

Abstract
Autophagy is a cellular process by which damaged organelles and dysfunctional proteins are degraded. Morusin is an anti-cancer drug isolated from the root bark of Morus alba. Morusin induces apoptosis in human prostate cancer cells by reducing STAT3 activity. In this study, we examined whether morusin induces autophagy and also examined the effects of autophagy on the morusin-induced apoptosis. Morusin induces LC3-II accumulation and ULK1 activation in HeLa cells. In addition, we found that induction of ULK1 Ser317 phosphorylation and reduction of ULK1 Ser757 phosphorylation occurred simultaneously during morusin-induced autophagy. Consistently, morusin induces autophagy by activation of AMPK and inhibition of mTOR activity. Next, we investigated the role of autophagy in morusin-induced apoptosis. Inhibition of autophagy by treating cells with the 3-methyladenine (3-MA) autophagic inhibitor induces high levels of morusin-mediated apoptosis, while treatment of cells with morusin alone induces moderate levels of apoptosis. Cell survival was greatly reduced when cells were treated with morusin and 3-MA. Taken together, morusin induces autophagy, which is an impediment for morusin-induced apoptosis, suggesting combined treatment of morusin with an autophagic inhibitor would increase the efficacy of morusin as an anti-cancer drug.
Keywords: Morusin, autophagy, apoptosis, AMP-activated protein kinase, anti-cancer drug
Introduction
Autophagy is a catabolic process that degrades cytoplasmic constituents and damaged organelles [1]. Autophagy-associated proteins are conserved from yeast to higher mammals [2]. The induction and progression of autophagy are tightly regulated by a variety of signals which are activated by nutrient starvation and cellular stress [3,4]. The mammalian target of rapamycin (mTOR) signaling pathway is essential in regulating the induction of autophagy. Under normal conditions, active mTOR phosphorylates S6K and 4E-BP1 for active protein synthesis and cell growth [5]. mTOR constitutively phosphorylates uncoordinated-51-like kinase 1 (ULK1) at Ser757 to block autophagy induction [6]. ULK1 is rapidly dephosphorylated upon nutrient deprivation or inhibition of target of rapamycin complex 1 (TORC1) by a variety of cellular stresses [7,8]. AMP-activated protein kinase (AMPK) is a highly conserved serine threonine protein kinase crucial for maintaining cellular energy homeostasis, since AMPK senses energy status and elicits cellular responses to promote adaptive changes in growth, differentiation, and autophagy [9]. Once activated, AMPK increases ATP production by stimulating glucose uptake and fatty acid oxidation, while inhibiting energy consuming processes such as protein synthesis by mTOR inhibition. AMPK also induces autophagy by ULK phosphorylation at Ser317 and Ser555. Coordinated phosphorylation and dephosphorylation of ULK1 is important for the induction of autophagy [6,7]. Activated ULK1 phosphorylates Beclin-1 at multiple sites including Ser15, thereby enhancing activity of the ATG14L-containing VPS34 complexes for ATG8/LC3 lipidation and autophagosome formation [10].
A variety of small molecule and natural compound anti-cancer drugs induce autophagy as well as apoptosis [11,12]. The root bark of the mulberry tree (Morus species, Moraceae) is used in traditional Chinese medicine for antiphlogistic, antipyretic, antiheadache, and diuretic effects [13]. Morusin was isolated from the branch and root bark of various species of Moraceae (Morus alba, Morus australis, and Morus nigra) [13-16], and was shown to exhibit various biological activities including inhibition of adipocyte differentiation [17], anti-diabetic activity [18], inhibition of platelet aggregation [13], scavenging activity against superoxide anion radicals [19], protection against NO-induced neuroblastoma cell death [20], and anti-microbial activity [21]. Morusin significantly affects the secretion and production of airway mucin and was proposed for use as a mucoregulator in inflammatory pulmonary diseases [20]. Notably, morusin possesses cytotoxicity against various human cancer cells including HT29, MCF, Hep3B, and HeLa cells [16,22]. Several recent papers report that morusin induces apoptosis through the regulation of multiple signaling pathways in various cancer cells. Morusin blocks STAT3 and NF-κB signaling resulting in caspase-3 activation and anti-invasive effects in liver, pancreatic, and prostate cancer cells [23-26]. Morusin also induces adipogenic protein expression and pro-apoptotic protein expression, while causing a decrease in anti-apoptotic protein expression in breast cancer cells [27]. In addition, morusin has the potential to kill human cervical cancer stem cells. Morusin inhibited the growth and migration of cancer stem cells through attenuation of NF-κB signaling which is up-regulated in cancer stem cells [28]. Cell death is orchestrated by various death signals. Autophagy is considered another death signal for apoptosis. Although the interplay between autophagy and apoptosis is known to control apoptosis, the autophagic targets for apoptosis induction have yet to be elucidated.
In this study, we characterized another function of morusin in the induction of autophagy and crosstalk between autophagy and apoptosis. We show that autophagy is induced shortly after morusin treatment and apoptosis induction follows the decline of autophagy. Morusin-induced autophagy appears to have a protective role against apoptosis by inhibiting induction of apoptosis. Simultaneous treatment of morusin with an autophagy inhibitor causes a strong induction of apoptosis. Therefore, combined treatment of morusin with an autophagic inhibitor could potentiate the efficiency of morusin as an anti-cancer drug.
