

Abstract
Heart failure with preserved ejection fraction (HFpEF) has similar prevalence and prognosis as HF with reduced EF, but there is no approved treatment for HFpEF. HFpEF is common in postmenopausal women, which suggests that the absence of estrogen (E2) plays a role in its pathophysiology. With the country's growing elderly population, the prevalence of HFpEF is rapidly increasing. This has triggered a renewed urgency in finding novel approaches to preventing and slowing the progression of HFpEF. In this review, we address the role of E2 in left ventricular diastolic function and how it impacts women with HFpEF as well as animal models. We also discuss the primary potential mechanisms that represent critical nodes in the mechanistic pathways of HFpEF and how new treatments could be developed to target those mechanisms.
Keywords: heart failure, estrogen, left ventricular diastolic dysfunction, mitochondrial function, cardiac remodeling, cardiac hypertrophy, Ca2+ homeostasis, titin isoforms
Introduction
It is estimated that by 2030, more than 50% of Americans will suffer from heart failure with preserved ejection fraction (HFpEF) that results from left ventricular diastolic dysfunction (LVDD).1 Cardiac function includes a systolic phase of contraction to pump blood from the left ventricle (LV) to the body and a diastolic phase of relaxation to fill blood into the LV. Thus, LVDD is associated with abnormalities in active LV relaxation and passive filling from myocardial stiffness accompanied by reduced or compensated cardiac output (CO).2 Older age, female gender, diabetes, obesity, hypertension, and cardiomyopathy are associated with a greater prevalence of HFpEF.
Accumulating evidence has demonstrated that postmenopausal women are susceptible to cardiovascular disease and have a higher incidence of LVDD than men of the same age, suggesting a close link between LVDD and estrogen (E2) deficiency.3,4 Estrogen replacement has been shown to improve diastolic function in women with LVDD and animal models of LVDD through mechanisms that involve an interaction with E2 receptors (ERs). There are three types of ERs: ERα, ERβ, and G protein-coupled ER (GPER or GPR30). The first two belong to nuclear transcriptional factors and exist in cardiac myocytes and cardiac fibroblasts. Both ERα and ERβ form homodimers and heterodimers to regulate downstream target genes through rapid nongenomic and longer-term genomic pathways,5 and both interact with E2 response element (ERE). GPR30, a plasma membrane protein, binds E2 with high affinity and is widely distributed in a variety of mammalian tissues including the heart.6 GPR30 maintains normal cardiac structure and function through a rapid nongenomic pathway mediated by activation of enzymes such as adenylyl cyclase, mitogen-activated protein kinases (MAPK), extracellular signal-regulated kinase (ERK)-1 and ERK-2, and phosphatidylinositol 3-kinase (PI3K). Recent studies show that ERs modulate cardiac mitochondrial biogenesis and energy production that is essential for regulating myocardial function.5–8
Increasing evidence indicates that loss of E2 leads to LVDD in animal models and postmenopausal women.9,10 In a study by Pines et al. comparing pre- and postmenopausal women, the latter displayed a significant reduction in diastolic function with insignificant alteration of ejection fraction and fraction shortening, but these changes were progressively aggravated during menopause without E2 replacement therapy (ERT).11 In contrast, postmenopausal women who received ERT showed significant increases in aortic flow accompanied by increased stroke volume and cardiac output, implicating that E2 significantly improves cardiac contractility.11 In addition, Manolio et al. reported that declined mitral flow velocity is restored to normal levels in postmenopausal women after receiving ERT,12 further indicating that ERT is important to maintain diastolic function in postmenopausal women. However, the underlying mechanisms regarding diastolic dysfunction remain unclear.
This review discusses the potential role of E2 in regulating cardiac diastolic function and the likely underlying mechanisms of action. It outlines pathological alteration of diastolic function relating to E2 deficiency in animal models and postmenopausal women, and it explores the underlying pathophysiological mechanism resulting from mitochondrial dysfunction, intracellular Ca2+ imbalance, and myocardial hypertrophy and remodeling.
