

Abstract
Compelling epidemiologic evidence indicates that obesity is a risk factor for human cancers, including breast. However, molecular mechanisms by which obesity could contribute to the development of breast cancer remain unclear. To understand the impact of obesity on breast cancer development, we used a mutant mouse that expresses a mutated thyroid hormone receptor β (denoted as PV) with haplodeficiency of the Pten gene (ThrbPV/PVPten+/- mice). We previously showed that adult nulliparous female ThrbPV/PVPten+/- mice developed extensive mammary hyperplasia and breast tumors. In this study, we induced obesity in ThrbPV/PVPten+/- mice by feeding them a high fat diet (HFD). We found HFD exacerbated the extent of mammary hyperplasia in ThrbPV/PVPten+/- mice. HFD elevated serum leptin levels but had no effect on the levels of serum thyroid stimulating hormone, thyroid hormones, and estrogens. Molecular analysis showed that the obesity-induced hyperplasia was mediated by the leptin/leptin receptor-JAK1-STAT3 pathway to increase key cell cycle regulators to stimulate mammary epithelial cell proliferation. Activated STAT3 signaling led to altered expression in the key regulators of epithelial-mesenchymal-transition (EMT) to augment invasiveness and migration of mammary proliferating epithelial cells. Moreover, treatment of HFD-ThrbPV/PVPten+/- mice with a STAT3 inhibitor, S3I-201, markedly reversed the obesity-induced mammary hyperplasia and reduced EMT signals to lessen cell invasiveness and migration. Our studies not only elucidated how obesity could contribute to mammary hyperplasia at the molecular level, but also, importantly, demonstrated that inhibition of the STAT3 activity could be a novel treatment strategy for obesity-induced breast cancer progression.
Keywords: Mammary carcinogenesis, JAK2-STAT3 signaling, obesity, STAT3 inhibitor, preclinical mouse model, thyroid hormone receptor β mutant
Introduction
Breast cancer is the most common neoplasia and the second-leading cause of cancer deaths in women of Western countries [1]. Genetic mutations, either inherited or sporadic, as well as dysregulation of ovarian hormone signaling are known to contribute to tumorigenesis of the breast. Obesity has long been recognized as a risk factor in breast cancer as it is associated with more advanced disease at the time of an initial cancer diagnosis and a poor prognosis [2-5]. Several reports indicate that leptin, whose synthesis and serum levels are increased with obesity, could impact breast cancer development and progression by functional crosstalk with different cell signaling molecules. Leptin, expressed in the mammary gland, binds to the leptin receptor to control the development and physiology of the mammary gland [6]. Moreover, studies have indicated that the binding of leptin to its receptor induces janus kinase (Jak/STAT) signaling to contribute to mammary carcinogenesis [7]. Leptin increases HER2 protein levels via the STAT3 signaling pathway to amplify responsiveness of breast cancer cells to growth factor stimulation [8]. These studies raise the possibility that leptin-activated Jak/STAT signaling could be considered molecular targets for a breast cancer treatment modality.
The availability of a unique mouse model of mammary tumors (ThrbPV/PVPten+/- mice) has allowed us to examine the impact of obesity on the development of breast tumors. The ThrbPV/PVPten+/- mouse expresses a potent dominant mutant of thyroid hormone receptor β (TRβ) (denoted TRβPV). TRβPV has a frameshift mutation in the C-terminal 14 amino acids. This results in the loss of the tumor suppressor functions of the wild type (WT) TRβ, thus causing TRβPV to function as an oncogene [9,10]. In addition to harboring the TRβPV mutation, the ThrbPV/PVPten+/- mouse is also deficient in one allele of the tumor suppressor Pten (phosphatase and tensin homolog deleted from chromosome 10). The tumor suppressor PTEN encodes a dual protein/lipid phosphatase, which counteracts the phosphatidylinositol 3-kinase (PI3K) signaling pathway [11]. Pten mutations have been implicated in the development of a variety of human neoplasia, including breast and thyroid carcinoma [12,13]. In the thyroid of ThrbPV/PVPten+/- mice, Pten haplodeficiency further promotes thyroid cancer progression. This is due to the decreased expression of the tumor suppressor, PTEN, resulting in further activation of the PI3K-AKT signaling in thyroid tumors of ThrbPV/PV mice [14]. In addition to promoting thyroid cancer progression, Pten haplodeficiency also augments mammary abnormalities found in ThrbPV/PV mice. Remarkably, while 36% of ThrbPV/PV mice exhibited mammary hyperplasia, ~77% of ThrbPV/PVPten+/- mice developed mammary hyperplasiaand mammary tumors [15], indicating increased sensitivity in the oncogenic effect of TRβPV by PTEN deficiency in the mammary gland.
Recently, we found that ThrbPV/PVPten+/- mice fed a high fat diet (HFD) develop obesity with a marked increase in body mass, body fat, adipocyte size, and elevated serum leptin levels. Extensive molecular analyses demonstrated that the increased serum leptin, via activated JAK-STAT3 signaling, further promotes thyroid carcinogenesis by increasing tumor growth and propel anaplastic changes [16]. Importantly, suppressing the JAK-STAT3 activity by S3I-201, a STAT3 inhibitor, ameliorates obesity-induced thyroid carcinogenesis [17]. As indicated by these recent findings, the ThrbPV/PVPten+/- mouse model is ideal for assessing how diet-induced obesity affects abnormal mammary gland development. Importantly, the ThrbPV/PVPten+/- mouse could also serve as a preclinical model to test whether inhibition of STAT3 activity downstream of leptin signaling could be beneficial in reducing the HFD diet-induced cancer risk in the mammary gland. Indeed, the present studies found that obesity exacerbated further mammary gland hyperplasia via activation of JAK-STAT3 signaling. Treatment of ThrbPV/PVPten+/- mice with a STAT3-specific inhibitor, S3I-201, decreased mammary cell proliferation and suppressed invasiveness of hyperplastic cells. Therefore, the present studies provide molecular evidence that obesity increased cancer risks in the mammary gland and that inhibition of the STAT3 activity could be beneficial in the obesity-associated tumor development.