Materials and methods
Cell culture and transfection
HEK293T, HeLa, HepG2, HCT116, A549, MCF7, Hep2 cells were cultured at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). U2OS and M2182 cells were cultured in RPMI 1640 supplemented with 10% FBS. MEFs were cultured in DMEM supplemented with 10% FBS, 1 mM pyruvate (Sigma Aldrich) and non-essential amino acid mixture (Invitrogen). HeLa cells stably expressing GFP-LC3 were cultured in DMEM supplemented with 10% FBS, 100 μg/ml penicillin/streptomycin and 1 μg/ml puromycin. Ulk1 wild-type and knockout mouse embryonic fibroblasts (MEFs) were kindly provided by Dr. JM Kim (Kyung Hee University School of Dentistry).
Antibodies and reagents
The following commercially available antibodies were used: Anti-AMPKα (2532), anti-pAMPKα T172 (2535), anti-ULK1 (8054S), anti-pULK1 S555 (5869S), anti-pULK1 S317 (12753S), anti-pULK1 S757 (6888S), anti-ATG12 (4180P), anti-ATG7 (8558P), anti-ATG5 (12994P), anti-ATG3 (3415P), anti-Beclin1 (3495P), anti-ATG16L1 (8089P), anti-LC3A/B (12741P), anti-mTOR (2983P), anti-4E-BP1 (9452S), anti-S6K (9202S), anti-pS6K (9234S), anti-cleaved caspase-3 (9661S), anti-PARP (9542S), anti-pSTAT3 (Y705) (9131), anti-STAT3 (9132), anti-Actin (3700S) antibodies were purchased from Cell Signaling Technology. Anti-p62 (ab56416) was purchased from Abcam. Morusin (root bark of Morus alba) (BP0961) was purchased from Biopurity Phytochemicals Ltd (Chendu, Sichuan, China). 3-methyladenine (M9281) was purchased from Sigma Aldrich. Compound C; AMPK inhibitor (171260) was purchased from CALBIOCHEM. Chloroquine (C6628) was purchased from Sigma Aldrich. Rapamycin from Streptomyces hygroscopicus; mTOR inhibitor (R0395) was obtained from Sigma Aldrich. The GFP-LC3 and GFP-RFP-LC3 expression plasmids were kindly provided by Dr. JM Kim (Kyung Hee University School of Dentistry).
RNAi
Transfection of small interfering RNA (siRNA) into the indicated cells were performed with RNAiMAXTM reagent (Invitrogen, 13778-075). Targeting siRNA and non-targeting control siRNA were synthesized and purified by Qiagen. The sequences were as follows: ULK1 siRNA, 5’-GCA CAG AGA CCG TGG GCAA-3’. Control siRNA, 5’-AAC TGT CAG TCA GTC GTA GTA-3’.
Western blot analysis
Western blotting was performed as described [29,30]. Briefly, cells were lysed with RIPA lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin). Cell lysates were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes, followed by detection with the indicated antibodies and ECL Western detection reagents (Intron).
Cell viability analysis
Cell viability was assessed by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Briefly, cells (1 × 103 cells/well) were seeded into 96-well plates and treated with morusin and/or 3-MA for 24 hours. Viable cells with MTT dye uptake were determined by measuring the optical density at 570 nm in an enzyme-linked immunosorbent assay reader. The Cell Viability Assay Kit (11465007001, Roche Diagnostics) was utilized and analysis was performed according to the manufacturer’s recommendations. Experiments were repeated three times, and the data is presented as the mean of triplicate wells ± SEM.
Confocal microscopy
Immunocytochemistry for HeLa cells was performed as described [30,31]. Briefly, HeLa and MEFs were seeded onto coverslips in 6-well plates. The cells were fixed in 3% paraformaldehyde for 15 minutes and permeabilized with 0.1% Triton X-100 in PBS for 5 minutes. Cells were blocked with 1X phosphate-buffered saline containing 2% bovine serum albumin for 20 min. Anti-LC3 antibody was used to stain cells for 1 hr. Samples were washed with PBS three times, and incubated with an Alexa Fluor 488-conjugated secondary antibody against a rabbit polyclonal anti-LC3 antibody. Confocal microscopy was performed with a Zeiss LSM700 microscope. Acquired images were processed with Adobe Photoshop.
Annexin V staining
Apoptotic cells were assessed by FACS analysis after annexin V-FITC and propidium iodide (PI) staining. Cells were washed twice in 1X phosphate-buffered saline and incubated with binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4) containing 0.5 mg annexin V-FITC (BD biosciences, 556420) for 10 min at room temperature. The cells were further incubated with binding buffer containing PI (BD bioscience, 556463) for 15 min on ice, before analysis by flow cytometry. Data were collected on a BD Canto-II, FACSCalibur (BD Biosciences), and analyzed with FlowJo software (Tree Star).
Colony formation assay
For clonogenic survival assays, cells were seeded into 60 mm plates (2,000 cells/plate). Cells were treated with the indicated dose of 3-MA and/or morusin, followed by visualization with Coomassie Brilliant Blue staining and counting of colonies.
