

Abstract
The membrane proximal external region (MPER) of HIV-1 gp41 is targeted by broadly neutralizing antibodies (bnAbs) 4E10 and 10E8. In this proof-of-concept study, we evaluated a novel multi-immunogen vaccine strategy referred to as Incremental, Phased Antigenic Stimulation for Rapid Antibody Maturation (IPAS-RAM) to induce 4E10/10E8-like bnAbs. Rabbits were immunized sequentially, but in a phased manner, with three immunogens that are progressively more native (gp41-28×3, gp41-54CT, and rVV-gp160DH12). Although nAbs were not induced, epitope-mapping analyses indicated that IPAS-RAM vaccination was better able to target antibodies towards the 4E10/10E8 epitopes than homologous prime-boost immunization using gp41-28×3 alone. MPER-specific rabbit monoclonal antibodies were generated, including 9F6. Although it lacked neutralizing activity, the target epitope profile of 9F6 closely resembled those of 4E10 and 10E8 (671NWFDITNWLWYIK683). B-cell repertoire analyses suggested the importance of co-immunizations for maturation of 9F6, which warrants further evaluation of our IPAS-RAM vaccine strategy using an improved priming immunogen.
Keywords: HIV-1, vaccine, gp41, MPER, rabbit, neutralizing antibody, antibody maturation, next-generation sequencing, NGS
INTRODUCTION
To date, dozens of human monoclonal antibodies (mAbs) have been isolated from virus-infected patients that can neutralize a large number of HIV-1 variants from multiple clades (Huang et al., 2014; 2012; Pejchal et al., 2011; Scheid et al., 2011; Walker et al., 2011; 2009; Wu et al., 2010; Zwick et al., 2001). These broadly neutralizing antibodies (bnAbs) target a few select conserved sites of vulnerability on viral envelope glycoproteins gp120 and gp41 (for reviews, see (Georgiev et al., 2013; Haynes et al., 2014; Kwong et al., 2013; Mascola and Haynes, 2013; Mascola and Montefiori, 2010; McCoy and Weiss, 2013; van Gils and Sanders, 2013)). One of these targets is the membrane proximal external region (MPER), a highly conserved domain of ~22 amino acid residues situated at the C-terminal end of the gp41 ectodomain. The MPER is thought to play a critical role during the fusion between viral and cellular membranes (Muñoz-Barroso et al., 1999; Salzwedel et al., 1999). It is targeted by bnAbs 2F5, Z13e1, 4E10 and 10E8 (Huang et al., 2012; Purtscher et al., 1994; Stiegler et al., 2001; Zwick et al., 2001). 4E10 and 10E8 are particularly notable as they have been shown to neutralize ~98% of the HIV-1 isolates tested (Huang et al., 2012). 4E10 and 10E8 epitopes lie within the C-terminal 13 residues of the MPER (671NWFDITNWLWYIK683) and their crystal structures have been determined (Cardoso et al., 2007; Huang et al., 2012).
Despite having short, linear, simple alpha-helical epitopes, efforts to develop a vaccine that can induce 4E10/10E8-like bnAbs have been unsuccessful (see (Banerjee et al., 2016; Habte et al., 2015) and references therein). Since all of the immunogens evaluated could bind 4E10/10E8, the failure to induce similar bnAbs is not because antigens could not assume the correct epitope structures. Rather, it is likely due to the difficulty in inducing high levels of MPER-directed antibodies that can bind the neutralizing epitopes in the context of a whole trimeric gp120/gp41 complex. Unfortunately, this problem cannot be remedied simply by using a trimeric envelope complex because the MPER is immunorecessive compared to other epitopes that elicit type-specific or non-neutralizing antibodies. An additional challenge in inducing MPER neutralizing antibodies is that the structure of the gp41 subunit is highly dynamic as it undergoes significant structural changes to mediate fusion between viral and cellular membranes (Melikyan, 2008). The conformation, orientation and accessibility of the MPER neutralizing epitopes likely vary significantly at different stages of the fusion process, about which little is known at the present time. Further, the orientation of the MPER relative to the membrane surface or to the rest of the native gp120/gp41 trimeric complex is unknown. These factors make it difficult to design small, sub-domain immunogens.