Materials and methods
Mice and treatment
The National Cancer Institute Animal Care and Use Committee approved the protocols for animal care and handling in the present study. Mice harboring the ThrbPV gene (ThrbPV/PV mice) were prepared via homologous recombination, and genotyping was carried out using the PCR method, as previously described [14]. Pten+/- mice were kindly provided by Dr. Ramon Parsons (Columbia University, NY, USA). ThrbPV/PVPten+/- mice were obtained from the breeding scheme as described by Guigon et al. [14]. The control low fat diet (LFD, 10% Kcal from fat) and the high fat diet (HFD, 60% Kcal from fat) were purchased from Research Diets (New Brunswick, NJ, USA). The female mice were randomly selected and divided into two groups fed with LFD or HFD diet. The mice were fed one or the other diet from the age of 8 weeks until reaching moribund state. S3I-201 (STAT3 inhibitor, Cat# S1155, Selleckchem, Huston, TX, USA) was dissolved in DMSO (0.05% DMSO) and was injected intraperitoneally (3 times a week at a dose 5 mg/kg/mouse) or vehicle (0.05% DMSO) starting at the age of 8 weeks. The mice were monitored until they became moribund with hunched posture and labored breathing due to thyroid cancer. After the mice were euthanized, the mammary tissues were dissected for whole-mount mammary gland staining, histopathologic analysis, and biochemical studies.
Whole-mount mammary gland staining and histologic analysis
Whole-mount mammary gland staining has been described previously [15]. The fourth mammary glands were spread on microscope slides, and fixed in a mix of glacial acetic acid (1 volume) and ethanol 100% (3 volumes) for 4 hours or longer. They were then hydrated in ethanol 70% for 15 minutes and distilled water and stained with carmine alum stain (1% carmine, 2.5% aluminum potassium sulfate) (Sigma, St Louis, MO, USA) overnight or longer, at room temperature. After staining, the slides were dehydrated through increasing ethanol concentrations (50%, 70%, 95% and 100%) for 5 minutes each, cleared in xylene (Sigma, St Louis, MO, USA) and mounted with Permount (Fisher Scientific, Hudson, NH, USA).
For histopathologic analysis, mammary glands were dissected and fixed in 10% neutral-buffered formalin (Sigma-Aldrich, St. Louis, MO, USA) and subsequently embedded in paraffin. Sections of 5-μm thickness were prepared and stained with hematoxylin and eosin (HistoServ, Germantown, MD, USA). For each animal, single random sections through the mammary glands were examined. Immunohistochemistry (IHC) was conducted as previously described with some modifications [18]. For the antigen retrieval step, slides were heated in 0.05% citraconic anhydride solution (Sigma-Aldrich; pH 7.4, St. Louis, MO, USA) at 98°C for 60 minutes, followed by treatment with anti-Ki-67 antibody (dilution 1:300) (Thermo Scientific, Cambridge, MA, USA) at 4°C overnight. The antigen signals were detected by treatment with the peroxidase substrate diaminobenzidine followed by counterstaining with Gill’s hematoxylin (Electron Microscopy Sciences, Hatfield, PA, USA). Relative positive cell ratio was quantified by using NIH IMAGE software (Image J 1.47).
Western blot analysis
Preparation of whole-cell lysates from mammary tissues has been described previously [18]. The protein sample (20 μg) was loaded and separated by SDS-PAGE. After electrophoresis, the protein was electrotransferred to a poly vinylidenedifluoride membrane (Immobilon-P; Millipore Corp., Billeria, MA, USA). The antibodies phosphorylated Rb (p-Rb, S780, 1:500 dilution), total JAK1 (1:1,000 dilution), p-STAT3 (1:500 dilution), total STAT3 (1:1,000 dilution), vimentin (1:1,000 dilution) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:1,000 dilution), and vimentin (1:1,000 dilution) were purchased from Cell Signaling Technology (Denver, MA, USA). Antibodies for MMP-2 (1:200 dilution), p-JAK1 (1:200 dilution) and E-cadherin (1:200) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibody for leptin receptor (1:1,000) was purchased from Abcam (Cambridge, MA, USA). Antibody for cyclin D1 (1:500 dilution) was purchased from Neomarkers (Thermo Scientific, Cambridge, MA, USA). The blots were stripped with Re-Blot Plus (Millipore, Billerica, MA, USA) and re-probed with rabbit polyclonal antibodies to GAPDH. Band intensities were quantified by using NIH IMAGE software (Image J 1.47).
Determination of serum thyroid stimulating hormone (TSH), total T3, T4 and Estrogen (E2)
Serum TSH levels were measured as previously described [19]. Serum levels of total T3 and T4 were determined by the use of radioimmunoassay (RIA) kits according to the company’s protocol (MP Biomedical, LLC cat. 06B256447 and 06B254029). Serum estradiol concentration was assayed by EIA/ELISA using the Estradiol Serum EIA kit (cat. KB30-H1, ArborAssay).
Statistical analysis
All data are expressed as mean ± standard errors, and Student’s t test was used to compare continuous variables. Statistical significance was set at P<0.05. GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA, USA) was used to draw graphs.
Results
HFD-induced mammary hyperplasia is sensitive to the effect of a STAT3 inhibitor in ThrbPV/PVPten+/- mice
We previously reported that HFD induces obesity and elevates serum leptin to exacerbate thyroid cancer progression in ThrbPV/PVPten+/- mice [16]. Recently we found that leptin activated JAK-STAT3 signaling in thyroid tumors is attenuated by treatment of ThrbPV/PVPten+/- mice with a STAT3 inhibitor, S3I-201 [17]. We therefore evaluated whether elevated serum leptin could also increase the extent of hyperplasia in the mammary gland of ThrbPV/PVPten+/- mice. We first assessed mammary gland morphology using whole-mount preparations to analyze the lobulo-alveolar development in nulliparous female mice (Figure 1Aa). All mammary glands analyzed could be assigned one of the four lobulo-alveolar scores: 1 = normal structure similar to young virgin mice (normal); 2 = increased number of ramifications and slightly increased alveolar size (mild hyperplasia); 3 = strongly increased number of ramifications with strongly increased alveolar size associated or not with moderate ductal dilatation (moderate hyperplasia); 4 = extreme alveolar size covering the space between the ducts accompanied by ductal dilation (severe hyperplasia). As shown in bar 5 of Figure 1Ab, 25% of LFD-ThrbPV/PVPten+/- mice at the age of 4-6 months exhibited moderate mammary hyperplasia (score = 3), but none showed severe hyperplasia (score of 4). As shown in bar 7, of the HFD-ThrbPV/PVPten+/- mice aged 4-6 months, 25% showed moderate hyperplasia (score = 3) and 33% displayed severe hyperplasia (score = 4). In contrast, HFD-diet had no apparent effect on the mammary gland development in Thrb+/+Pten+/+ mice (WT-mice; compare bar 1 to 3). Treatment of LFD-ThrbPV/PVPten+/- mice with a STAT3 inhibitor, S3I-201, had no apparent effect on the mammary gland development (compare bar 6 versus 5). However, treatment of HFD-ThrbPV/PVPten+/- mice with S3I-201 led to the decrease in the extent of hyperplasia from severe (score = 4) to moderate (score = 3) in 33% of mice (compare bar 8 with 7).