Results
Morusin increases autophagy by enhancement of autophagy at initiation steps
Morusin induces apoptosis by downregulating STAT3-regulated gene expression, followed by activation of caspase-3 [23-25]. Since autophagy is another mechanism of cell death, and the crosstalk between autophagy and apoptosis was previously established, we tested whether morusin also induces autophagy. We tested various cancer cell lines for the accumulation of phosphatidylethanolamine-conjugated LC3-II upon morusin treatment, to rule out cell-type specificity of morusin effects. In all cell lines tested, treatment of morusin increased the level of LC3-II in a dose-dependent manner (Figure 1A). In addition, LC3 puncta accumulated upon morusin treatment, although fold increase is variable depending on the cell line (Figure 1B and 1C). Morusin-induced LC3-II accumulation was inhibited by co-treatment with 3-MA, an autophagy inhibitor (Figure 1D). Morusin-induced accumulation of LC3 puncta was also inhibited by 3-MA administration (Figure 1E and 1F). These results indicate that morusin can induce autophagy.
Figure 1.

Morusin induces autophagy. A. Morusin induces LC3-II accumulation in a dose-dependent manner. Various cancer cells were treated with increasing amounts of morusin, and LC3-II and p62 levels were analyzed after 6 hr of treatment. B. Morusin induces accumulation of LC3 puncta. Various cancer cells were treated with either DMSO or morusin (30 uM), and endogenous LC3 puncta were detected by immunostaining using an anti-LC3 antibody. C. The number of LC3 puncta was counted in three different arbitrary areas, and is shown on the graph. Data are presented as the mean ± SEM. D. The autophagy inhibitor 3-MA inhibits LC3-II accumulation induced by morusin. HeLa cells were treated with either DMSO or morusin in combination with or without 3-MA, and levels of LC3 and p62 were detected by immunoblotting. E. Inhibition of morusin-induced LC3 puncta by 3-MA treatment. HeLa cells expressing GFP-LC3 were treated with either DMSO or morusin for the indicated times in the presence or absence of 3-MA. F. The number of LC3 puncta was counted in three different arbitrary areas, and is shown on the graph. Data are presented as the mean ± SEM.
Autophagy consists of several sequential steps including induction, autophagosome formation, autolysosome fusion and degradation [32]. An increase in the number of LC3-containing autophagosomes upon morusin treatment can be due to either increased autophagic activity at an early stage or reduced turnover of autophagosomes at a late stage. As a preliminary study to identify the molecular target(s) of morusin in autophagy activation, autophagy flux was investigated using a GFP-RFP-LC3 expression strategy [33]. As shown in Figures 1E and 2A, morusin treatment induced accumulation of LC3 puncta which was inhibited by 3-MA treatment. In GFP-RFP-LC3 expressing cells treated with morusin, LC3 puncta emitting red fluorescence from RFP-LC3 with no or weak green fluorescence from GFP-LC3 (Figure 2A, arrow; Figure 2B, right panel) as well as LC3 puncta emitting both red and green fluorescence were observed (Figure 2A, arrowhead), while all the LC3 puncta emitted both red and green fluorescence in cells treated with chloroquine (CQ), an inhibitor of autolysosomal fusion (Figure 2A, arrowhead; Figure 2B, right panel). In addition, accumulation of LC3-II was further potentiated upon co-treatment with morusin and chloroquine (Figure 2C). These results suggest that morusin may increase LC3-II levels by induction of LC3-II formation at an early step, and not by blocking degradation of autophagosomes at a later step.
Figure 2.
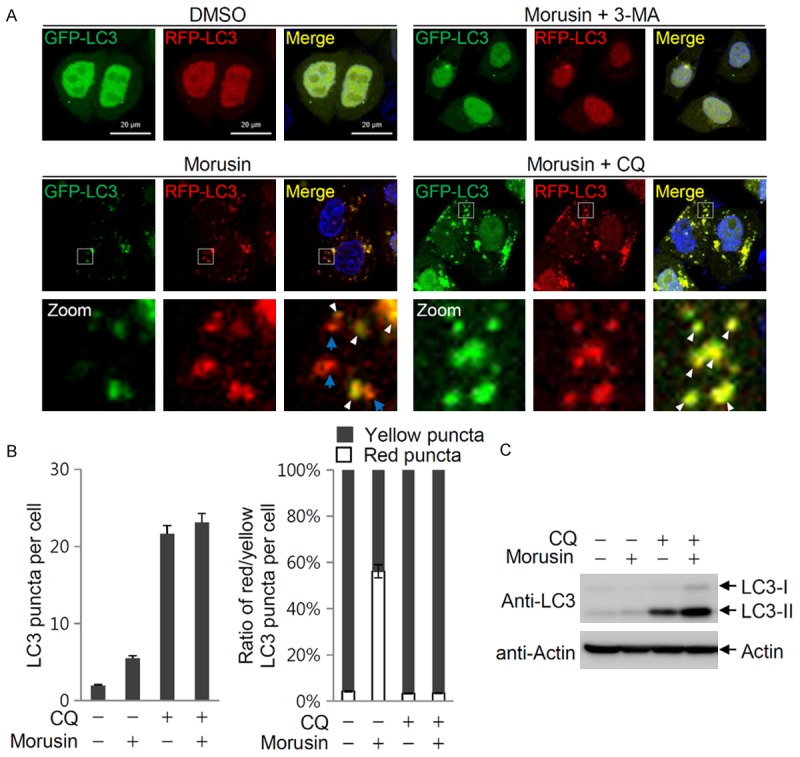
Morusin induces autophagy at early stages. A. Morusin induces autophagy at the initiation step. HeLa cells expressing GFP-RFP-LC3 were treated with morusin alone or in combination with 3-MA or chloroquine (CQ). Images of LC3 puncta containing green and red fluorescence were obtained with a confocal fluorescent microscope at an excitation wavelength of 488 nm and 543 nm, respectively. In the zoomed images, the arrows indicate red LC3 puncta with weak green fluorescence, and the arrowheads indicate red and green puncta colocalization. B. The number of LC3 puncta was counted in three different arbitrary areas, and is shown on the graph. Data are presented as the mean ± SEM. Morusin and CQ additively induce LC3-II accumulation (left panel). The relative ratio of yellow and red puncta is presented (right panel). C. The levels of LC3-II in cells treated with both morusin and CQ was compared to levels in cells treated with either drug alone.
