

Abstract
Ductal plate malformations are a heterogenous group of congenital fibrocystic liver diseases resulting from insult to the ductal plate at various stages of embryogenesis. As a result various biliary malformations, cysts, hamartomas and congenital hepatic fibrosis may be seen. We present a radiological pictorial of ductal plate malformations, accurate diagnosis of which is important for clinical management.
Keywords: Biliary, choledochal cysts, ductal plate, polycystic liver
Introduction
Ductal plate malformations (DPMs), also known as fibropolycystic liver diseases, represent a unique spectrum of pathological abnormalities that are caused by insult to the embryonic ductal plate development at various stages. This results in the formation of congenital cystic lesions of the biliary tract that involve the intra as well as extrahepatic bile ducts. The importance of detecting these DPMs at an early stage is their predisposition for pancreatitis, cholangitis, lithiasis, and malignancy. The purpose of this pictorial essay is to acquaint the readers with imaging features in DPMs.
Embryology
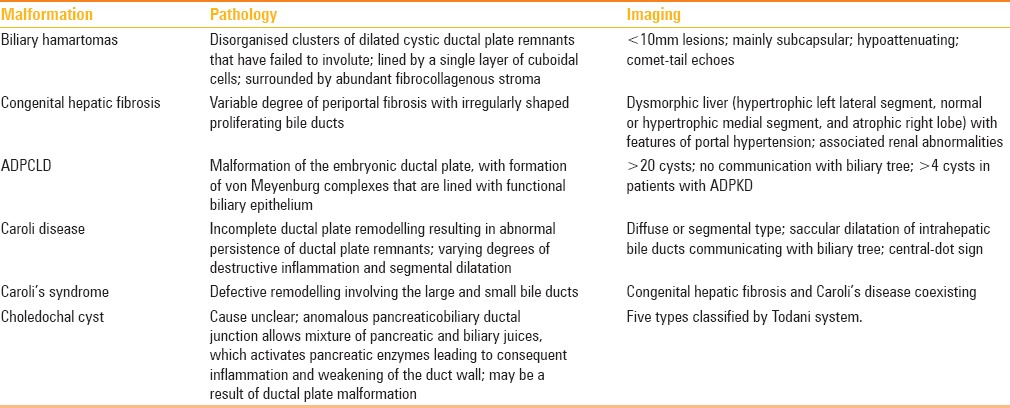
Ductal plate is defined as a double-layered cylindrical structure of bile duct epithelium that surroundsthe portal ramifications by the eighth gestational week. After approximately the12th gestational week, remodelling of ductal plate begins, and maturity is attained by the end of gestation or early postnatal period [Figure 1]. Biliary ducts are normally formed from remodelling and partial involution of these cylindrical ductal plates. Insufficient remodelling and resorption leads to DPM.[1] The timing of defective development determines the resulting clinicopathologic disorder. Insult to the small interlobular ducts leads to congenital hepatic fibrosis or biliary hamartomas; autosomal dominant polycystic liver disease due to medium-sized interlobular ducts; Caroli disease; and choledochal cyst due to the defective development of large-sized interlobular ducts[1,2,3,4,5] [Figures 2 and 3]. A brief illustration of embryology and imaging in different types of ductal plate malformations is tabulated in Table 1.
Figure 1 (A-E).
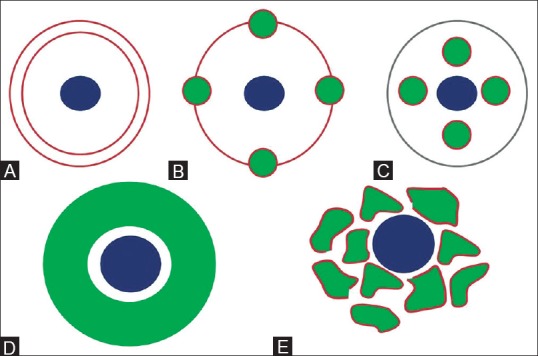
Schematic line diagram showing development of biliary tract. (A) During 8th gestational week, ductal plate (brown), becomes apparent in mesenchyme surrounding portal vein radicle (blue). (B) By 12th gestational week, remodelling starts and parts of ductal plate fuse and are reabsorbed. (C) Unfused portions constitute definitive bile ducts (green). (D) Ductal plate malformation in which continuous dilated duct encircles the portal vein radicle; and (E) interrupted circle of ectatic bile ducts
Figure 2.
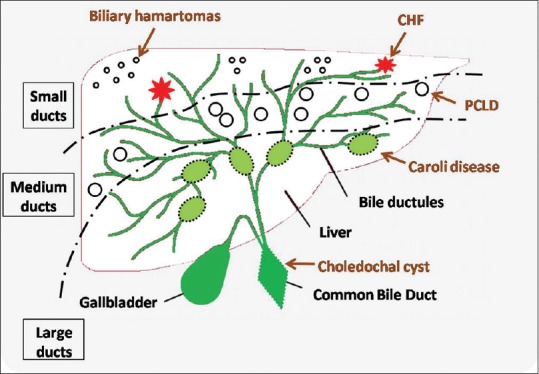
Schematic line diagram showing the types of ductal plate malformations depending on the duct size affected
Figure 3 (A-D).