Figure 1.
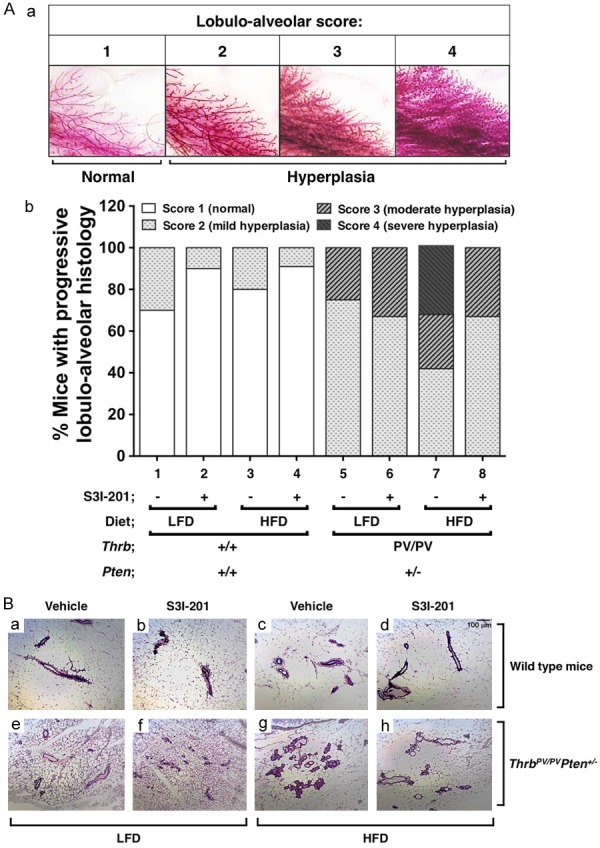
Effects of obesity and S3I-201 on lobulo-alveolar development and morphology of the mammary glands of wild type (WT) and ThrbPV/PVPten+/- mice. (A) Histopathology of mammary glands of WT and ThrbPV/PVPten+/- mice fed with low fat diet (LFD) or high fat diet (HFD), and treated with vehicle or S3I-201. The histopathological changes were given a score based on the rating scale in (a). The distribution of scores is shown in (b). The genotypes of mice are as marked. (B) Effects of S3I-201 on obesity-induced aberrant lobulo-alveolar development in HFD-ThrbPV/PVPten+/- mice. Hematoxylin and eosin-stained sections of mouse mammary tissue from LFD-WT with/without S3I-201 (a, b) and HFD-WT with or without S3I-201 (e and f), and a LFD-ThrbPV/PVPten+/- with/without S3I-201 (c and d) and HFD-ThrbPV/PVPten+/- with/without S3I-201 (g and h) mice.
We further evaluated pathohistology by H&E staining of mammary gland sections (Figure 1B). Consistent with the whole mount analysis (Figure 1Ab), no discernable changes were observed in the histology of mammary gland between LFD- and HFD-treated WT mice (compare panel a to e, Figure 1B). In contrast, in the mammary gland of HFD-ThrbPV/PVPten+/- mice, moderate ductal proliferation was apparent (compare panel g with c). While treatment with S3I-201 had no effects on the histology of the mammary gland in the LFD-WT mice (compare panel b with a) or HFD-WT mice (compare panel f with e), treatment of HFD-ThrbPV/PVPten+/- mice with S3I-201 lessened the ductal proliferation phenotype (compare panel h with g).
That HFD led to increased cell proliferation was further confirmed by immunohistochemical analysis using Ki67 as a proliferation marker (Figure 2A). Compared with the negative controls (panels a and c, Figure 2A), no apparent positive-stained signals were detected in the mammary gland sections of WT mice whether S3I-201 was present or not (panels b and d). In contrast, clear positive-stained signals were apparent in the sections of vehicle-treated HFD-ThrbPV/PVPten+/- mice (panel f). The stained signals were decreased in the mammary sections of treated HFD-ThrbPV/PVPten+/- mice (panel h). The Ki67-positive stained cells were counted, and the data are shown in Figure 2B. It was clear that more cells were stained with Ki67 in the mammary tissue of vehicle-treated HFD-ThrbPV/PVPten+/- mice than in the HFD-WT mice (compare bar 3 with bar 1, Figure 2B). While we detected no significant effect on the Ki67 positive-stained cells by S3I-201 treatment in the wild type mice (compare bar 2 with bar 1), S3I-201 reducedKi67 positive-stained cells in the mammary gland of HFD-ThrbPV/PVPten+/- mice (compare bar 4 with bar 3). Taken together, these results indicate that the HFD-induced obesity leads to increased cell proliferation in ThrbPV/PVPten+/- mice, and that the increases were inhibited by S3I-201.
Figure 2.