Activation of ULK1 by morusin
Since morusin appears to induce autophagy at early steps, the levels of key regulators of autophagy at early steps were examined following morusin treatment. Immunoblotting showed that ULK1 phosphorylation was induced upon morusin treatment, while the levels of other proteins were not significantly altered (Figure 3A). Time-course analysis of ULK1 and LC3-II levels indicated that LC3-II began to accumulate soon after morusin treatment and ULK1 phosphorylation gradually increased in a time-dependent manner (Figure 3B). To determine whether ULK1 is responsible for morusin-mediated induction of autophagy, the effects of ULK1 depletion on morusin-induced autophagy were investigated. Morusin-induced accumulation of LC3-II was observed in HeLa cells, but not in ULK1 depleted cells (Figure 3C). Of note, ULK1 depletion resulted in accumulation of LC3-II in response to ammonia [34,35], but LC3-II levels were not increased by morusin treatment. In addition, the increase in LC3 puncta formation induced by morusin treatment was not observed in ULK1 depleted cells (Figure 3D). ULK1-dependent induction of autophagy in morusin treated cells was further confirmed in Ulk1 knockout mouse embryonic fibroblasts (MEFs). In wild-type MEFs, dose- and time-dependent ULK1 activation and LC3-II accumulation were observed upon morusin treatment, but not in Ulk1 knockout MEFs (Figure 3E and 3F). Consistent with the results from mammalian cells (Figure 3C), the basal level of LC3-II was increased in Ulk1 KO MEFs, but LC3-II levels were not increased by morusin treatment. In addition, morusin induced accumulation of LC3 puncta in wild-type MEFs, but not in Ulk1 knockout MEFs (Figure 3G). These results indicate that morusin-induced autophagy is mediated by ULK1 activation.
Figure 3.

ULK1 is activated upon morusin treatment. A. ULK1 activation by morusin treatment. HeLa cells treated with either DMSO or morusin were subjected to immunoblotting with the indicated antibodies. B. Time course of ULK1 activation. HeLa cells were collected at the indicated time points following administration of morusin (30 uM), and subjected to immunoblotting with anti-ULK1 and anti-LC3-II antibodies. C. ULK1 depletion impairs morusin-induced LC3-II accumulation. HeLa cells either depleted or undepleted of ULK1 were treated with morusin, and subjected to immunoblotting with anti-LC3 and anti-ULK1 antibodies. D. ULK1 depletion impairs morusin-induced LC3 puncta accumulation. HeLa cells either depleted or undepleted of ULK1 were treated with morusin, and subjected to immunostaining with an anti-LC3 antibody. E. Wild-type MEF or Ulk1-KO MEF was treated with increasing amounts of morusin, and cells were collected after 6 hr of morusin treatment for immunoblotting. F. Wild-type MEF or Ulk1-KO MEF was treated with morusin (30 uM), and cells were collected at the indicated time points for immunoblotting. G. ULK1 knockout impairs morusin-induced accumulation of LC3 puncta. Wild-type MEF or Ulk1-KO MEF was treated with morusin, and subjected to immunostaining with an anti-LC3 antibody.
Morusin activates AMPK and inhibits mTOR activity
Since ULK1 activation is regulated by AMPK and mTOR, we investigated whether morusin affects AMPK and/or mTOR for ULK1 activation. Immunoblotting indicated that S6K phosphorylation was markedly reduced by morusin treatment, suggesting that morusin inhibits mTOR activity for ULK1 activation (Figure 4A). ULK1 is constitutively regulated by mTOR-mediated inhibitory phosphorylation at Ser757 under normal conditions, and ULK1 is phosphorylated at Ser317 and Ser555 by activated AMPK under stressed conditions [6,8]. To address the upstream signaling events for morusin-mediated ULK1 activation, the phosphorylation status of ULK1 and AMPK upon morusin treatment was analyzed by immunoblotting with phospho-specific anti-ULK1 antibodies. Immunoblotting indicated that AMPK was activated due to Thr172 phosphorylation of the AMPKα subunit in morusin-treated cells. Consequently, ULK1 phosphorylation at Ser317, a target site of AMPK phosphorylation, was induced by morusin treatment. On the other hand, ULK1 phosphorylation at Ser757, a target site of mTOR phosphorylation, was observed under normal conditions, but phosphorylation gradually decreased upon morusin treatment (Figure 4B). These results indicated that morusin treatment simultaneously causes the induction of stimulating phosphorylation (Ser317) and reduction of inhibitory phosphorylation (Ser757) of ULK1. Further analysis of cells treated with increasing amounts of morusin indicated that morusin treatment resulted in AMPK activation, induction of ULK1 Ser317 phosphorylation, and reduction of ULK1 Ser757 phosphorylation in a dose-dependent manner (Figure 4C). To address whether morusin reduces ULK1 Ser757 phosphorylation independent of mTOR inhibition by AMPK, the levels of ULK1 Ser757 phosphorylation were determined in the presence of compound C, an AMPK inhibitor. ULK1 Ser555 phosphorylation was induced by morusin treatment, which was inhibited by co-treatment with compound C. However, ULK1 Ser757 phosphorylation was slightly reduced by treatment of compound C alone, and further reduced by morusin treatment (Figure 4D, lane 4). This result indicated that morusin directly inhibits mTOR activity independent of AMPK-mediated mTOR inhibition. mTOR inhibition by morusin treatment was also confirmed by the lack of 4E-BP1 phosphorylation as well as ULK1 Ser757 phosphorylation, which was comparable to the results obtained with rapamycin-treated cells as a positive control (Figure 4E). Collectively, morusin activates ULK1 by induction of Ser317/Ser555 phosphorylation and reduction of Ser757 phosphorylation through AMPK activation and mTOR inhibition, respectively.