Taken together, the major challenge in developing an MPER-based vaccine is designing immunogens and/or developing vaccine strategies that both force the immune system to focus antibody responses towards the MPER and also guide antibody evolution so that mature antibodies bind neutralizing epitopes on trimeric envelope spikes on the virus particles. Considering that antibody maturation will have to occur during a relatively short timeframe, we postulated that inducing bnAbs against the MPER would be impossible to accomplish with a single immunogen using typical vaccine approaches. To address this problem, we devised a novel vaccine strategy, referred to as Incremental, Phased Antigenic Stimulation for Rapid Antibody Maturation, or IPAS-RAM. The basic concept is to prime the immune system using a small MPER-derived peptide to stimulate a broad spectrum of antibodies against the MPER, then selectively amplify those that bind the native structure by boosting with progressively more native immunogens. Although a number of studies recently reported sequential immunization with different immunogens, they used only a single immunogen during each immunization (Briney et al., 2016; Escolano et al., 2016; Haynes et al., 2012; Sok et al., 2016; Tian et al., 2016). What makes our IPAS-RAM strategy unique is that the immune system is exposed to different, but related, immunogens simultaneously in a phased manner, such that B cells stimulated by a smaller immunogen can concurrently engage common epitopes on a larger, more native immunogen. By repeating this process using incrementally more native antigens, we hypothesized that the immune system can better target MPER neutralizing epitopes and that antibodies could undergo the maturation process more efficiently.
In this proof-of-concept study, we evaluated the IPAS-RAM vaccine strategy using three immunogens in rabbits: An MPER-based polypeptide, a membrane-bound gp41 mini-protein, and a full-length gp160. We hypothesized that a peptide-based priming antigen would be highly effective in eliciting antibodies against the MPER, and that boosting with progressively larger and more “native” antigens would enable select antibodies to mature into bnAbs capable of binding gp41 as it appears in the native trimer. Although we did not succeed in eliciting neutralizing antibodies, results of our study demonstrate proof-of-principle for the IPAS-RAM vaccine strategy and identify ways to improve it.
RESULTS
Immunogens
To focus antibody responses towards the MPER, we generated an immunogen designated gp41-28×3, which consists of three tandem copies of the C-terminal 28 a.a. of gp41 ectodomain (Fig. 1A). The immunogen was produced initially as a fusion protein (HR1-HR2-28×3) by adjoining 28×3 to the HR1-HR2 regions of gp41, which forms a six-helix bundle (6HB) (Shi et al., 2010). This was done because we had observed that HR1-HR2 6HB allows high level protein expression in E. coli (Fig. 1B; unpublished data). The HR1-HR2 portion was subsequently removed by thrombin digestion (Fig. 1C). Three 28-mer peptides were linked together to increase its immunogenicity without requiring conjugation to a heterologous carrier protein. All of the bnAbs tested (2F5, Z13e1, 4E10 and 10E8) bound gp41-28×3 (Fig. 1D). However, 10E8 binding was about ~50-fold weaker than 2F5 and ~10-fold weaker than Z13e1 and 4E10, suggesting that the conformation of the 10E8 epitope may not be optimal.
Fig. 1. Immunogens used in the study.