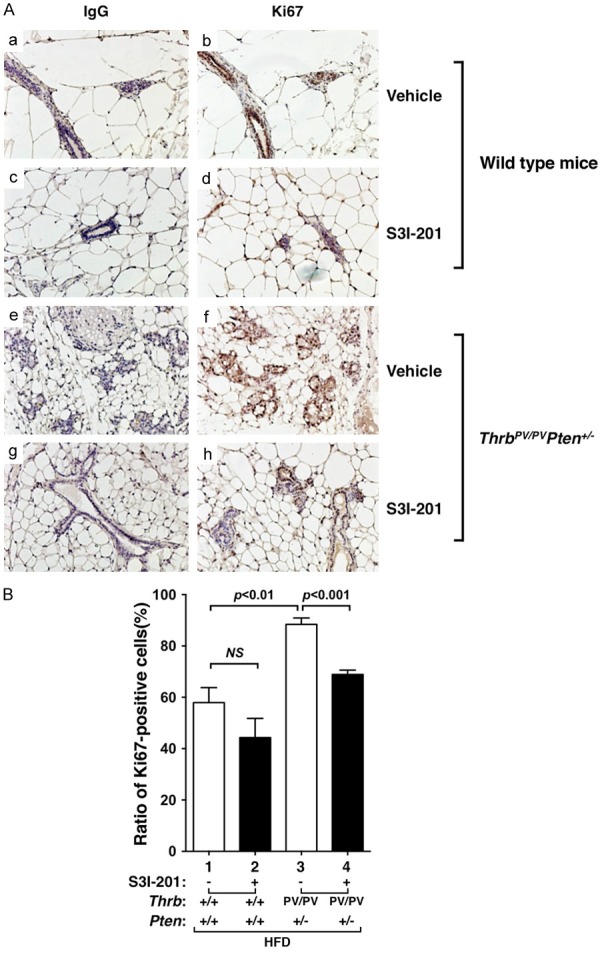
Effects of S3I-201 on cell proliferation in mammary tissue sections from HFD-WT and HFD-ThrbPV/PVPten+/- mice. (A) Representative microphotographs of immunohistochemical analysis of Ki67 on mammary tissue sections of WT mice treated with vehicle (a, b) or S3I-201 (c and d) or ThrbPV/PVPten+/- mice treated with vehicle (e and f) or treated with S3I-201 (g and h). (a, c, e, and g) were the negative controls from using anti-IgG antibodies and (b, d, f, and h) were from using anti-Ki67 antibodies. (B) The Ki67-positively stained cells were counted and the data expressed as percentage of Ki67-positive cells versus total cells. The data are expressed as mean ± SE (n = 3 slides). The p values are shown.
S3I-201 decreases the mammary cell proliferation by attenuation of the HFD-induced activated STAT3 signaling in obese ThrbPV/PVPten+/- mice
The findings that S3I-201 inhibited mammary cell proliferation of HFD-ThrbPV/PVPten+/- mice prompted us to examine the STAT3 activity in the mammary gland. We first evaluated the effect of HFD. Similar to the effect observed in the thyroid [17], HFD activated the STAT3 activity by increasing the phosphorylation of STAT3 at Y705 in the mammary gland of ThrbPV/PVPten+/- mice (Figure 3Aa, lanes 8-10 versus 5-7), while no effects were observed in WT mice (panel a, lanes 3-4 versus 1-2). Neither LFD nor HFD affected total STAT3 protein levels in WT mice (panel b, lanes 1-4) or ThrbPV/PVPten+/- mice (panel b, lanes 5-10). The increased STAT3 activity was evident by the increased p-STAT3/total STAT3 ratios shown in Figure 3Ba in that a significant 2.3-fold increase was found (bar 4 versus 3). Consistent with increased cell proliferation, the cyclin D1 protein level was elevated in the mammary gland of HFD-ThrbPV/PVPten+/- mice (Figure 3Ac, lanes 8-10; also see Figure 3Bb), but only minor increases were detected in WT mice (Figure 3Ac, lanes 1-4; also Figure 3Bb). Activation of cyclin D1 by HFD led to increased phosphorylation of retinoblastoma [p-Rb (S780)] without changing the total Rb protein levels (Figure 3Ae). The ratios of p-Rb versus total Rb were ~4-fold higher in the mammary gland of HFD-treated than LFD-treated ThrbPV/PVPten+/- mice (bar 4, Figure 3Bc). Increased p-Rb (S780) drives cell cycle progression from the G1 phase to the S phase to increase cell proliferation [28]. These data indicate that HFD increased STAT3 signaling to increase mammary cell proliferation.
Figure 3.

Effects of HFD and S3I-201 on protein levels of key regulators of the STAT3 signaling pathway in mammary glands from WT and ThrbPV/PVPten+/- mice. (A) Western blot analysis of p-STAT3 (Y705), total STAT3, cyclin D1, phosphorylated retinoblastoma (p-Rb; S780), total Rb, the leptin receptor, p-JAK1, and total JAK1 in mammary glands from WT mice and ThrbPV/PVPten+/- mice fed with LFD (lanes 1, 2, 5, 6, and 7) or with HFD (lanes 3, 4, 8, 9, and 10). Two or 3 mice were used for WT mice and ThrbPV/PVPten+/- mice, respectively. GAPDH was used as a loading control (g and J). (B) The band intensities of the proteins detected in (A) were quantified and graphed. The open bars represent mice fed with LFD and filled bars represent mice fed with HFD. The data, shown as mean ± SE, were analyzed by Student’s t test. (C) Western blot analysis of p-STAT3 (Y705), t-STAT3, cyclin D1, p-Rb (S780), total Rb, the leptin receptor, p-JAK1 (Y1022/1023), and total JAK1 in mammary glands from WT or ThrbPV/PVPten+/- mice treated with vehicle (lanes 1, 2, 5, 6, and 7) or with S3I-201 (lanes 3, 4, 8, 9, and 10). Two or 3 mice were used from WT and ThrbPV/PVPten+/- mice, respectively. GAPDH was used as a loading control (g and J). (D) The band intensities of the proteins detected in (C) were quantified and graphed. The open bars represent mice treated with vehicle and filled bars represent mice treated S3I-201. The data, shown as mean ± SE, were analyzed by Student’s t test. NS = not significant.
We next evaluated whether the inhibition of HFD-induced cell proliferation by S3I-201 shown in Figures 1 and 2 was mediated by inhibition of STAT3 activity. Figure 3C shows that, indeed, S3I-201 decreased STAT3 signaling by reducing the protein abundance of p-STAT3 (Y705) (panel a), cyclin D1 (panel c), and p-Rb (S780) (panel d). The ratio of p-STAT3 (Y705) versus total STAT3 and the ratio of p-Rb (S780) versus total Rb were clearly decreased by S3I-201 in the mammary gland of HFD-ThrbPV/PVPten+/- mice (bar 4 in Figure 3Da and 3Dc, respectively). Thus, these data indicate that inhibition of STAT3 activity lessened HFD-induced hyperplasia in the mammary gland.