Figure 4.
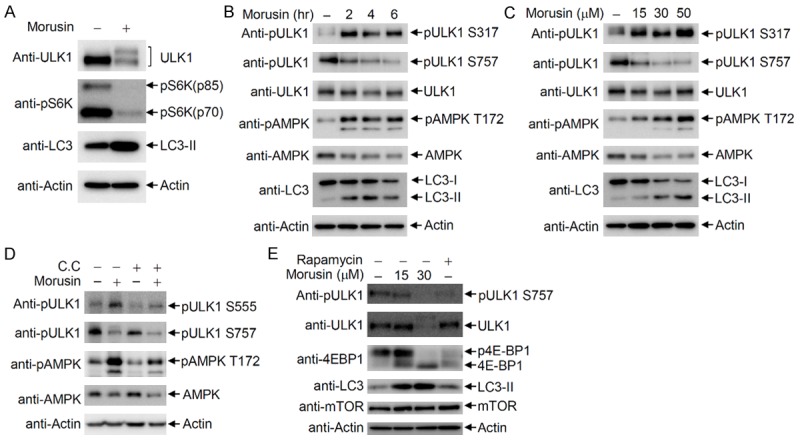
Morusin activates AMPK and inhibits mTOR activity. A. ULK1 activation and mTOR inhibition by morusin. HeLa cells were treated with either DMSO or morusin, and subjected to immunoblotting with anti-ULK1, anti-LC3, and anti-phospho S6K antibodies. B and C. AMPK activation by morusin in a time- and dose-dependent manner. B. HeLa cells were treated with either DMSO or morusin, and cells were collected at the indicated time points for immunoblotting with the indicated antibodies. C. HeLa cells were treated with either DMSO or increasing amounts of morusin, and cells were collected after 6 hr of treatment for immunoblotting with the indicated antibodies. D. Inhibition of morusin-induced ULK1 phosphorylation at Ser555 upon compound C treatment. ULK1 phosphorylation levels at Ser555 and Ser757 in cells treated with both morusin and compound C were compared to levels in cells treated with either drug alone. E. Inhibition of mTOR activity by morusin. HeLa cells were treated with morusin or rapamycin, and collected cells were subjected to immunoblotting with the indicated antibodies.
Morusin-induced autophagy enhances cell survival by inhibiting apoptosis
To investigate the effects of autophagy on morusin-induced apoptosis, a time-course analysis of autophagic and apoptotic markers following morusin treatment was performed (Figure 5A). Immunoblotting indicated that AMPK activation (phosphorylation at Thr172) was observed as an early response to morusin treatment (lane 2), which was followed by transient activation of apoptosis, which includes events such as cleavage of PARP and caspase-3 (lane 3). ULK1 was gradually activated, with de-phosphorylated faster-migrating ULK1 first observed and followed by a gradual increase in phosphorylated slowly-migrating ULK1. Induction of apoptosis was transiently inhibited at the time point of appearance of slowly-migrating ULK1 (lane 4), and apoptosis was induced again at later times during morusin treatment (lanes 7-9). These results suggest that the induction of autophagy transiently inhibits induction of apoptosis. To confirm the inhibitory effects of autophagy on apoptosis induction, apoptosis levels were determined following inhibition of autophagy. Immunoblotting indicated that cleavage of PARP and caspase-3 were increased by morusin treatment, and were potentiated by blockage of autophagy through treatment with 3-MA or chloroquine (Figure 5B and 5C). In addition, morusin-induced apoptosis was determined by FACS analysis after annexin V-FITC and propidium iodide (PI) staining. Morusin treatment increased annexin V-positive cells, which was markedly potentiated by administration of 3-MA (Figure 5D). These results indicate that morusin-induced apoptosis is negatively regulated by autophagy.
Figure 5.