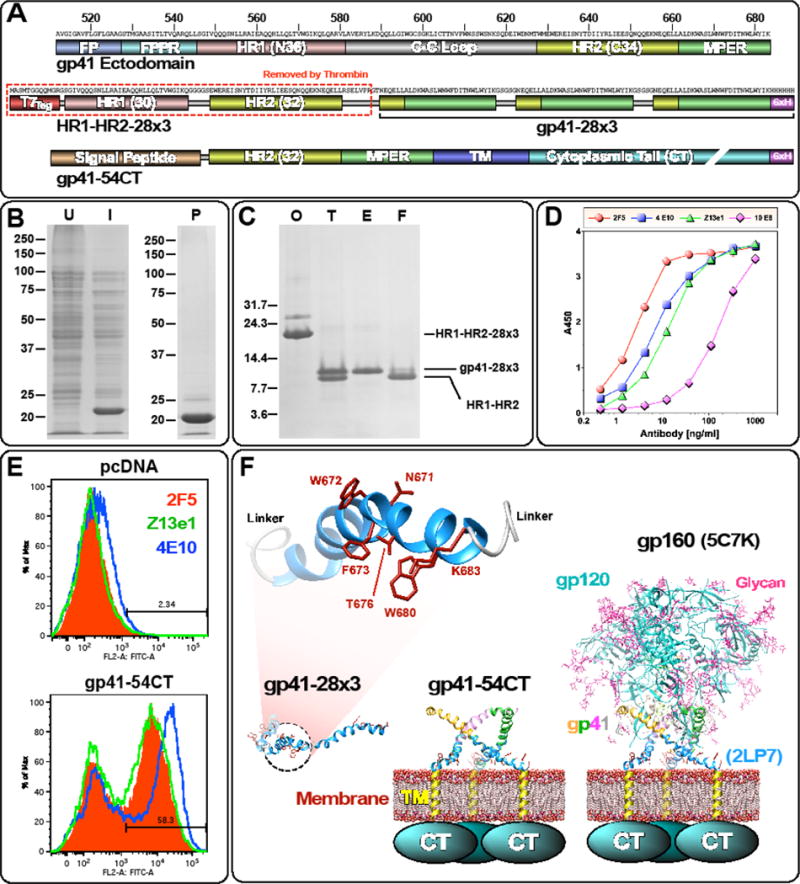
(A) Schematic diagrams of gp41-28×3 and gp41-54CT. The entire gp41 ectodomain is shown on the top as a reference. To generate gp41-28×3, the T7Tag and HR1-HR2 portion of HR1-HR2-28×3 is removed with thrombin. (B) Expression and purification of HR1-HR2-28×3. Coomassie stained SDS-PAGE gels (U: uninduced; I: induced; P: purified). (C) Cleavage of HR1-HR2-28×3 with thrombin (O: original sample; T: thrombin cleaved; E: eluted from Ni-NTA column; F: flow through). (D) ELISA of gp41-28×3 with 2F5, 4E10, Z13e1 and 10E8. (E) Flow cytometry analyses of cell surface expression of gp41-54CT. 2F5, Z13e1 and 4E10 were used to probe the antigen. (F) Structural models of three immunogens are shown to illustrate their relative size. NMR structure of 28-mer peptide (2LP7; (Reardon et al., 2014)) and BG505 SOSIP gp140 structure (5C7K; (Kong et al., 2015)) were used. Structures were rendered using Chimera (Pettersen et al., 2004).
The conformation, orientation, and/or accessibility of the neutralizing epitopes on the MPER are likely affected by the membrane surface as well as other proximal gp41 domains (Irimia et al. 2016; Montero et al., 2012; Rujas et al., 2015). To present the MPER in a more native-like setting, we generated a second immunogen designated gp41-54CT. It comprises the C-terminal 54 a.a. of the gp41 ectodomain that includes the HR2 domain and the MPER, along with the transmembrane domain and the cytoplasmic tail (CT). We hypothesized that this immunogen would selectively amplify a subset of antibodies induced by gp41-28×3 that could bind the MPER in the context of the membrane surface and HR2. HEK-293T cells transfected with the plasmid encoding gp41-54CT could be detected using 2F5, Z13e1 and 4E10 by flow cytometry analyses (Fig. 1E), indicating cell surface expression of the protein and correct conformation of neutralizing epitopes. Both gp41-54CT and gp41-28×3 are based on M group consensus sequence (Gao et al., 2005). For the final boost, we generated a recombinant vaccinia virus expressing the full-length gp160 of the HIV-1DH12 strain (rVV-gp160DH12). A schematic diagram showing the relative sizes of the three immunogens are illustrated in Fig. 1F.