Mechanisms by Which E2 Deficiency Accelerates LVDD
Influence of E2-Mediated Mitochondrial Metabolism on LVDD
Decreased cardiac performance stems from dysregulation of cardiac energy production in postmenopausal women. Mitochondria are the primary energy source to regulate cardiac function, and E2 has been shown to impact mitochondrial function (Figure 1). Previous studies show that E2 increases protein components and decreases oxidative stress in the cerebral mitochondria of ovariectomized rats.13 E2-deprived rats fed a high-fat diet show a dynamic alteration of mitochondrial function paralleled with decreased cardiac function.14 One study proposed an underlying molecular mechanism in breast cancer cells, showing that in serum-depleted medium, E2 stimulation causes relocation of ERα into mitochondria, where it interacts with mitochondrial ribonuclease P complex to control gene expression and translation.15 E2-ER complex regulates mitochondrial function and biogenesis by binding and activating the ERE on the nuclear respiratory factor-1 (NRF-1), which activates downstream molecules such as mitochondrial transcription factor to turn on gene expression.16,17 It has also been shown that GPR30 activation by its agonist G-1 inhibited mitochondrial permeability transition pore opening in mouse hearts subjected to ischemia-reperfusion.6 Further, E2 (5 μg/rat, 3 times/wk) has been shown to reverse ovariectomy (OVX)-induced mitochondrial structure damage, energy reduction, and increased reactive oxygen species.18
Figure 1.
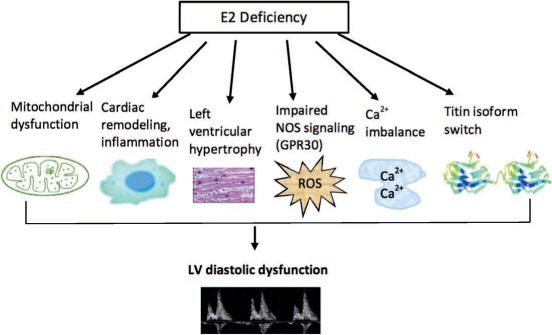
Mechanisms of action of estrogen (E2) in regulating left ventricular (LV) diastolic function. Loss of E2 leads to aggravation of mitochondrial dysfunction, inflammation and cardiac remodeling, LV hypertrophy, inhibition of nitric oxide signaling, Ca2+ imbalance, and titin isoform switch, all of which contribute to reduced diastolic relaxation despite normal ejection fractions. These critical mechanistic nodes are potential targets for the development of personalized therapies that treat diastolic dysfunction in postmenopausal women. NOS: nitric oxide synthase; ROS: reactive oxygen species; GPR30: G protein-coupled receptor 30
In a study by Chen et al., loss of ovarian E2 in cTnT-Q92 transgenic mice attenuated myocardial structure, energy production, and oxidative stress and resulted in downregulation of PPARα, PGC-1α, and NRF-1.19 Importantly, E2 replacement improved diastolic dysfunction in this model and showed a decreased E/A ratio and prolonged isovolumic relaxation time,19 both of which are significantly correlated with myocardial ATP levels. This further implicates that LVDD is closely dependent on mitochondrial energy production.19 In addition, fatty acid transporter proteins and AMP-activated protein kinase (AMPK) activity were increased by E2, suggesting that E2 regulates fatty acid and glucose metabolism, both of which may contribute to modified mitochondrial function. Recently, E2 was shown to restore AMPK activity in the hearts of OVX rats on high-fat diets.20 Taken together, the current studies suggest that ovarian E2 plays an important role in maintaining mitochondrial biogenesis and function through PPARα, PGC1α, NRF-1, and AMPK-mediated downstream signaling. Loss of ovarian E2 is associated with abnormal mitochondrial structure and function, oxidative stress, and diastolic dysfunction.