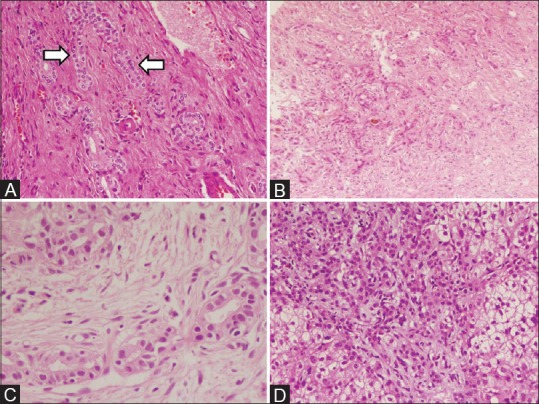
Histopathology images showing spectrum of ductal plate malformations (A) Caroli's disease, (B) congenital hepatic fibrosis, (C) biliary atresia, (D) polycystic liver disease
Table 1.
Ductal plate malformations: Embryology and imaging
Von Meyernburg Complex
Von Meyernburg complex (VMC), also known as multiple biliary hamartomas or biliary microhamartomas, result from the failure of involution of embryonic bile ducts, with a prevalence of 1–5%.[2] It is named after Hans von Meyenburg, who first described this entity in 1918.[3] The lesions are scattered, multiple, uniform, and usually smaller than 10 mm, especially in the subcapsular location. The lesions are predominantly cystic, however, rarely can be solid or mixed.[4,5] They donot communicate with the biliary tree. On ultrasound, biliary hamartomas are seen as tiny hyperechoic, hypoechoic, or mixed lesionswith comet-tail echoes. On computed tomography (CT), the lesions are small and hypoattenuating. On magnetic resonance imaging (MRI), the lesions show T1 hypo/T2 hyperintense signal with no diffusion restriction. MRCP may actually show the exact number and delineation of the lesions and lack of communication with the biliary tree [Figure 4]. No enhancement is seen on post-contrast scans, however uncommonly; homogeneous enhancement or a peripheral rim enhancement may be seen which represents compressed hepatic parenchyma. Rarely, a mural nodule can be seen due to fibrocollagenous stroma. The differential diagnosis includes simple hepatic cysts, microabscesses, metastatic lesions, peribiliary cysts, and Caroli's disease.[4,5] Simple hepatic cysts and metastatic lesions are rarely uniform in size, attenuation, and signal intensity. Microabscesses show diffusion restriction and have typical clinical presentation. Peribiliary cysts are classically seen predominantly in the hilum and along the larger portal tracts [Figure 5]. Caroli's disease demonstrate enhancing central dot sign of portal radicles. VMCs may coexist with simple hepatic cysts or polycystic liver and kidney diseases. Simple hepatic cysts are usually larger than 10 mm in diameter on imaging and are round in shape.
Figure 4 (A and B).
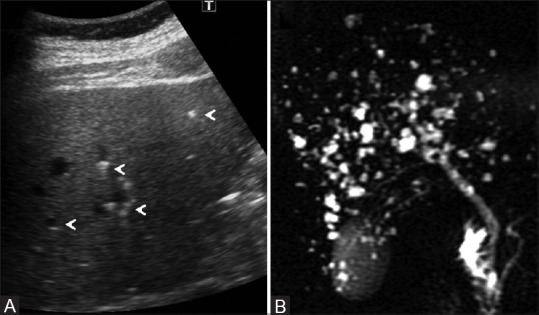
Von Meyenburg complexes. (A) Abdominal ultrasound images showed scattered hyperechoic and hypoechoic submillimetric lesions in liver showing comet-tail echoes (arrowheads); (B) thick-section single shot fast spin-echo heavily T2-weighted MR image shows no communication with biliary tree
Figure 5.