Immunization and evaluation of antibody responses
To evaluate IPAS-RAM vaccine strategy, rabbits (R1, R2 and R3) were first immunized with gp41-28×3 only (Fig. 2A). Four weeks later, a combination of gp41-28×3 and gp41-54CT was administered, with the latter delivered by DNA electroporation. Instead of immunizing with just gp41-54CT, gp41-28×3 was also included because we postulated that immunizing with both immunogens would preferentially stimulate antibody responses towards epitopes present on both immunogens (i.e. the C-terminal 28 a.a.). Similarly, on week 11, a combination of gp41-54CT and rVV-gp160DH12 was administered. After the first immunization, the antibody titers reached >103 and the titers increased by about 100 fold after the second immunization (Fig. S1A). However, because significant increases in antibody responses were not observed after the third immunization, a fourth immunization was given on week 29 using gp41-28×3 and rVV-gp160DH12. The long resting period was given so that antiviral immune responses against the vaccinia virus vector induced after the third immunization would subside. Antibody responses increased only slightly after the fourth immunization.
Fig. 2. Immunization schedule and linear epitope mapping analyses.
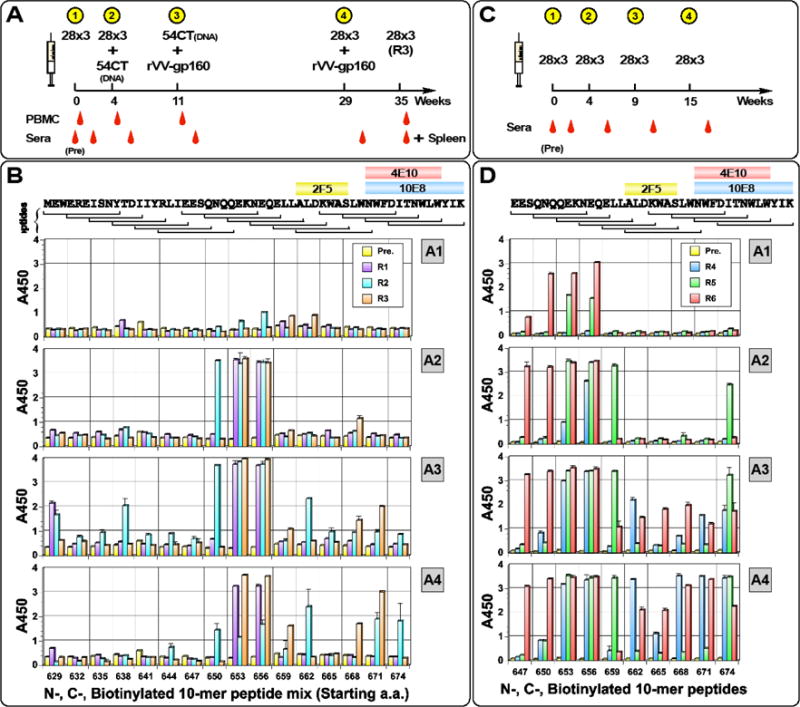
(A) Timeline for IPAS-RAM immunization and sampling. Rabbits were immunized on weeks 0, 4, 11 and 29. Pre-immune, as well as post-immune sera (two weeks post each immunization) were taken. PBMC were collected four days after immunization (except after 4th) for antibody repertoire analyses. To generate hybridomas, Rabbit R3 was immunized intravenously on week 35 with gp41-28×3 and spleen was collected four days later. (B) Immunogenic, linear epitopes were identified by ELISA using overlapping “10-mer” peptides. Serum samples collected two weeks after first (A1), second (A2), third (A3) and fourth (A4) immunizations were analyzed. A mixture of N- and C-terminally biotinylated peptides spanning the C-terminal 54 a.a. of gp41 ectodomain was used (BGGXXXXXXXXXX and XXXXXXXXXXGGKB, respectively; B=biotin, X=gp41 sequence). Pre-immune serum was used as a negative control. Horizontal brackets on top indicate the sequence for each peptide and core-binding epitopes for bnAbs 2F5, 4E10 and 10E8 are shown. The numbers on the X-axis indicate the starting a.a. position of “10-mer” peptides. Timeline (C) and epitope mapping analyses (D) for the homologous prime-boost immunization group with gp41-28×3.