E2 Deficiency Induces Cardiac Remodeling in LVDD
Estrogen deficiency has been proposed to initiate cardiac inflammation and fibrosis followed by cardiac remodeling that subsequently results in LVDD.21 Loss of ovarian E2 by OVX induced a progressive increase of macrophage filtration and monocyte chemoattractant protein-1 (MCP-1) mRNA levels in the heart with aortic constriction (AC) from day 3 to day 7, and they returned to control levels by day 28; myocardial fibrosis with upregulated transforming growth factor (TGF)-β1 was observed at day 28. Concurrently, diastolic dysfunction occurred with an elevated LV end-diastolic pressure (LVEDP) and LV filling pressure.21 All of the results were attenuated by E2. Current studies further support that the inflammatory process initially contributes to the development of diastolic dysfunction, and suppression of inflammation reverses pathological and functional phenotypes; for example, MCP-1 neutralizing antibody or intercellular adhesion molecule-1 antibody were shown to inhibit macrophage infiltration, fibroblast proliferation, TGF-β upregulation, and myocardial fibrosis in AC rats.22 Importantly, the same antibodies were shown to prevent AC-induced diastolic dysfunction.22
Another investigation by Kuwahara showed that fibroblast activation in AC rats increased to maximum levels at day 3, and myocardial fibrosis and hypertrophy occurred by day 28.23 At that point, echocardiography indicated normal LV ejection fraction but reduced transmitral E/A ratio, and hemodynamic studies showed an increase of LVEDP without systolic dysfunction. The TGF-β mRNA expression was increased at day 3, peaked at day 7, and remained slightly elevated at day 28. Blocking of TGF-β by anti-TGF-β neutralizing antibody before AC inhibited fibrosis activation, collagen mRNA induction, and myocardial fibrosis.22 Interestingly, this antibody reversed diastolic dysfunction without affecting blood pressure and systolic function, suggesting that TGF-β plays a major role in the pathogenesis of myocardial fibrosis and diastolic dysfunction.22 Taken together, these data suggest that cardiac inflammation occurs at the early stage of E2 deficiency, and in turn, activation of profibrotic up-mediated signaling pathway (which contributes to cardiac remodeling) plays an important role in E2 deficiency-induced diastolic dysfunction. Inhibition of inflammation and fibrosis that occurs with E2 deficiency may be a potential therapeutic target to control LVDD with normal systolic function in the stressed heart.
E2 Deficiency Enhances Left Ventricular Hypertrophy in LVDD
Left ventricular hypertrophy (LVH) generally develops in response to pressure overload and progressively leads to loss of cardiac function.23 Clinical and basic studies have revealed that loss of E2 enhances LVH, and replacement of E2 prevents development of LVH in postmenopausal women and OVX female animal models.10,24 However, the underlying molecular mechanisms are incompletely understood. The reninangiotensin system (RAS) plays an important role in the development of cardiac hypertrophy, and inhibition of RAS is commonly used in the treatment of cardiac hypertrophy and heart failure. Previous studies show that E2 deficiency induced by OVX increases angiotensin II type 1 receptor (AT1R) expression and angiotensin-converting enzyme (ACE) activity, while E2 replacement downregulates AT1R expression and ACE activity in OVX rats and postmenopausal women.25,26 Either E2 or losartan, an antagonist of AT1R, inhibits OVX-induced cardiac hypertrophy and recovers force development in response to Ca2+ and isoproterenol in isolated LV papillary muscles.25 It is thought that inhibition of angiotensin II-induced cardiac hypertrophy by E2 is mediated by ERβ and not ERα since inhibition of only ERβ attenuates myocyte hypertrophy and reduces protein synthesis.27 Furthermore, this effect may be mediated by the calcineurin-NFAT (nuclear factor of activated T-cells) signaling pathway. Replenishment of E2 also attenuates prohypertrophic TGFβ signaling pathway through downregulation of miRNA levels such as miRNA-455 in OVX-induced transverse AC mice.28,29 E2 downregulates prohypertrophic (class I) histone deacetylases (HDACs) but upregulates antihypertrophic (class II) HDACs in angiotensin II-stimulated neonatal rat cardiomyocytes and angiotensin II-administrated mice.30 Either E2 or ERβ agonist inhibits HDAC 2 expression and phosphorylation and decreases prohypertrophic genes such as β-MHC and GATA4 induced by angiotensin II.