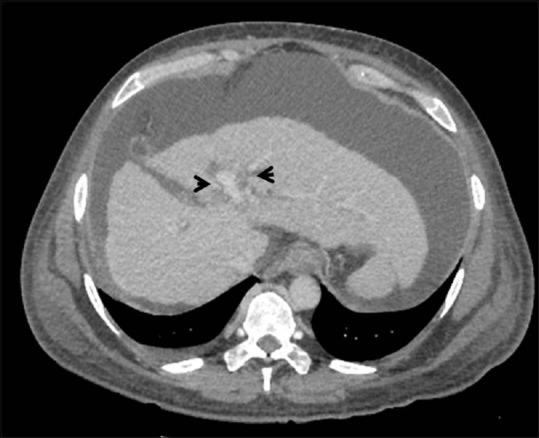
Peribiliary cysts. Axial contrast-enhanced CT image showing cystic lesions (arrowheads) in peribiliary location on both sides of portal vein with associated features of cirrhosis and portal hypertension
Congenital Hepatic Fibrosis
Congenital hepatic fibrosis is a dynamic progressive fibrotic process involving the liver and is histologically characterized by a variable degree of periportal fibrosis and irregularly shaped proliferating bile ducts. Patients with congenital hepatic fibrosis typically have dysmorphic liver on imaging (hypertrophic left lateral segment, normal or hypertrophic medial segment, and atrophic right lobe), portal hypertension, renal abnormalities (particularly autosomal recessive polycystic kidney disease), and other associated ductal plate malformation such as biliary hamartomas or Caroli's disease (i.e., Caroli's syndrome) [Figure 6].[6] The medial segment in congenital hepatic fibrosis is usually normal or enlarged contrary to cirrhosis due to other causes where atrophy of medial segment is seen.[7] Periportal high signal intensity is seen on T2-weighted images due to periportal fibrosis and proliferating small biliary ductules,[8] and especially T2-weighted half-Fourier acquisition single-shot turbo spin-echo (HASTE) images better depict these pathological features. Other findings that can be seen on imaging are portal vein thrombosis, cavernoma formation, and benign regenerative nodules.
Figure 6 (A and B).
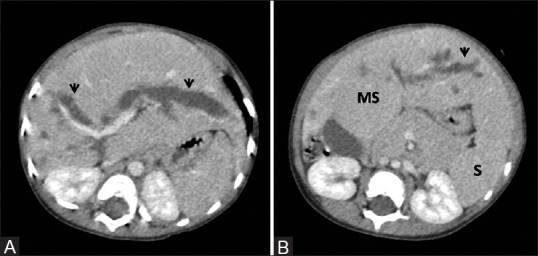
Congenital hepatic fibrosis with Caroli's disease. (A, B) Contrast-enhanced portal venous phase images depicting marked dilatation of intrahepatic biliary radilces (arrowheads) with associated findings of congenital hepatic fibrosis – atrophy of right lobe and hypertrophy of medial segment (MS) of left lobe with signs of portal hypertension, namely, splenomegaly (s)
Autosomal Dominant Polycystic Liver Disease
Polycystic liver disease (PCLD) can be hereditary or nonhereditary. Hereditary PCLD can occur in isolation or be associatedwith autosomal dominant polycystic kidney disease (ADPKD). Isolated polycystic liver disease has been associated with the genes SEC63 and PRKCSH whereas PKD1 and PKD2 genes are implicated for ADPKD.[9] The prevalence of hepatic cysts is reported to be 58–75% in females and 42–62% in males with ADPKD.[10] These cysts are a result of malformation of the embryonic ductal plate, with formation of von Meyenburg complexes lined with functional biliary epithelium.[11] Two types of cysts may be seen in the liver in hereditary PCLD, namely, intrahepatic and peribiliary cysts.[12] The presence of more than 20 liver cysts is considered to be hereditary polycystic liver and replace over 50% of the hepatic parenchyma [Figure 7].[13] In patients with ADPKD, presence of four to six cysts in the liver is suggestive of polycystic liver disease [Figure 8]. On ultrasound, cysts appear anechoic with well-defined thin walls. CT scans show homogeneous, water-attenuation, nonenhancing lesions; on MRI, the cysts show T1-hypointense, T2-hyperintense signal. Various complications may be seen in liver cystsin PCLD such as infection, intracystic hemorrhge, rupture, cholecystic jaundice due to compression of bile ducts, mass effect on the hepatic veins, and inferior vena cava and portal hypertension. The main differential diagnoses of PCLD are simple liver cysts, biliary hamartomas, and cysts associated with ADPKD. Simple liver cyst is a benign entity seen in normal aging liver in normal individuals, which is usually not associated with complications such as infection or hemorrhage. In patients with ADPKD, the cysts in liver are scattered diffusely, also known as hepatobiliary cysts (intrahepatic and peribiliary cysts), smaller in size, and associated with complications. Clinically, these patients present with symptoms of renal disease, unlike in cases of isolated PCLD which is most often asymptomatic. Extrarenal manifestations such as intracranial aneurysms and valvular heart disease are more common in patients with ADPKD.[14,15]
Figure 7 (A and B).
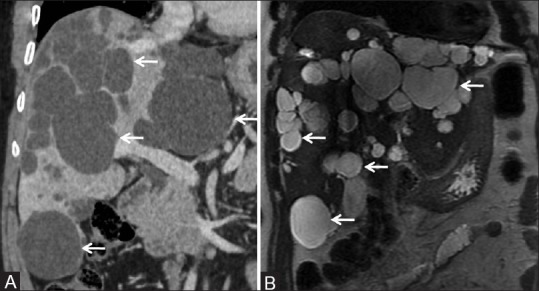
Isolated PCLD: (A) Coronal CT image showing numerous fluid attenuating cystic lesions (arrows) in almost entire liver parenchyma. (B) Coronal T2-weighted MR image showing T2 hyperintense cystic lesions (arrows) in liver parenchyma in a case of polycystic liver disease
Figure 8 (A and B).