To identify immunogenic linear epitopes, ELISAs were conducted with overlapping 10-mer peptide sets biotinylated either at the N- or C-terminal ends as we previously reported (Banerjee et al., 2016; Habte et al., 2015). After the first immunization, very little response was detected (Fig. 2B). However, after the second immunization, strong antibody responses were detected against peptides in the cluster II immunodominant region just upstream of the 2F5 epitope (peptides 653 and 656; also peptide 650 for rabbit R2). After the third immunization with gp41-54CT and rVV-gp160DH12, the cluster II region still remained immunodominant. However, antibody responses appeared against other linear epitopes. R2 showed good binding to peptide 662ALDKWASLWN671 containing the 2F5 epitope. This rabbit also showed low level reactivity against other C-terminal peptides (665, 668, 671 and 674). Antiserum from R3 bound to peptides 668SLWNWFDITN677 and 671NWFDITNWLW680 that contain parts of the 4E10 and 10E8 epitopes. R1 and R2 also recognized additional peptides within the HR2 domain (peptides 629 and 638). Except for peptides 653 and 656, antibody responses against peptides exhibited animal-to-animal variation. The fourth immunization with gp41-28×3 and rVV-gp160DH12 further enhanced anti-MPER antibodies in R2 (peptides 668 and 671) and R3 (peptides 671 and 674). Interestingly, antibody responses against many of the upstream peptides (629, 638, 650, 653 and 656) diminished significantly for R2.
To examine whether antibodies induced by the IPAS-RAM vaccine strategy are different from those induced by a typical homologous prime-boost immunization, another group of rabbits (R4, R5 and R6) was immunized with gp41-28×3 (Fig. 2C). Strong antibody responses were induced after the first immunization (~104 to ~105; Fig. S1B). Since these rabbits received the same vaccination as the animals in the IPAS-RAM group, the higher antibody titers suggest animal-to-animal variations. Antibody titers continued to increase with each successive immunization (Fig. S1B). Not surprisingly, 10-mer peptides 653 and 656 were most immunogenic (Fig. 2D), which might be the reason why they were also immunogenic in the IPAS-RAM group. After the fourth immunization, high-level antibodies were detected against peptide 674 all three animals. R4 and R6 mounted strong antibody responses against the 671 peptide that contains 4E10/10E8 epitopes. Overall, the gp41-28×3 homologous prime-boost immunization induced more robust antibody responses against gp41 MPER than the IPAS-RAM vaccine. Interestingly, antibodies from rabbit R6 reacted strongly to peptides 647 even though gp41-28×3 does not extend this far out into the N-terminus. One likely possibility is that antibody responses against 656NEQE659 could be cross-reacting against 651NQQE654. Another possibility is that our gp41-28×3 preparation could have minor contamination of the HR1-HR2 fragment or undigested HR1-HR2-28×3.
Antibody responses against the C-terminal 13 a.a. residues of gp41 MPER
Despite the overwhelming immunodominance of the cluster II region, antibody responses were induced towards the C-terminal end of the gp41 ectodomain, where 4E10/10E8 epitopes are located, in five of the six rabbits. However, no serum neutralizing activity was detected (data not shown). Regardless, we were interested in determining whether antibodies against the C-terminal 13 a.a. were qualitatively different between the two vaccine groups. We examined which amino acid residues were targeted by ELISA using two panels of alanine mutant peptides. First, we used peptides that were biotinylated at the C-terminal K683 (Fig. 3A and 3B) as previously described (Banerjee et al., 2016; Habte et al., 2015). As noted previously, the D674A mutation affects the helical conformation of the peptide (Banerjee et al., 2016; Habte et al., 2015); thus, results should be interpreted with some caution. For R2, the four residues that were most affected besides D674 were F673, N677, W678 and L679. Residues important in R3 were N671, W672, F673 and N677; the first three of which are absolutely critical for 10E8. Of all the immunogens we evaluated to date (Banerjee et al., 2016; Habte et al., 2015), this is the first time we targeted all three of these residues. For R4, the pattern was similar to R3, except that W6762 was not targeted. The epitope targeting profile for R5 was quite unique in that antibodies targeted W680 and Y681. The overall pattern for R6 resembled that of R2, except for not targeting T676 and N677.