We next sought to understand whether the upstream regulators of the STAT3 activity were affected by HFD in the mammary gland of ThrbPV/PVPten+/- mice. We found that the leptin receptor was expressed in the mammary epithelial cells of WT and ThrbPV/PVPten+/- mice (Figure 3Af). The expression was not affected by HFD in either WT mice (panel f, lanes 1-4) or ThrbPV/PVPten+/- mice (panel f, lanes 5-10). However, phosphorylated JAK1 (Y1022/1023) was elevated in the mammary gland of HFD-ThrbPV/PVPten+/- mice (Figure 3Ah, lanes 8-10 versus lanes 5-7) without changing the total JAK1 levels. In fact, the ratio of p-JAK1 versus total JAK1 was 1.5-fold higher in HFD-ThrbPV/PVPten+/- mice than in LFD-ThrbPV/PVPten+/- mice (Figure 3Bd). Since S3I-201 is a specific inhibitor of STAT3 [17], no changes in the protein abundance of the leptin receptor p-JAK1and total JAK1 were found by S3I-201 (Figure 3Ch and 3Ci, and 3Dd).
Increased mammary cell proliferation in HFD-ThrbPV/PVPten+/- mice is not mediated by serum thyroid hormone, thyroid stimulating hormone, and estrogens
Prospective studies for potential association of serum thyroid stimulating hormone (TSH) levels with breast cancer risk have been investigated [20-23]. One small study reported a negative association [20], while the others showed no significant association [21-23]. We therefore carried out thyroid function tests to determine the effect of HFD on serum TSH and thyroid hormone levels of ThrbPV/PVPten+/- mice. Figure 4A shows that HFD did not change serum TSH levels of WT mice (Figure 4A, lanes 1-4). Consistent with previous findings, ThrbPV/PVPten+/- mice exhibited dysregulation of the pituitary-thyroid axis with markedly elevated TSH [14]. However, HFD did not affect the serum TSH levels in ThrbPV/PVPten+/- mice (Figure 4Aa, lane 7 versus 5). Moreover, serum TSH levels in WT and ThrbPV/PVPten+/- mice were not affected by S3I-201 (lanes 2, 4, 6, and 8 versus lanes 1, 3, 5, and 7, respectively).
Figure 4.
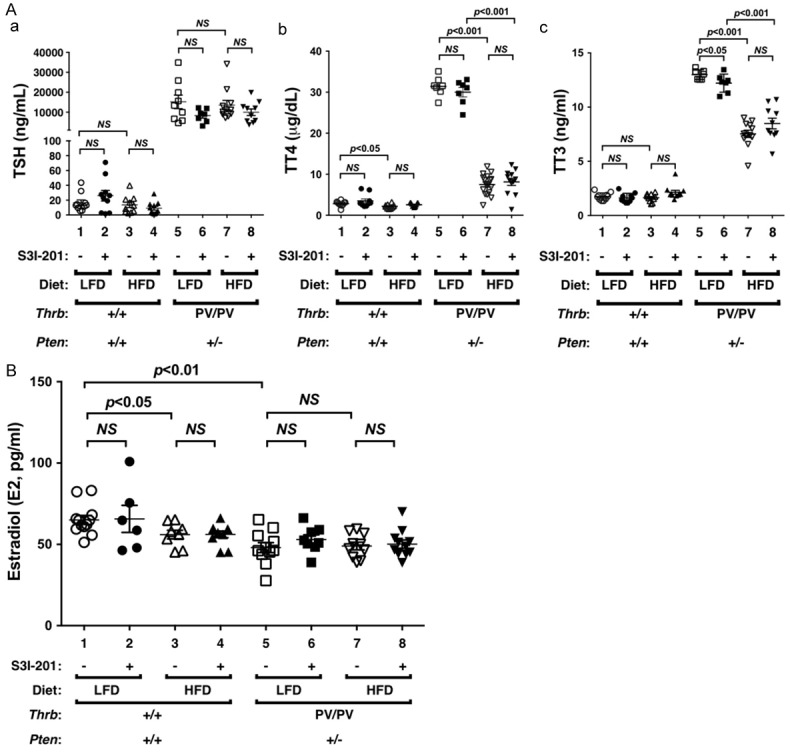
Effects of HFD and S3I-201 on the serum levels of TSH, thyroid hormones, and estrogens in WT and ThrbPV/PVPten+/- mice. (A) Thyroid function tests of WT and ThrbPV/PVPten+/- mice fed with LFD or HFD and treated with vehicle or with S3I-201. Levels of TSH (Aa, n = 7-11), total T4 (Ab, n = 7-18), total T3 (Ac, n = 7-12) were determined as described in Methods and Materials. The genotypes are marked. (B) E2 was determined as described in Methods and Materials (n = 6-12).
We next evaluated serum thyroid hormone levels in WT and ThrbPV/PVPten+/- mice. The dysregulation of the pituitary-thyroid axis leads to an elevated serum total T4 levels in spite of non-suppressible TSH in ThrbPV/PVPten+/- mice [14]. HFD led to a marginal decrease in total T4 (3%) in WT mice (Figure 4Ab, lane 3 versus 1), but a marked decrease of total T4 (76%) by HFD was detected in ThrbPV/PVPten+/- mice (lane 7 versus 5). However, S3I-201 did not affect the total T4 in WT and ThrbPV/PVPten+/- mice fed with LFD or HFD (compare lanes 2, 4, 6, and 8 with lanes 1, 3, 5, and 7, respectively). Figure 4Ac shows that consistent with previous findings [14], total T3 was higher in LFD-ThrbPV/PVPten+/- mice than in LFD-WT mice (lane 5 versus 1). HFD lowered total T3 (42%) in HFD-ThrbPV/PVPten+/- mice (lane 7 versus 5). However, S3I-201 did not affect the total T3 in WT and ThrbPV/PVPten+/- mice fed with LFD or HFD (compare lanes 2, 4, 6, and 8 with lanes 1, 3, 5 and 7, respectively).
These findings indicate that there was no association of serum TSH levels with HFD-induced hyperplasia of mammary epithelial cells. Still, since lower thyroid hormone levels were found in HFD-ThrbPV/PVPten+/- mice, it is reasonable to conclude that obesity-induced mammary hyperplasia was not due to thyroid hormones.