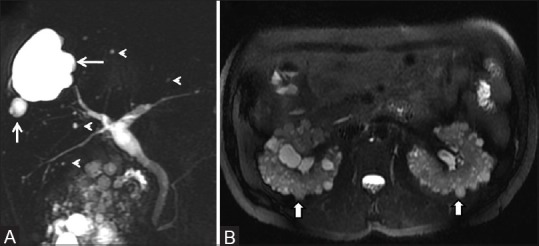
ADPKD with PCLD. (A) Single-shot fast spin-echo heavily T2-weighted MR images showing multiple T2 hyperintense cysts (arrow) and biliary hamartomas (arrowheads) in liver (B) Axial fat-saturated T2-weighted MR showing numerous cysts in bilateral kidneys (arrows)
Caroli's Disease
Caroli's disease was first described by Caroli et al. in 1958.[16] It corresponds to type V choledochal cyst as classified by Todani et al.[17] The disease results from incomplete ductal plate remodelling at the level of large intrahepatic bile ducts which results in abnormal persistence of ductal plate remnants.[18] This entity is further divided into two subtypes: Simple Caroli's disease and complex type associated with congenital hepatic fibrosis also known as Caroli's syndrome. It has an autosomal recessive inheritance. On imaging, Caroli disease can be diffuse, lobar, or segmental type. Diffuse type is seen as saccular or fusiform dilatation of intrahepatic bile ducts without any evidence of obstruction. The central dot sign is typical and represents malformed biliary cysts enveloping the portal radicle [Figure 9]. Irregular bile duct walls, strictures, and stones may be present. Extrahepatic ductal dilatation may be seen in Caroli's disease due to recurrent episodes of cholangitis and stone passage, and this incidence may range 26–53%.[18,19] Cholangitis, cirrhosis, and cholangiocarcinoma are known complications. The exact incidence of cholangiocarcinoma in Caroli's disease is not known, however, there is a 100-fold increase in the risk compared to general population.[20] Sometimes, Caroli's disease may be difficult to differentiate from PCLD; in suchcases, hepatocyte specific MR contrast agent shows communication between the cysts and biliary tree in cases of Caroli's disease. Segmental or lobar types of Caroli's disease are managed by sectionectomy or lobectomy. Diffuse type is managed by biliodigestive anastomosis. Liver transplant is advocated in patients with Caroli's syndrome associated with portal hypertension and in cases with refractory recurrent cholangitis in diffuse Caroli's disease.[21]
Figure 9.
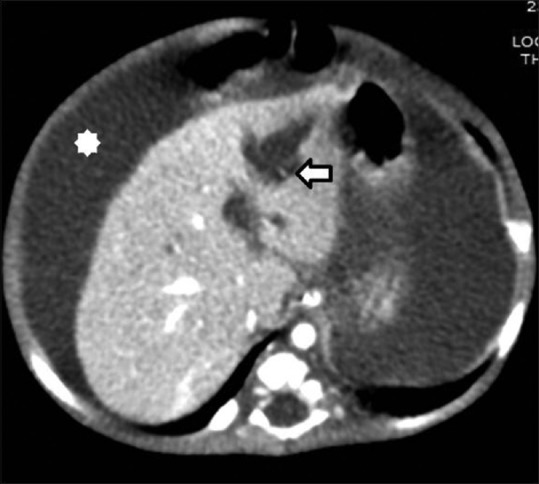
Segmental Caroli's disease. Contrast-enhanced CT image showing segmental cystic dilatation in left hepatic lobe with “central-dot sign”(arrow). Associated features of portal hypertension seen namely ascites (asterix)
If the remodelling is defective involving the large as well as small bile ducts, features of both congenital hepatic fibrosis and Caroli's disease are present. This condition has been termed “Caroli's syndrome.” The two conditions may represent different stagesof the same disease.[22] Mutation of PKHD1 gene is responsible for Caroli's syndrome.[23] Immunohistochemically, the biliary cells in Caroli's disease are positive for MUC1 glycoprotein.[24]
Various renal disorders may be seen in association with Caroli's disease including ADPKD, ARPKD, medullary sponge kidney, and medullary cystic disease. Imaging features of ARPKD include enlarged kidney with radiallyarranged tubules throughout the renal parenchyma (fan-shaped pattern), multiple renal cystic, and tubular lesions located predominantly at the medulla, corticomedullary junction with sparing of the peripheral cortex, associated with multiple renal calculi.[25]
Choledochal Cyst
Choledochal cysts are uncommon anomalies characterized by dilatation of intrahepatic or extra-hepatic biliary ducts or both. Some authors believe it to be the result of ductal plate malformation and others report that it is related to an anomalous common channel causing pancreatic enzymes to reflux, resulting in progressive bile duct dilatation.[26,27] Choledochal cysts are classified into five basic types according to the Todanisystem [Figures 10 and 11] [Table 2].[28] Recently, authors have described combined dilatation of cystic duct and common bile duct as new variant of Type I or Type VI variety of choledochal cyst [Figure 12]. However, cystic duct dilatation may be seen coexisting in various combinations with other types, as described in the literature.[29,30,31,32] MRCP is an excellent modality to demonstrate choledochal cyst. On heavily T2-weighted sequences, they are characterized by a hyperintense tubular, fusiform, or cystic dilatation of the bile ducts. Various complications may be associated with choledochal cyst such asintraductal stone formation, gallstones, cholangitis, pancreatitis, cholangiocarcinoma, gallbladder cancer, and biliary peritonitis as a consequence of cyst rupture.[26] If left untreated, the risk of cholangiocarcinoma increases with an incidence as high up to 20–30% by the second decade of life.[33]
Figure 10.