HFpEF is closely associated with hypertension and vascular stiffening. Estrogen is also known to influence blood pressure and vascular stiffening. In fact, mice with a global knockout of ERβ are hypertensive with increased vascular stiffening.31 Thus, HFpEF in postmenopausal women is likely driven by the combination of hypertension and loss of estrogen. TGF-β is one molecular candidate thought to mediate vascular stiffening since administration of neutralizing antibodies to TGF-β can attenuate diastolic dysfunction in a rodent model of pressure overload.32 It is possible that estrogen loss after menopause removes the inhibition of TGF-β, resulting in increased vascular stiffening that in turn aggravates diastolic dysfunction in HFpEF.
Mitogen-activated protein kinases—including ERK1/2, p38, and JNK—also play an important role in the development of cardiac hypertrophy. Hemodynamic studies found that OVX significantly accentuated pressure overload-induced LV hypertrophy and LVDD, showing an increase in LVEDP and rate of relaxation that was accompanied by activation of p38 MAPK, Akt, and nitric oxide synthase (NOS).33 Taken together, E2 plays an antihypertrophic role through modulation of multiple cellular signaling molecules such as AT1R, NFAT, HDAC, and MAPK, some of which are driven by ERβ. Thus, activation of ERβ offers a potential therapeutic intervention for LVH and LVDD.
Impairment of Nitric Oxide Pathway: Role of GPR30 in LVDD
To better understand the mechanisms underlying E2 deficiency-induced diastolic dysfunction, mRen2.Lewis rats were ovariectomized to establish an animal model that mimics the cardiac phenotype of postmenopausal woman.34 The study found that G1, a selective agonist of GPR30, is able to abolish abnormal cardiac structure, myocardial fibrosis, and diastolic dysfunction induced by OVX with an insignificant alteration of blood pressure.35 G1 also suppressed angiotensin II-mediated hypertrophy and increased antihypertrophic ANP gene, whereas G15, an antagonist of GPR30, abolished the effects of E2 and G1 on this animal model.35 Another study showed that E2-intact mRen2.Lewis rats with a high-salt diet exhibit early diastolic dysfunction compared to those on a normal-salt diet.36 In contrast, OVX rats on either a normal- or high-salt diet show impaired relaxation with evidence of elevated LV filling pressures or pseudonormalization that was attenuated by G1. In addition, researchers observed a decrease in nitric oxide and increased superoxides in OVX mRen2.Lewis rats that could be reversed with tetrahydrobiopterin (BH4), a major cofactor of nNOS catalytic subunit. BH4 also abolished diastolic dysfunction induced by loss of ovarian E2.37 Inhibition of nNOS by the selective nNOS inhibitor L-VNIO has been shown to reduce myocardial reactive oxygen species and elevate nitrite concentrations, mitigate cardiac remodeling, and improve diastolic function.38 This further suggests that cardiac-derived NOS may act as a prominent pathway in diastolic dysfunction induced by OVX in mRen2.Lewis rats. Estrogen receptor GPR30 activation, supplementation of BH4, and inhibition of NOS may provide insight into pharmaceutical strategies for treating diastolic dysfunction in postmenopausal women.
Altered Cellular Ca2+ from E2 Deficiency Contributes to LVDD
Alteration of cellular calcium homeostasis has been shown to influence cardiac systolic and diastolic performance. The process of cardiac diastole, including myocardial relaxation, requires reuptake of Ca2+ from cytosol into sarcoplasmic reticulum (SR) by the calcium-handling protein SR Ca2+ ATPase (SERCA2a) in the presence of ATP. Previous studies found that deprivation of ovarian E2 reduced the expression and activities of both SER-CA2a and ATPase, resulting in a decrease of SR Ca2+ reuptake in cardiomyocytes.39 Additionally, the density and protein level of cardiac b1-adrenergic receptor, cardiac sympathetic response, and myofibrillar Ca2+ sensitivity were increased. Thus, the loss of ovarian E2 leads to a prolonged and impaired relaxation process in cardiomyocytes. However, E2 replacement prevents intra-cellular Ca2+ alteration and improves myocardial relaxation.40 Taken together, deprivation of ovarian E2 leads to an imbalance of intracellular Ca2+ that results in LVDD with preserved ejection fraction.