Morusin-induced autophagy enhances cell survival by inhibition of cell death. (A) Time-course induction of autophagy and apoptosis by morusin. HeLa cells were treated with morusin (30 uM), and cells were collected at the indicated time points for immunoblotting. (B and C) Induction of morusin-mediated apoptosis in the absence of autophagy. HeLa cells were treated with morusin (30 uM), and cells were collected at the indicated time points for immunoblotting in the presence or absence of 3-MA (B) or CQ (C). (D) Analysis of morusin-induced apoptosis using fluorescence-activated cell sorting (FACS) analysis. HeLa cells were treated with either morusin alone or in combination with 3-MA, and apoptotic cells were assessed by FACS analysis after annexin V-FITC and propidium iodide (PI) staining. Apoptotic cells (double-positive for PI and annexin V) are presented on the graph. (E) Inhibition of STAT3 signaling upon combined treatment of morusin and 3-MA. The levels of STAT3 phosphorylation at Tyr705 and AMPKα phosphorylation at Thr172 in cells treated with both morusin and 3-MA were compared to levels in cells treated with either drug alone. (F) Inhibition of STAT3 signaling upon ULK1 depletion and morusin treatment. The levels of STAT3 phosphorylation at Tyr705 and AMPKα phosphorylation at Thr172 in cells subjected to both morusin treatment and ULK1 depletion were compared to levels in cells either treated with morusin or depleted of ULK1. (G) Reduction of cell viability by blocking morusin-induced autophagy. HepG2 cells were treated with morusin and/or 3-MA, and cell viability was determined by measuring MTT metabolic activity. Mock-depleted and ULK1-depleted HeLa cells were treated with either DMSO or morusin, and subjected to the MTT assay. Data are presented as the mean ± SEM. (H) Inhibition of colony formation by blocking morusin-induced autophagy. Cells were treated with morusin and/or 3-MA, and the number of colonies were counted following staining of cells with Coomassie Brilliant Blue. The number of colonies obtained from three independent experiments are presented as a graph. One representive image is shown. Data are presented as the mean ± SEM.
To examine the crosstalk between morusin-induced autophagy and apoptosis at the molecular level, STAT3 phosphorylation at Tyr705 (a marker for STAT3 activation) was compared with or without autophagy inhibition. STAT3 phosphorylation at Tyr705 was decreased by both pharmacological inhibition of autophagy by 3-MA treatment and genetic inhibition of autophagy by ULK1 depletion compared to cells treated with morusin alone (Figure 5E, lane 4 and Figure 5F, lane 4). These results indicate that the increase in apoptosis upon autophagy inhibition is mediated by inhibition of STAT3 signaling. To assess the cellular outcome of crosstalk between apoptosis and autophagy, cell viability was determined with the MTT assay and colony forming assay. Morusin treatment reduced cell viability due to the induction of apoptosis, which was enhanced by co-treatment with the autophagy inhibitor 3-MA. Similarly, autophagy-defective ULK1 depleted cells were more sensitive to morusin than wild-type HeLa cells (Figure 5G). Consistent with these results, the number and size of colonies were reduced by morusin treatment, and further decreased upon treatment with both morusin and 3-MA (Figure 5H). Collectively, morusin induces both autophagy and apoptosis, and morusin-induced autophagy inhibits morusin-induced apoptosis.
Discussion
Apoptosis and autophagy are evolutionarily conserved catabolic processes important for organismal homeostasis. A variety of death stimuli are able to activate both pathways in which the functional role of either pathway is dependent on complicated patterns of crosstalk between apoptosis and autophagy [36-38]. Although it is clear that crosstalk between apoptosis and autophagy are key factors in determining the cellular outcomes of death-associated pathologies such as cancers, much is unknown about the interplay between autophagy and apoptosis at the molecular level, since autophagy can act both as a cell survival and cell death process [39,40]. In this study, we demonstrate that morusin activates both autophagy and apoptosis, and morusin-induced autophagy inhibits the induction of apoptosis. Morusin induced AMPK activation and inhibited mTOR activity, by which ULK1 was activated to induce autophagy (Figures 3 and 4). To demonstrate the inhibitory role of autophagy on morusin-induced apoptosis, autophagy was pharmacologically blocked at an early and late stage with 3-MA and chloroquine, respectively. Blockage of autophagy at any step potentiated morusin-induced apoptosis (Figure 5B and 5C). A similar result was observed with genetic disruption of autophagy by ULK1 depletion (Figure 5F and 5G). Therefore, autophagy per se is an inhibitory process for morusin-induced apoptosis.
Cisplatin is one of the best known anti-cancer drugs used as first-line chemotherapy for advanced epithelial cancers. Cisplatin treatment induces both autophagy and apoptotic cell death [41-43]. Cisplatin-triggered activation of AMPK can induce an autophagic response that protects tumor cells from cisplatin-mediated cell death. In cells co-treated with melphalan, bortezomib, and rapamycin, however, AMPK was activated and contributed to synergistic induction of apoptosis by phosphorylation of Beclin-1 at Ser93/96 and cleavage of Beclin-1 [44]. The multidrug-induced AMPK activation resulted in reduction of autophagy through Beclin-1 cleavage and amplification of mitochondrion-mediated apoptosis through the cleaved C-terminal fragment of Beclin-1 [44]. Although AMPK was activated by cisplatin, morusin or multidrug treatment, it appears that the effects of autophagy on drug-induced apoptosis are not the same and cannot be explained by AMPK activation alone. A time-course analysis of AMPK following morusin treatment indicated that AMPK activation occurred in two stages, with early activation of AMPK involved in autophagy induction and late re-activation possibly participating in induction of apoptosis. Morusin treatment induced AMPK activation at an early stage of autophagy initiation (Figures 4B and 5A). Afterwards, AMPKα and the activated AMPK (phosphorylated AMPKα at Thr172) levels gradually decreased (Figure 5A, lanes 2-8). However, AMPK was markedly activated again at the time of apoptosis induction (Figure 5A, lane 9), implying an association of AMPK with the induction of apoptosis. Consistently, AMPK activation was further enhanced by pharmacological inhibition and genetic disruption of autophagy (Figure 5E and 5F). Further studies are required for deciphering the functional roles of AMPK and identifying molecular targets in morusin-mediated induction of apoptosis. Knowledge of drug-specific mechanisms of cell death can lead to efficient cancer treatment with anti-cancer drugs.