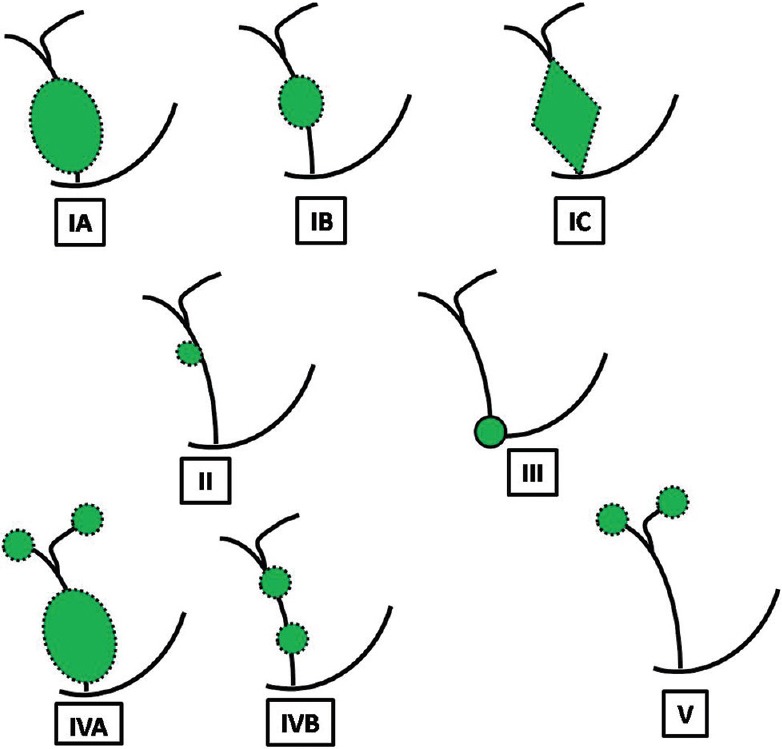
Todani classification of choledochal cyst. Type IA is cystic dilatation; IB – focal saccular dilatation; IC – smooth fusiform dilatation of entire extrahepatic bile duct; Type II is diverticulum of extrahepatic bile duct; Type III is dilatation of distal common bile duct confined to wall of duodenum (choledochocele); Type IVA is multiple sites of dilatation of both extra and intrahepatic biliary tree; Type IVB is multiple sites of dilatation of extrahepatic bile duct only (string-of-beads appearance); Type V is saccular dilatation of only intrahepatic biliary tree (Caroli's disease)
Figure 11 (A-F).
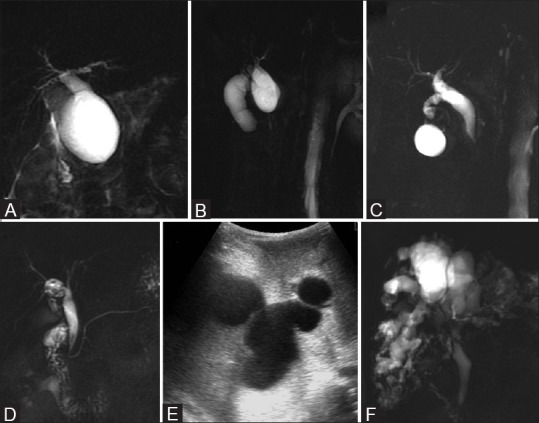
Choledochal cyst. (A) Type IA – marked cystic dilatation of entire extrahepatic bile duct (B) Type IB – focal saccular dilatation (C) Type IC –smooth fusiform dilatation of entire extrahepatic bile duct (D) Type II –discrete diverticulum arising from lateral wall of common hepatic duct (E) Type IVA –multiple sites of dilatation of both extrahepatic and intrahepatic biliary tree (F) Type V –multiple sites of saccular or cystic dilatation of only intrahepatic biliary tree (Caroli disease)
Table 2.
Todani classification of choledochal cyst

Figure 12.