Titin Involvement in LVDD Associated with E2 Loss
Sliding sarcomere proteins generate diastolic force in the stretched cardiac muscle. Titin is a giant elastic sarcomere protein and a prominent contributor to diastolic force that mediates the passive and active properties of cardiac muscle.41 Titin isoforms contain both N2B (stiff type) and N2BA (compliant type), and research shows that the mechanisms underlying cardiac diastolic dysfunction include the change of isoform expression and phosphorylation.41 Recent studies found that loss of ovarian E2 increases myocardial stiffness, elevating the ratio of N2BA to N2B titin isoform40 and matrix metalloproteinase 2 and decreasing maximum active tension.
Conclusion
Estrogen exhibits multiple mechanistic roles that affect various aspects of diastolic function. In this review, we discussed the impact of E2 on regulation of mitochondrial function, cardiac remodeling, cardiac hypertrophy, Ca2+ homeostasis, and titin isoform switches, all of which affect diastolic function. Further investigation of E2's underlying mechanisms can produce valuable leads in drug discovery and pharmaceutical strategies to combat diastolic heart failure in postmenopausal women. Since many of the specified mechanisms mentioned above are driven by only a single E2 receptor, it may be prudent to target one receptor for therapy and avoid the noncardiac effects of general E2 treatment. This approach will support the development of personalized medicine, in which the specific defective aspect of diastolic function can be treated with a specific ER agonist for a given patient.
Key Points
Estrogen has emerged as an important regulator of diastolic cardiac function in women.
Mechanisms by which loss of estrogen in menopause impacts diastolic function include: changes in mitochondrial metabolic function; induction of cardiac remodeling; enhancement of left ventricular hypertrophy; impairment of nitric oxide pathway/role of GPR30; alteration of cellular Ca2+; and alteration of titin.
Targeting restoration of estrogen and its signaling provides novel therapeutic strategies for managing and treating diastolic heart failure in postmenopausal women.
Conflict of Interest Disclosure
The authors have completed and submitted the Methodist DeBakey Cardiovascular Journal Conflict of Interest Statement and none were reported.
References
- 1. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM.. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006. July 20; 355( 3): 251– 9. [DOI] [PubMed] [Google Scholar]
- 2. Zile MR, Baicu CF, Gaasch WH.. Diastolic heart failure--abnormalities in active relaxation and passive stiffness of the left ventricle. N Engl J Med. 2004. May 6; 350( 19): 1953– 9. [DOI] [PubMed] [Google Scholar]
- 3. Gokce M, Karahan B, Erdol C, Kasap H, Ozdemirci S.. Left ventricular diastolic function assessment by tissue Doppler echocardiography in relation to hormonal replacement therapy in post-menopausal women with diastolic dysfunction. Am J Ther. 2003. Mar-Apr; 10( 2): 104– 11. [DOI] [PubMed] [Google Scholar]
- 4. Voutilainen S, Hippelainen M, Hulkko S, Karppinen K, Ventila M, Kupari M.. Left ventricular diastolic function by Doppler echocardiography in relation to hormonal replacement therapy in healthy postmenopausal women. Am J Cardiol. 1993. March 1; 71( 7): 614– 7. [DOI] [PubMed] [Google Scholar]
- 5. Gupte AA, Pownall HJ, Hamilton DJ.. Estrogen: an emerging regulator of insulin action and mitochondrial function. J Diabetes Res. 2015; 2015: 916585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bopassa JC, Eghbali M, Toro L, Stefani E.. A novel E2 receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2010. January; 298( 1): H16– 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang J, Green PS, Simpkins JW.. Estradiol protects against ATP depletion, mitochondrial membrane potential decline and the generation of reactive oxygen species induced by 3-nitroproprionic acid in SK-N-SH human neuroblastoma cells. J Neurochem. 2001. May; 77( 3): 804– 11. [DOI] [PubMed] [Google Scholar]