We next evaluated the effect of HFD on the serum estradiol (E2) levels and found that while HFD lowered the E2 (14%) in WT mice (Figure 4B, lane 3 versus 1), E2 levels were not affected by HFD in E2 in ThrbPV/PVPten+/- mice (lane 7 versus 5). S3I-201 did not affect the E2 in WT and ThrbPV/PVPten+/- mice treated with LFD or HFD (compare lanes 2, 4, 6, and 8 with lanes 1, 3, 5, and 7, respectively). Since estrogen receptors (ER) play a critical role in the pathogenesis of breast cancer, we also determined the ERα protein levels in the mammary epithelial cells of WT and ThrbPV/PVPten+/- mice. We found that HFD did not change ERα protein levels in the mammary cells of WT and ThrbPV/PVPten+/- mice. Moreover, S3I-201 had no detectable effects on ERα protein levels (data not shown). Taken together, these results indicate that ERα/E2 signaling did not significantly contribute to obesity-induced proliferation of mammary epithelial cells in HFD-ThrbPV/PVPten+/- mice.
S3I-201 suppresses epithelial-mesenchymal transition by altering expression of key regulators in mammary gland of HFD-ThrbPV/PVPten+/- mice
The developing mammary gland displays many of the properties associated with tumor progression, such as invasion, migration, re-initiation of cell proliferation, and resistance to apoptosis [24]. For example, terminal end buds are a proliferating epithelial cell mass that invades into the stroma, resembling the invasiveness of tumor cells. Evidence has been presented that activated Jak-STAT3 signaling leads to altered expression of key regulators in the epithelial-mesenchymal transition (EMT) to promote cell invasiveness [25]. We therefore evaluated the effects of HFD on key regulators of EMT in WT and ThrbPV/PVPten+/- mice. HFD lowered E-cadherin protein levels in the mammary gland of WT mice (Figure 5Aa, lanes 3&4 versus 1&2; 5Ba, bar 2 versus 1) as well as in ThrbPV/PVPten+/- mice (Figure 5Aa, lanes 8-10 versus 5-7; 5Ba, bar 2 versus 1). Vimentin protein levels were not changed in the mammary gland of HFD-WT mice (Figure 5Ab, lanes 1-4; 5Bb, bar 2 versus 1), but were elevated in the mammary gland of HFD-ThrbPV/PVPten+/- mice (Figure 5Ab, lanes 8-10 versus 5-7; 5Bb, bar 4 versus 3). Neither pro-MMP2 nor active-MMP2 was changed by HFD in the mammary gland of WT mice (Figure 5Ac, Lanes 1-4). However, HFD decreased pro-MMP2 protein levels with corresponding increased protein levels of active-MMP2 in the mammary gland of HFD-ThrbPV/PVPten+/- mice (Figure 5Ac, lanes 8-10 versus 5-7; 5Bc and 5Bd, bar 4 versus 3). These data indicate that HFD induced alteration of key regulators of EMT to increase invasiveness of mammary cells to facilitate lobulo-alveolar remodeling. We next examined whether the inhibition of STAT3 activity by S3I-201 could decrease EMT by reverting the protein levels of key signaling regulators. We found that the protein levels of E-cadherin were higher in the mammary gland of S3I-201-treated HFD-ThrbPV/PVPten+/- mice than vehicle-treated HFD-ThrbPV/PVPten+/- mice (Figure 5Ca, lanes 8-10 versus 5-7; 5Da, bar 4 versus 3). Vimentin was lower in S3I-201-treated HFD-ThrbPV/PVPten+/- mice than control HFD-ThrbPV/PVPten+/- mice (Figure 5Cb, lanes 8-10 versus 5-7; 5Db, bar 4 versus 3). Importantly, pro-MMP2 protein levels were higher in S3I-201-treated HFD-ThrbPV/PVPten+/- mice than control HFD-ThrbPV/PVPten+/- mice (Figure 5Cc, lanes 8-10 versus 5-7; 5Dc, bar 4 versus 3). In contrast, active-MMP2 protein levels were lower in S3I-201-treated HFD-ThrbPV/PVPten+/- mice than control HFD-ThrbPV/PVPten+/- mice (Figure 5Cc, lanes 8-10 versus 5-7; 5Dd, bar 4 versus 3). These results indicate that inhibition of STAT3 activity by S3I-201 attenuated EMT as evidenced by elevated E-cadherin and decreased vimentin and MMP2 protein levels, thereby suppressing lobulo-alveolar remodeling.
Figure 5.

Effects of HFD and S3I-201 on the protein levels of EMT regulators in mammary glands from of WT and ThrbPV/PVPten+/- mice. (A) Western blot analysis of E-cadherin, vimentin, and pro- and active-MMP2 in mammary tissues fed with LFD (lanes 1, 2, 5, 6, and 7) or HFD (lanes 3, 4, 8, 9, and 10) from WT or ThrbPV/PVPten+/- mice. Two WT and ThrbPV/PVPten+/- mice were used. GAPDH was used as a loading control (d). (B) The band intensities of the protein detected in (A) were quantified and graphed. The open bars represent mice treated with LFD and filled bars represent those treated with HFD. The data, shown as mean ± SE, were analyzed by Student’s t test. (C) Western blot analysis of E-cadherin, vimentin, pro- and active-MMP2 in mammary tissues treated with vehicle (lanes 1, 2, 5, 6, and 7) or with S3I-201 (lanes 3, 4, 8, 9, and 10) from WT or ThrbPV/PVPten+/- mice. Two WT and ThrbPV/PVPten+/- mice were used. GAPDH was used as a loading control (d). (D) The band intensities of the protein detected in (C) were quantified and graphed. The open bars represent mice treated with vehicle and filled bars represent those treated with S3I-201. The data, shown as mean ± SE, were analyzed by Student’s t test.
Discussion
Recent epidemiological studies have shown that obesity is associated with breast cancer risks [3-5]. Extensive analyses have also provided compelling evidence to demonstrate a link between obesity and clinical pathological manifestations of breast cancer, such as tumor size, lymph node positivity, and regional and/or distant stage at diagnosis [5]. However, evidence to establish a direct causal relationship between obesity and breast cancer development and progression remains to be established at the molecular level.