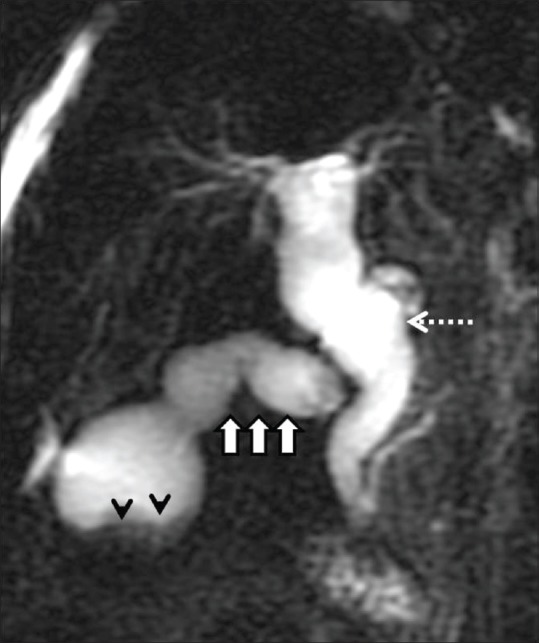
Type VI choledochal cyst. Two-dimensional MRCP image showing diffuse dilatation of the common bile duct (dashed arrow), dilated and tortuous cystic duct (arrows) suggestive of coexistence of Type I and Type VI choledochal cyst. Incidental note is made of cholelithiasis (arrowheads)
Association with Biliary Atresia
DPM-like lesions along the portal tracts may also be seen in cystic biliary atresia.[34] The triangular cord sign, intrahepatic bile duct dilatation, and anechoic cysts at porta hepatis might suggest cystic biliary atresia [Figure 13].[35]
Figure 13 (A-D).
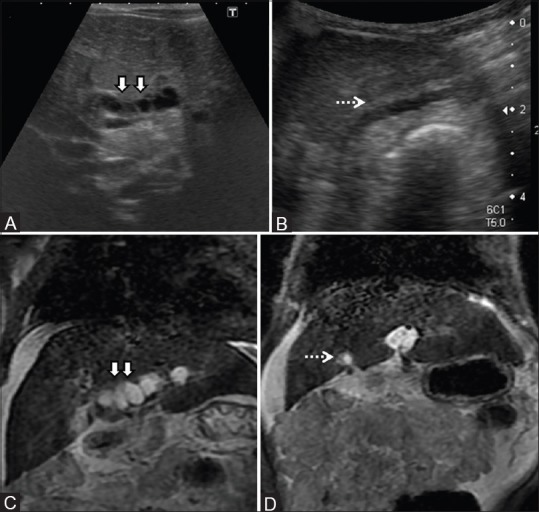
Cystic Biliary atresia with DPM. (A, B) Ultrasound images showing periportal cysts (arrows) and irregular contracted gallbladder (dashed arrow) in a case of cystic biliary atresia. (C, D) Coronal T2-weighted MR confirming the above findings and depicting hyperintenseperiportal cysts (arrows) with small atretic gallbladder (dashed arrow). Note is made of changes of chronic liver disease with ascites
Association with Mesenchymal Hamartoma
Ductal plate malformations are also a part of tissue abnormalities seen in the cystic variant of mesenchymal hamartomas [Figure 14].[36] Pathologically, previous studies have shown that cystic variant of mesenchymal hamartomas consists of abnormally dilated biliary structures in the form of cystic and tortuous biliary ducts or abnormal proliferation of small bile ducts.[37,38]
Figure 14.
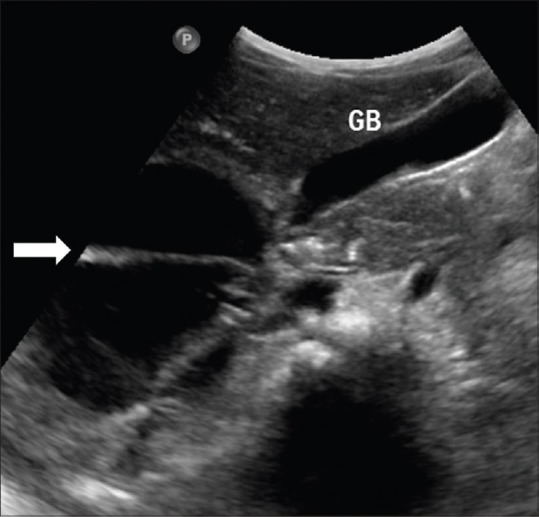
Mesenchymal hamartoma. Ultrasound image of a child showing complex cystic lesion (arrow) with thick septations and fine internal echoes in a proven case of mesenchymal hamartoma
Conclusion
Awareness of the embryopathogenesis and characteristic imaging features plays an important role in correct noninvasive diagnosis of DPMs. In addition, it is important to consider DPMs in the context of associated renal manifestations, particularly ARPKD. Sometimes, more than one form of DPMs can coexist in the same patient, and it should not deter a radiologist in reaching a correct diagnosis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
Dr. Shiv Kumar Sarin, Senior Professor and Director, ILBS.
