deficient advanced ovarian cancer. J. Clin. Oncol. 2009;27:5500. [Google Scholar]
- 32.Sui H, Shi C, Yan Z, Li H. Combination of erlotinib and a PARP inhibitor inhibits growth of A2780 tumor xenografts due to increased autophagy. Drug Des Devel Ther. 2015;9:3183–90. doi: 10.2147/DDDT.S82035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang D, Li C, Zhang Y, Wang M, Jiang N, Xiang L, Li T, Roberts TM, Zhao JJ, Cheng H, Liu P. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol Oncol. 2016;142:548–56. doi: 10.1016/j.ygyno.2016.07.092. [DOI] [PMC free article] [PubMed] [Google Scholar]