In the present studies, using the model of the ThrbPV/PVPten+/- mouse, we showed that HFD led to extensive hyperplasia in the mammary epithelial cells as evidenced by the accelerated lobulo-alveolar development and intense expression of the Ki67 proliferation marker. Similar to obese patients [26-28], HFD-ThrbPV/PVPten+/- mice exhibited elevated serum leptin levels [16]. However, serum TSH, total T4, total T3, and estradiol levels were not significantly higher in ThrbPV/PVPten+/- mice fed with HFD than in mice fed with LFD. Thus, these hormones did not play a significant role in the obesity-induced extensive mammary gland hyperplasia. Molecular analyses showed that the elevated leptin, via the leptin receptor-JAK1-STAT3 signaling, increased mammary cell proliferation. Importantly, we also uncovered that this signaling in the mammary gland of HFD-ThrbPV/PVPten+/- mice led to the changes in the expression of several key regulators of EMT to facilitate the invasion/migration of the proliferating epithelial cell mass into the stroma. Thus, this study has elucidated one molecular pathway by which leptin could act through JAK1-STAT3 signaling to increase cell proliferation and augment EMT. Our findings have provided direct molecular evidence to link obesity to breast cancer risks.
That STAT3 was the active effector in the leptin-mediated breast cancer risks suggests an opportunity for therapeutic intervention. Indeed, we tested whether STAT3 could be used as a potential molecular target. Treatment of HFD-ThrbPV/PVPten+/- mice with a STAT3 inhibitor, S3I-201, lessened the extent of hyperplasia (see the proposed molecular model in Figure 6). Cell proliferation was decreased as evidenced by reduced expression of key cell cycle regulators and proliferation marker Ki67. Inhibition of STAT3 activity by S3I-201 reverted the expression patterns of key regulators to lessen the extent of EMT, thereby dampening the invasiveness of proliferating mammary epithelial cells (Figure 6). These results support the idea that STAT3 inhibitors could be used to inhibit mammary hyperplasia, thereby minimizing potential subsequent progression to breast cancer. These findings are of particular significance because increased expression and activated STAT3 were detected in malignant specimens from breast cancer patients [29,30]. Currently, several STAT3 inhibitors are being tested in phase I and phase II clinical trials in patients with solid tumors such as brain tumors and melanomas [31,32], and various types of leukemias and lymphomas [33]. However, clinical trials to assess the effectiveness of STAT3 inhibitors in the treatment of breast cancer are very limited [32]. The molecular pathway elucidated in the present studies by using the preclinical mouse model could pave the way to expand clinical trials to include breast cancer patients with obesity.
Figure 6.
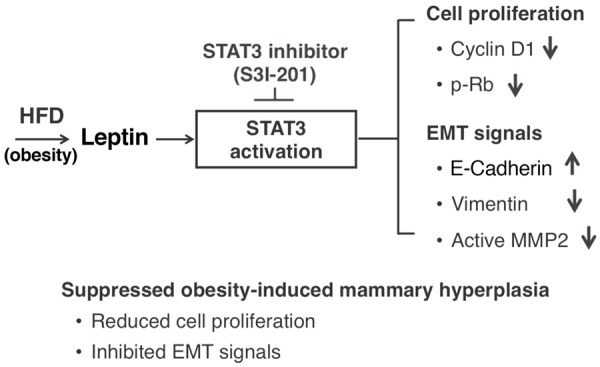
Schematic representation of the effect of S3I-201 in obesity-induced extensive mammary hyperplasia. Obesity-induced aberrant activation of the STAT3 pathway contributes to the mammary hyperplasia via increased cell proliferation and EMT signals in ThrbPV/PVPten+/- mice. HFD induces elevated leptin to activate STAT3 signaling, resulting in increased cyclin D1, p-Rb, vimentin, and active MMP2 and decreased E-cadherin. All of these STAT3 downstream effectors are reversed by S3I-201 to attenuate STAT3 signaling, leading to reduced cell proliferation and decreased EMT signals.
Acknowledgements
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Disclosure of conflict of interest
None.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Rose DP, Vona-Davis L. Influence of obesity on breast cancer receptor status and prognosis. Expert Rev Anticancer Ther. 2009;9:1091–1101. doi: 10.1586/era.09.71. [DOI] [PubMed] [Google Scholar]
- 3.Vrieling A, Buck K, Kaaks R, Chang-Claude J. Adult weight gain in relation to breast cancer risk by estrogen and progesterone receptor status: a meta-analysis. Breast Cancer Res Treat. 2010;123:641–649. doi: 10.1007/s10549-010-1116-4. [DOI] [PubMed] [Google Scholar]
- 4.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 5.Neuhouser ML, Aragaki AK, Prentice RL, Manson JE, Chlebowski R, Carty CL, Ochs-Balcom HM, Thomson CA, Caan BJ, Tinker LF, Urrutia RP, Knudtson J, Anderson GL. Overweight, Obesity, and Postmenopausal Invasive Breast Cancer Risk: A Secondary Analysis of the Women’s Health Initiative Randomized Clinical Trials. JAMA Oncol. 2015;1:611–621. doi: 10.1001/jamaoncol.2015.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin Y, Li Q. Expression and function of leptin and its receptor in mouse mammary gland. Sci China C Life Sci. 2007;50:669–675. doi: 10.1007/s11427-007-0077-2. [DOI] [PubMed] [Google Scholar]
- 7.Wagner KU, Schmidt JW. The two faces of Janus kinases and their respective STATs in mammary gland development and cancer. J Carcinog. 2011;10:32. doi: 10.4103/1477-3163.90677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giordano C, Vizza D, Panza S, Barone I, Bonofiglio D, Lanzino M, Sisci D, De Amicis F, Fuqua SA, Catalano S, Ando S. Leptin increases HER2 protein levels through a STAT3-mediated up-regulation of Hsp90 in breast cancer cells. Mol Oncol. 2013;7:379–391. doi: 10.1016/j.molonc.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JW, Zhao L, Willingham M, Cheng SY. Oncogenic mutations of thyroid hormone receptor beta. Oncotarget. 2015;6:8115–8131. doi: 10.18632/oncotarget.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guigon CJ, Cheng SY. Novel oncogenic actions of TRbeta mutants in tumorigenesis. IUBMB Life. 2009;61:528–536. doi: 10.1002/iub.