References
- 1.Terada T, Kitamura Y, Nakanuma Y. Normal and abnormal development of the human intrahepatic biliary system: A review. Tohoku J Exp Med. 1997;181:19–32. doi: 10.1620/tjem.181.19. [DOI] [PubMed] [Google Scholar]
- 2.Santiago I, Loureiro R, Curvo-Semedo L, Marques C, Tardáguila F, Matos C, et al. Congenital cystic lesions of the biliary tree. AJR Am J Roentgenol. 2012;198:825–35. doi: 10.2214/AJR.11.7294. [DOI] [PubMed] [Google Scholar]
- 3.von Meyenburg H. Über die Zystenleber. Beiträge zur pathologischen Anatomie und zur allgemeinen Pathologie. Jena. 1918;64:477–532. [Google Scholar]
- 4.Zheng RQ, Zhang B, Kudo M, Onda H, Inoue T. Imaging findings of biliary hamartomas. World J Gastroenterol. 2005;28(11):6354–9. doi: 10.3748/wjg.v11.i40.6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung YC, Tan CF, Wan YL, Lui KW, Tsai CC. MRI of multiple biliary hamartomas. Br J Radiol. 1997;70:527–9. doi: 10.1259/bjr.70.833.9227236. [DOI] [PubMed] [Google Scholar]
- 6.Brancatelli G, Federle MP, Vilgrain V, Vullierme MP, Marin D, Lagalla R. Fbropolycystic liver disease: CT and MR imaging findings. Radiographics. 2005;25:659–70. doi: 10.1148/rg.253045114. [DOI] [PubMed] [Google Scholar]
- 7.Zeitoun D, Brancatelli G, Colombat M, Federle MP, Valla D, Wu T, et al. Congenital hepatic fibrosis: CT findings in 18 adults. Radiology. 2004;231:109–16. doi: 10.1148/radiol.2311030108. [DOI] [PubMed] [Google Scholar]
- 8.Akhan O, Karaosmanoğlu AD, Ergen B. Imaging findings in congenital hepatic fibrosis. Eur J Radiol. 2007;61:18–24. doi: 10.1016/j.ejrad.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Polycystic Liver Disease. Gastroenterol Hepatol. 2015;11:542–4. [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan DE, Lockhart ME, Canon CL, Holcombe MP, Bynon JS. Polycystic liver disease: Multimodality imaging for complications and transplant evaluation. Radiographics. 2006;26:1655–68. doi: 10.1148/rg.266065013. [DOI] [PubMed] [Google Scholar]
- 11.Bistritz L, Tamboli C, Bigam D, Bain VG. Polycystic liver disease: Experience at a teaching hospital. Am J Gastroenterol. 2005;100:2212–7. doi: 10.1111/j.1572-0241.2005.50258.x. [DOI] [PubMed] [Google Scholar]
- 12.Itai Y, Ebihara R, Eguchi N, Saida Y, Kurosaki Y, Minami M, et al. Hepatobiliary cysts in patients with autosomal dominant polycystic kidney disease: Prevalence and CT findings. AJR Am J Roentgenol. 1995;164:339–42. doi: 10.2214/ajr.164.2.7839965. [DOI] [PubMed] [Google Scholar]
- 13.Hansman MF, Ryan JA, Jr, Holmes JH, 4th, Hogan S, Lee FT, Kramer D, et al. Management and long-term follow-up of hepatic cysts. Am J Surg. 2001;181:404–10. doi: 10.1016/s0002-9610(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 14.Qian Q. Isolated polycystic liver disease. Adv Chronic Kidney Dis. 2010;17:181–9. doi: 10.1053/j.ackd.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian Q, Li A, King BF, Kamath PS, Lager DJ, Huston J, 3rd, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37:164–71. doi: 10.1053/jhep.2003.50006. [DOI] [PubMed] [Google Scholar]
- 16.Caroli J, Soupault R, Kossakowski J, Plocker L, Paradowska M. La dilatation polykystiquecongénitale des voiesbiliairesintrahépatiques: Essai de classification. Sem Hop. 1958;34:488–95. [PubMed] [Google Scholar]
- 17.Todani T, Watanabe Y, Narusue M, Tabuchi K, Okajima K. Congenital bile duct cysts: Classification, operative procedures, and review of thirty-seven cases including cancer arising from choledochal cyst. Am J Surg. 1977;134:263–9. doi: 10.1016/0002-9610(77)90359-2. [DOI] [PubMed] [Google Scholar]
- 18.Levy AD, Rohrmann CA, Jr, Murakata LA, Lonergan GJ. Caroli's disease: Radiologic spectrum with pathologic correlation. AJR Am J Roentgenol. 2002;179:1053–7. doi: 10.2214/ajr.179.4.1791053. [DOI] [PubMed] [Google Scholar]
- 19.Barros JL, Polo JR, Sanabia J, Garcia-Sabrido JL, Gomez-Lorenzo FJ. Congenital cystic dilatation of the intrahepatic bile ducts (Caroli's disease): Report of a case and review of the literature. Surgery. 1979;85:589–92. [PubMed] [Google Scholar]