Autophagy inhibitors have been successfully developed for clinical trials for various cancers [45,46]. From studies over the past few years, it has become apparent that apoptosis and autophagy are inter-connected to ensure the elaborate regulation of cellular homeostasis and responses to various stresses. Consistent with this idea, a variety of anti-cancer drugs and phytochemicals induce both cellular processes of autophagy and apoptosis simultaneously or sequentially. Given that autophagy can act in either a cyto-protective or pro-apoptotic manner, combined treatment of anti-cancer drugs with inhibitors or activators of autophagy could provide more efficient protocols for chemotherapy.
Acknowledgements
This work was supported by a grant from the Basic Science Research Program (2014R1A2A1A11053690 to C.Y.C.) through the National Research Foundation of Korea funded by the Korea government.
Disclosure of conflict of interest
None.
References
- 1.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–472. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 2.Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–865. doi: 10.4161/auto.6555. [DOI] [PubMed] [Google Scholar]
- 3.Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. 2015;517:302–310. doi: 10.1038/nature14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014;24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7:643–644. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A. 2011;108:4788–4793. doi: 10.1073/pnas.1100844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–750. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, Wang X, He C, Pan H. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838. doi: 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Law BY, Chan WK, Xu SW, Wang JR, Bai LP, Liu L, Wong VK. Natural small-molecule enhancers of autophagy induce autophagic cell death in apoptosis-defective cells. Sci Rep. 2014;4:5510. doi: 10.1038/srep05510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ko HH, Yu SM, Ko FN, Teng CM, Lin CN. Bioactive constituents of Morus australis and Broussonetia papyrifera. J Nat Prod. 1997;60:1008–1011. doi: 10.1021/np970186o. [DOI] [PubMed] [Google Scholar]
- 14.Abbas GM, Abdel Bar FM, Baraka HN, Gohar AA, Lahloub MF. A new antioxidant stilbene and other constituents from the stem bark of Morus nigra L. Nat Prod Res. 2014;28:952–959. doi: 10.1080/14786419.2014.900770. [DOI] [PubMed] [Google Scholar]
- 15.Geng C, Yao S, Xue D, Zuo A, Zhang X, Jiang Z, Ma Y, Chen J. New isoprenylated flavonoid from Morus alba. Zhongguo Zhong Yao Za Zhi. 2010;35:1560–1565. [PubMed] [Google Scholar]
- 16.Dat NT, Binh PT, Quynh le TP, Van Minh C, Huong HT, Lee JJ. Cytotoxic prenylated flavonoids from Morus alba. Fitoterapia. 2010;81:1224–1227. doi: 10.1016/j.fitote.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Yang ZG, Matsuzaki K, Takamatsu S, Kitanaka S. Inhibitory effects of constituents from Morus alba var. multicaulis on differentiation of 3T3-L1 cells and nitric oxide production in RAW264.7 cells. Molecules. 2011;16:6010–6022. doi: 10.3390/molecules16076010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abd El-Mawla AM, Mohamed KM, Mostafa AM. Induction of biologically active flavonoids in cell cultures of morus nigra and testing their hypoglycemic efficacy. Sci Pharm. 2011;79:951–961. doi: 10.3797/scipharm.1101-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ko HH, Wang JJ, Lin HC, Wang JP, Lin CN. Chemistry and biological activities of constituents from Morus australis. Biochim Biophys Acta. 1999;1428:293–299. doi: 10.1016/s0304-4165(99)00084-7. [DOI] [PubMed] [Google Scholar]
- 20.Lee HJ, Lyu da H, Koo U, Nam KW, Hong SS, Kim KO, Kim KH, Lee D, Mar W. Protection of prenylated flavonoids from Mori Cortex Radicis (Moraceae) against nitric oxide-induced cell death in neuroblastoma SH-SY5Y cells. Arch Pharm Res. 2012;35:163–170. doi: 10.1007/s12272-012-0118-7. [DOI] [PubMed] [Google Scholar]
- 21.Sohn HY, Son KH, Kwon CS, Kwon GS, Kang SS. Antimicrobial and cytotoxic activity of 18 prenylated flavonoids isolated from medicinal plants: Morus alba L. , Morus mongolica Schneider, Broussnetia papyrifera (L.) Vent, Sophora flavescens Ait and Echinosophora koreensis Nakai. Phytomedicine. 2004;11:666–672. doi: 10.1016/j.phymed.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 22.Lee JC, Won SJ, Chao CL, Wu FL, Liu HS, Ling P, Lin CN, Su CL. Morusin induces apoptosis and suppresses NF-kappaB activity in human colorectal cancer HT-29 cells. Biochem Biophys Res Commun. 2008;372:236–242. doi: 10.1016/j.bbrc.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 23.Kim C, Kim JH, Oh EY, Nam D, Lee SG, Lee J, Kim SH, Shim BS, Ahn KS. Blockage of STAT3 signaling pathway by morusin induces apoptosis and inhibits invasion in human pancreatic tumor cells. Pancreas. 2016;45:409–419. doi: 10.1097/MPA.0000000000000496. [DOI] [PubMed] [Google Scholar]