- 8. O'Lone R, Knorr K, Jaffe IZ, . et al. Estrogen receptors alpha and beta mediate distinct pathways of vascular gene expression, including genes involved in mitochondrial electron transport and generation of reactive oxygen species. Mol Endocrinol. 2007. June; 21( 6): 1281– 96. [DOI] [PubMed] [Google Scholar]
- 9. Alecrin IN, Aldrighi JM, Caldas MA, Gebara OC, Lopes NH, Ramires JA.. Acute and chronic effects of oestradiol on left ventricular diastolic function in hypertensive postmenopausal women with left ventricular diastolic dysfunction. Heart. 2004. July; 90( 7): 777– 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scheuer J, Malhotra A, Schaible TF, Capasso J.. Effects of gonadectomy and hormonal replacement on rat hearts. Circ Res. 1987. July; 61( 1): 12– 9. [DOI] [PubMed] [Google Scholar]
- 11. Pines A, Fisman EZ, Drory Y, . et al. Menopause-induced changes in Doppler-derived parameters of aortic flow in healthy women. Am J Cardiol. 1992. April 15; 69( 12): 1104– 6. [DOI] [PubMed] [Google Scholar]
- 12. Manolio TA, Furberg CD, Shemanski L, . et al. Associations of postmenopausal estrogen use with cardiovascular disease and its risk factors in older women. The CHS Collaborative Research Group. Circulation. 1993. November; 88( 5 Pt 1): 2163– 71. [DOI] [PubMed] [Google Scholar]
- 13. Stirone C, Duckles SP, Krause DN, Procaccio V.. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. 2005. October; 68( 4): 959– 65. [DOI] [PubMed] [Google Scholar]
- 14. Sivasinprasasn S, Sa-Nguanmoo P, Pratchayasakul W, Kumfu S, Chattipakorn SC, Chattipakorn N.. Obese-insulin resistance accelerates and aggravates cardiometabolic disorders and cardiac mitochondrial dysfunction in E2-deprived female rats. Age (Dordr). 2015; 37( 2): 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanchez MI, Shearwood AM, Chia T, Davies SM, Rackham O, Filipovska A.. E2-mediated regulation of mitochondrial gene expression. Mol Endocrinol. 2015. January; 29( 1): 14– 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem. 2008. December 15; 105( 6): 1342– 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM.. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol Endocrinol. 2008. March; 22( 3): 609– 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rattanasopa C, Phungphong S, Wattanapermpool J, Bupha-Intr T.. Significant role of estrogen in maintaining cardiac mitochondrial functions. J Steroid Biochem Mol Biol. 2015. March; 147: 1– 9. [DOI] [PubMed] [Google Scholar]
- 19. Chen Y, Zhang Z, Hu F, . et al. 17beta-estradiol prevents cardiac diastolic dysfunction by stimulating mitochondrial function: a preclinical study in a mouse model of a human hypertrophic cardiomyopathy mutation. J Steroid Biochem Mol Biol. 2015. March; 147: 92– 102. [DOI] [PubMed] [Google Scholar]
- 20. Bendale DS, Karpe PA, Chhabra R, Shete SP, Shah H, Tikoo K.. 17-β Oestradiol prevents cardiovascular dysfunction in post-menopausal metabolic syndrome by affecting SIRT1/AMPK/H3 acetylation. Br J Pharmacol. 2013. October; 170( 4): 779– 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mori T, Kai H, Kajimoto H, . et al. Enhanced cardiac inflammation and fibrosis in ovariectomized hypertensive rats: a possible mechanism of diastolic dysfunction in postmenopausal women. Hypertens Res. 2011. January 20; 34( 4): 496– 502. [DOI] [PubMed] [Google Scholar]
- 22. Kuwahara F, Kai H, Tokuda K, . et al. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension. 2004. April; 43( 4): 739– 45. [DOI] [PubMed] [Google Scholar]
- 23. Nediani C, Formigli L, Perna AM, . et al. Early changes induced in the left ventricle by pressure overload. An experimental study on swine heart. J Mol Cell Cardiol. 2000. January; 32( 1): 131– 42. [DOI] [PubMed] [Google Scholar]
- 24. Cleland JG, Swedberg K, Poole-Wilson PA.. Successes and failures of current treatment of heart failure. Lancet. 1998. August; 352 Suppl 1: SI19– 28. [DOI] [PubMed] [Google Scholar]