- 20.Taylor AC, Palmer KR. Caroli's disease. Eur J Gastroenterol Hepatol. 1998;10:105–8. doi: 10.1097/00042737-199802000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Lendoire J, Barros Schelotto P, Alvarez Rodríguez J, Duek F, Quarin C, Garay V, et al. Bile duct cyst type V (Caroli's disease): Surgical strategy and results. HPB. 2007;9:281–4. doi: 10.1080/13651820701329258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yonem O, Bayraktar Y. Clinical characteristics of Caroli's syndrome. World J Gastroenterol. 2007;13:1934–7. doi: 10.3748/wjg.v13.i13.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergmann C, Senderek J, Sedlacek B, Pegiazoglou I, Puglia P, Eggermann T, et al. Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1) J Am Soc Nephrol. 2003;14:76–89. doi: 10.1097/01.asn.0000039578.55705.6e. [DOI] [PubMed] [Google Scholar]
- 24.Terada T. Human fetal ductal plate revisited: II. MUC1, MUC5AC, and MUC6 are expressed in human fetal ductal plate and MUC1 is expressed also in remodelling ductal plate, remodeled ductal plate and mature bile ducts of human fetal livers. Int J Clin Exp Pathol. 2013;6:571–85. [PMC free article] [PubMed] [Google Scholar]
- 25.Tzoufi M, Rogalidou M, Drimtzia E, Sionti I, Nakou I, Argyropoulou M, et al. Caroli's disease: Description of a case with a benign clinical course. Ann Gastroenterol. 2011;24:129–33. [PMC free article] [PubMed] [Google Scholar]
- 26.Venkatanarasimha N, Thomas R, Armstrong EM, Shirley JF, Fox BM, Jackson SA. Imaging features of ductal plate malformations in adults. Clin Radiol. 2011;66:1086–93. doi: 10.1016/j.crad.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 27.Kim MJ, Han SJ, Yoon CS, Kim JH, Oh JT, Chung KS, et al. Using MR cholangiopancreatography to reveal anomalous pancreaticobiliary ductal union in infants and children with choledochal cysts. AJR Am J Roentgenol. 2002;179:209–14. doi: 10.2214/ajr.179.1.1790209. [DOI] [PubMed] [Google Scholar]
- 28.Todani T, Watanabe Y, Fujii T, Toki A, Uemura S, Koike Y. Congenital choledochal cyst with intrahepatic involvement. Arch Surg. 1984;119:1038–43. doi: 10.1001/archsurg.1984.01390210042010. [DOI] [PubMed] [Google Scholar]
- 29.Michaelides M, Dimarelos V, Kostantinou D, Bintoudi A, Tzikos F, Kyriakou V, et al. A new variant of Todani type I choledochal cyst. Imaging evaluation. Hippokratia. 2011;15:174–7. [PMC free article] [PubMed] [Google Scholar]
- 30.Bhoil R, Sood S, Sood RG, Singla G, Bakshi S. A variant of type VI choledochal cyst: Combined dilatation of cystic duct and common bile duct. J Ultrasound. 2015;19:71–2. doi: 10.1007/s40477-015-0186-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serena Serradel AF, Santamaría Linares E, Herrera Goepfert R. Cystic dilatation of the cystic duct: A new type of biliary cyst. Surgery. 1991;109:320–2. [PubMed] [Google Scholar]
- 32.Sethi S, Upreti L, Verma AK, Puri SK. Choledochal cyst of the cystic duct: Report of imaging findings in three cases and review of literature. Indian J Radiol Imaging. 2015;25:315–20. doi: 10.4103/0971-3026.161468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Todani T, Toki A. [Cancer arising in choledochal cyst and management] Nihon Geka Gakkai Zasshi. 1996;97:594–8. [PubMed] [Google Scholar]
- 34.Low Y, Vijayan V, Tan CE. The prognostic value of ductal plate malformation and other histological parameters in EHBA; An immunohistochemical study. J Pediatr. 2001;139:320–2. doi: 10.1067/mpd.2001.117003. [DOI] [PubMed] [Google Scholar]
- 35.Zhou LY, Guan BY, Li L, Xu ZF, Dai CP, Wang W, et al. Objective differential characteristics of cystic biliary atresia and choledochal cysts in neonates and young infants: Sonographic findings. J Ultrasound Med. 2012;31:833–41. doi: 10.7863/jum.2012.31.6.833. [DOI] [PubMed] [Google Scholar]
- 36.Desmet VJ. Ludwig symposium on biliary disorders-part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80–9. doi: 10.4065/73.1.80. [DOI] [PubMed] [Google Scholar]
- 37.Chang HJ, Jin SY, Park C, Park YN, Jang JJ, Park CK, et al. Mesenchymal hamartomas of the liver: Comparison of clinicopathologic features between cystic and solid forms. J Korean Med Sci. 2006;21:63–8. doi: 10.3346/jkms.2006.21.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cook JR, Pfeifer JD, Dehner LP. Mesenchymal hamartoma of the liver in the adult: Association with distinct clinical features and histological changes. Hum Pathol. 2002;33:893–8. doi: 10.1053/hupa.2002.127442. [DOI] [PubMed] [Google Scholar]