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eng C. Role of PTEN, a lipid phosphatase upstream effector of protein kinase B, in epithelial thyroid carcinogenesis. Ann N Y Acad Sci. 2002;968:213–221. doi: 10.1111/j.1749-6632.2002.tb04337.x. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 13.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 14.Guigon CJ, Zhao L, Willingham MC, Cheng SY. PTEN deficiency accelerates tumour progression in a mouse model of thyroid cancer. Oncogene. 2009;28:509–517. doi: 10.1038/onc.2008.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guigon CJ, Kim DW, Willingham MC, Cheng SY. Mutation of thyroid hormone receptor-beta in mice predisposes to the development of mammary tumors. Oncogene. 2011;30:3381–3390. doi: 10.1038/onc.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim WG, Park JW, Willingham MC, Cheng SY. Diet-induced obesity increases tumor growth and promotes anaplastic change in thyroid cancer in a mouse model. Endocrinology. 2013;154:2936–2947. doi: 10.1210/en.2013-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park JW, Han CR, Zhao L, Willingham MC, Cheng SY. Inhibition of STAT3 activity delays obesity-induced thyroid carcinogenesis in a mouse model. Endocr Relat Cancer. 2016;23:53–63. doi: 10.1530/ERC-15-0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu X, Zhao L, Park JW, Willingham MC, Cheng SY. Synergistic signaling of KRAS and thyroid hormone receptor beta mutants promotes undifferentiated thyroid cancer through MYC up-regulation. Neoplasia. 2014;16:757–769. doi: 10.1016/j.neo.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furumoto H, Ying H, Chandramouli GV, Zhao L, Walker RL, Meltzer PS, Willingham MC, Cheng SY. An unliganded thyroid hormone beta receptor activates the cyclin D1/cyclin-dependent kinase/retinoblastoma/E2F pathway and induces pituitary tumorigenesis. Mol Cell Biol. 2005;25:124–135. doi: 10.1128/MCB.25.1.124-135.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuijpens JL, Nyklictek I, Louwman MW, Weetman TA, Pop VJ, Coebergh JW. Hypothyroidism might be related to breast cancer in post-menopausal women. Thyroid. 2005;15:1253–1259. doi: 10.1089/thy.2005.15.1253. [DOI] [PubMed] [Google Scholar]
- 21.Hellevik AI, Asvold BO, Bjoro T, Romundstad PR, Nilsen TI, Vatten LJ. Thyroid function and cancer risk: a prospective population study. Cancer Epidemiol Biomarkers Prev. 2009;18:570–574. doi: 10.1158/1055-9965.EPI-08-0911. [DOI] [PubMed] [Google Scholar]
- 22.Tosovic A, Bondeson AG, Bondeson L, Ericsson UB, Malm J, Manjer J. Prospectively measured triiodothyronine levels are positively associated with breast cancer risk in postmenopausal women. Breast Cancer Res. 2010;12:R33. doi: 10.1186/bcr2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tosovic A, Becker C, Bondeson AG, Bondeson L, Ericsson UB, Malm J, Manjer J. Prospectively measured thyroid hormones and thyroid peroxidase antibodies in relation to breast cancer risk. Int J Cancer. 2012;131:2126–2133. doi: 10.1002/ijc.27470. [DOI] [PubMed] [Google Scholar]
- 24.Wiseman BS, Werb Z. Stromal effects on mammary gland development and breast cancer. Science. 2002;296:1046–1049. doi: 10.1126/science.1067431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong H, Hong J, Du W, Lin YW, Ren LL, Wang YC, Su WY, Wang JL, Cui Y, Wang ZH, Fang JY. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J Biol Chem. 2012;287:5819–5832. doi: 10.1074/jbc.M111.295964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zareifar S, Shorafa S, Haghpanah S, Karamizadeh Z, Adelian R. Association of Serum Leptin Level with Obesity in Children with Acute Lymphoblastic Leukemia. Iran J Ped Hematol Oncol. 2015;5:116–124. [PMC free article] [PubMed] [Google Scholar]
- 27.Thanakun S, Izumi Y. Effect of Periodontitis on Adiponectin, C-Reactive Protein and Immunoglobulin G to Porphyromonas Gingivalis in Overweight or Obese Thai People. J Periodontol. 2015:1–16. doi: 10.1902/jop.2015.150583. [DOI] [PubMed] [Google Scholar]
- 28.Kurek K, Baniukiewicz A, Swidnicka-Siergiejko A. Buried bumper syndrome: a rare complication of percutaneous endoscopic gastrostomy. Wideochir Inne Tech Maloinwazyjne. 2015;10:504–507. doi: 10.5114/wiitm.2015.54056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Xiao Q, Bai X, Yu Z, Sun M, Zhao H, Mi X, Wang E, Yao W, Jin F, Zhao L, Ren J, Wei M. Activation of STAT3 is involved in malignancy mediated by CXCL12-CXCR4 signaling in human breast cancer. Oncol Rep. 2014;32:2760–2768. doi: 10.3892/or.2014.3536. [DOI] [PubMed] [Google Scholar]
- 30.Diaz N, Minton S, Cox C, Bowman T, Gritsko T, Garcia R, Eweis I, Wloch M, Livingston S, Seijo E, Cantor A, Lee JH, Beam CA, Sullivan D, Jove R, Muro-Cacho CA. Activation of stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin Cancer Res. 2006;12:20–28. doi: 10.1158/1078-0432.CCR-04-1749. [DOI] [PubMed] [Google Scholar]
- 31.Dutzmann J, Daniel JM, Bauersachs J, Hilfiker-Kleiner D, Sedding DG. Emerging translational approaches to target STAT3 signalling and its impact on vascular disease. Cardiovasc Res. 2015;106:365–74. doi: 10.1093/cvr/cvv103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santoni M, Massari F, Del Re M, Ciccarese C, Piva F, Principato G, Montironi R, Santini D, Danesi R, Tortora G, Cascinu S. Investigational therapies targeting signal transducer and activator of transcription 3 for the treatment of cancer. Expert Opin Investig Drugs. 2015;24:809–824. doi: 10.1517/13543784.2015.1020370. [DOI] [PubMed] [Google Scholar]
- 33.O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311–328. doi: 10.1146/annurev-med-051113-024537. [DOI] [PMC free article] [PubMed] [Google Scholar]