- 25. Ribeiro RF Jr, Pavan BM, Potratz FF, . et al. Myocardial contractile dysfunction induced by ovariectomy requires AT1 receptor activation in female rats. Cell Physiol Biochem. 2012; 30( 1): 1– 12. [DOI] [PubMed] [Google Scholar]
- 26. van Eickels M, Schreckenberg R, Doevendans PA, Meyer R, Grohe C, Schluter KD.. The influence of oE2-deficiency and ACE inhibition on the progression of myocardial hypertrophy in spontaneously hypertensive rats. Eur J Heart Fail. 2005. December; 7( 7): 1079– 84. [DOI] [PubMed] [Google Scholar]
- 27. Pedram A, Razandi M, Lubahn D, Liu J, Vannan M, Levin ER.. Estrogen inhibits cardiac hypertrophy: role of E2 receptor-beta to inhibit calcineurin. Endocrinology. 2008. July; 149( 7): 3361– 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu C, Dong S, Li Y.. Effects of miRNA-455 on cardiac hypertrophy induced by pressure overload. Int J Mol Med. 2015. April; 35( 4): 893– 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iorga A, Li J, Sharma S, . et al. Rescue of Pressure Overload-Induced Heart Failure by Estrogen Therapy. J Am Heart Assoc. 2016. January 22; 5( 1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pedram A, Razandi M, Narayanan R, Dalton JT, McKinsey TA, Levin ER.. Estrogen regulates histone deacetylases to prevent cardiac hypertrophy. Mol Biol Cell. 2013. December; 24( 24): 3805– 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pelzer T, Loza PA, Hu K, . et al. Increased mortality and aggravation of heart failure in estrogen receptor-beta knockout mice after myocardial infarction. Circulation. 2005. March 28: 111( 12): 1492– 8. [DOI] [PubMed] [Google Scholar]
- 32. Kuwahara F, Kai H, Tokuda H, . et al. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002. July 2; 106( 1): 130– 5. [DOI] [PubMed] [Google Scholar]
- 33. Bhuiyan MS, Shioda N, Fukunaga K.. Ovariectomy augments pressure overload-induced hypertrophy associated with changes in Akt and nitric oxide synthase signaling pathways in female rats. Am J Physiol Endocrinol Metab. 2007. December; 293( 6): E1606– 14. [DOI] [PubMed] [Google Scholar]
- 34. Zhao Z, Wang H, Jessup JA, Lindsey SH, Chappell MC, Groban L.. Role of estrogen in diastolic dysfunction. Am J Physiol Heart Circ Physiol. 2014. March 1; 306( 5): H628– 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang H, Jessup JA, Lin MS, Chagas C, Lindsey SH, Groban L.. Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res. 2012. April 1; 94( 1): 96– 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jessup JA, Lindsey SH, Wang H, Chappell MC, Groban L.. Attenuation of salt-induced cardiac remodeling and diastolic dysfunction by the GPER agonist G-1 in female mRen2.Lewis rats. PLoS One. 2010. November 3; 5( 11): e15433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jessup JA, Zhang L, Presley TD, . et al. Tetrahydrobiopterin restores diastolic function and attenuates superoxide production in ovariectomized mRen2.Lewis rats. Endocrinology. 2011. June; 152( 6): 2428– 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jessup JA, Zhang L, Chen AF, . et al. Neuronal nitric oxide synthase inhibition improves diastolic function and reduces oxidative stress in ovariectomized mRen2.Lewis rats. Menopause. 2011. June; 18( 6): 698– 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thawornkaiwong A, Pantharanontaga J, Wattanapermpool J.. Hypersensitivity of myofilament response to Ca2+ in association with maladaptation of estrogen-deficient heart under diabetes complication. Am J Physiol Regul Integr Comp Physiol. 2007. February; 292( 2): R844– 51. [DOI] [PubMed] [Google Scholar]
- 40. Bupha-Intr T, Oo YW, Wattanapermpool J.. Increased myocardial stiffness with maintenance of length-dependent calcium activation by female sex hormones in diabetic rats. Am J Physiol Heart Circ Physiol. 2011. May; 300( 5): H1661– 8. [DOI] [PubMed] [Google Scholar]
- 41. Fukuda N, Terui T, Ishiwata S, Kurihara S.. Titin-based regulations of diastolic and systolic functions of mammalian cardiac muscle. J Mol Cell Cardiol. 2010. May; 48( 5): 876– 81. [DOI] [PubMed] [Google Scholar]