- 24.Lim SL, Park SY, Kang S, Park D, Kim SH, Um JY, Jang HJ, Lee JH, Jeong CH, Jang JH, Ahn KS, Lee SG. Morusin induces cell death through inactivating STAT3 signaling in prostate cancer cells. Am J Cancer Res. 2015;5:289–299. [PMC free article] [PubMed] [Google Scholar]
- 25.Lin WL, Lai DY, Lee YJ, Chen NF, Tseng TH. Antitumor progression potential of morusin suppressing STAT3 and NFkappaB in human hepatoma SK-Hep1 cells. Toxicol Lett. 2015;232:490–498. doi: 10.1016/j.toxlet.2014.11.031. [DOI] [PubMed] [Google Scholar]
- 26.Wan LZ, Ma B, Zhang YQ. Preparation of morusin from Ramulus mori and its effects on mice with transplanted H22 hepatocarcinoma. Biofactors. 2014;40:636–645. doi: 10.1002/biof.1191. [DOI] [PubMed] [Google Scholar]
- 27.Li H, Wang Q, Dong L, Liu C, Sun Z, Gao L, Wang X. Morusin suppresses breast cancer cell growth in vitro and in vivo through C/EBPbeta and PPARgamma mediated lipoapoptosis. J Exp Clin Cancer Res. 2015;34:137. doi: 10.1186/s13046-015-0252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Guo H, Yang L, Dong L, Lin C, Zhang J, Lin P, Wang X. Morusin inhibits human cervical cancer stem cell growth and migration through attenuation of NF-kappaB activity and apoptosis induction. Mol Cell Biochem. 2013;379:7–18. doi: 10.1007/s11010-013-1621-y. [DOI] [PubMed] [Google Scholar]
- 29.Ahn JW, Kim S, Na W, Baek SJ, Kim JH, Min K, Yeom J, Kwak H, Jeong S, Lee C, Kim SY, Choi CY. SERBP1 affects homologous recombination-mediated DNA repair by regulation of CtIP translation during S phase. Nucleic Acids Res. 2015;43:6321–6333. doi: 10.1093/nar/gkv592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi DW, Na W, Kabir MH, Yi E, Kwon S, Yeom J, Ahn JW, Choi HH, Lee Y, Seo KW, Shin MK, Park SH, Yoo HY, Isono K, Koseki H, Kim ST, Lee C, Kwon YK, Choi CY. WIP1, a homeostatic regulator of the DNA damage response, is targeted by HIPK2 for phosphorylation and degradation. Mol Cell. 2013;51:374–385. doi: 10.1016/j.molcel.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 31.Lee D, Park SJ, Sung KS, Park J, Lee SB, Park SY, Lee HJ, Ahn JW, Choi SJ, Lee SG, Kim SH, Kim DH, Kim J, Kim Y, Choi CY. Mdm2 associates with Ras effector NORE1 to induce the degradation of oncoprotein HIPK1. EMBO Rep. 2012;13:163–169. doi: 10.1038/embor.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang XJ, Chen S, Huang KX, Le WD. Why should autophagic flux be assessed? Acta Pharmacol Sin. 2013;34:595–599. doi: 10.1038/aps.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A. 2011;108:11121–11126. doi: 10.1073/pnas.1107969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheong H, Lindsten T, Thompson CB. Autophagy and ammonia. Autophagy. 2012;8:122–123. doi: 10.4161/auto.8.1.18078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Klionsky DJ. The regulation of autophagy-unanswered questions. J Cell Sci. 2011;124:161–170. doi: 10.1242/jcs.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubinstein AD, Kimchi A. Life in the balance - a mechanistic view of the crosstalk between autophagy and apoptosis. J Cell Sci. 2012;125:5259–5268. doi: 10.1242/jcs.115865. [DOI] [PubMed] [Google Scholar]
- 38.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, Adams PD, Anderson KI, Gottlieb E, Sansom OJ, Ryan KM. P53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 40.Choi KS. Autophagy and cancer. Exp Mol Med. 2012;44:109–120. doi: 10.3858/emm.2012.44.2.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008;74:631–640. doi: 10.1038/ki.2008.214. [DOI] [PubMed] [Google Scholar]
- 42.Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J Cell Mol Med. 2009;13:3644–3654. doi: 10.1111/j.1582-4934.2009.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim HS, Hwang JT, Yun H, Chi SG, Lee SJ, Kang I, Yoon KS, Choe WJ, Kim SS, Ha J. Inhibition of AMP-activated protein kinase sensitizes cancer cells to cisplatin-induced apoptosis via hyper-induction of p53. J Biol Chem. 2008;283:3731–3742. doi: 10.1074/jbc.M704432200. [DOI] [PubMed] [Google Scholar]
- 44.Song X, Kim SY, Zhang L, Tang D, Bartlett DL, Kwon YT, Lee YJ. Role of AMP-activated protein kinase in cross-talk between apoptosis and autophagy in human colon cancer. Cell Death Dis. 2014;5:e1504. doi: 10.1038/cddis.2014.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gewirtz DA. The challenge of developing autophagy inhibition as a therapeutic strategy. Cancer Res. 2016;76:5610–5614. doi: 10.1158/0008-5472.CAN-16-0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manic G, Obrist F, Kroemer G, Vitale I, Galluzzi L. Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol. 2014;1:e29911. doi: 10.4161/mco.29911. [DOI] [PMC free article] [PubMed] [Google Scholar]