Fig. 3. Detailed epitope mapping analyses of immune sera.
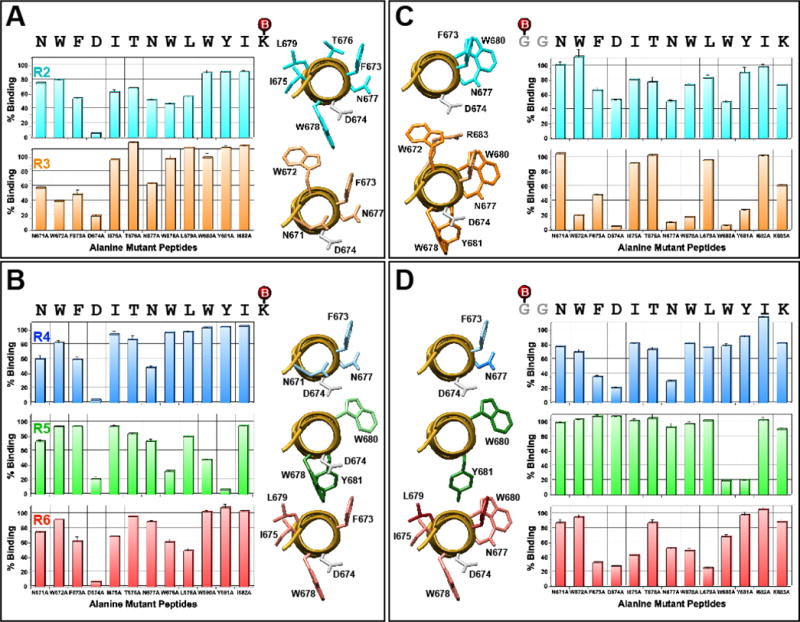
ELISAs were conducted using a set of C-(panels A and B) or N-terminally (panels C and D) biotinylated 13-mer alanine mutant peptides to assess impact of the mutation on antibody binding in comparison to the unmutated peptide. Serum samples collected after the fourth immunization were used. Peptides were biotinylated (shown as red spheres) at the primary amine of C-terminal lysine or N-terminal glycine. Graphic views of residues that affected binding when mutated are shown. An axial view of N671 to R683 segment of the 28-mer peptide co-crystalized with 10E8 is shown (PDB: 4G6F). Residues that showed <70% binding are shown. Residues that are more critical (<35%) are shown in darker tone. D674 is shown in grey.
Because peptides that were biotinylated at the C-terminal K683 might not allow binding of some antibodies (e.g. those that approach the peptide from the C-terminal end), the analyses were repeated using peptides that were biotinylated at the N-terminus with a two glycine spacer (Fig 3C and 3D). As expected, somewhat different patterns were observed, especially for R3, which showed marked difference (>40%) for residues N671, N677, W678, W680, Y681 and K683. For rabbits R4, R5 and R6, however, many of the targeted residues identified using one peptide set are simply a subset of residues identified using the other peptide set. For example, for R6, the important residues were F673, I675, W678 and L679 using peptides biotinylated at the C-terminus while additional N677 and W680 were identified using peptides biotinylated at the N-terminus.
Generation and characterization of MPER-specific monoclonal antibodies
Although target epitope profiling with whole serum provides some sense of which residues are critical for antibody binding, it does not provide clear information due to the polyclonal nature of antibodies. To better evaluate MPER-directed antibodies in R3 that might be targeting the 671NWF673 residues, and to gain insights into how we might improve immunogens and/or vaccine strategies, MPER-specific mAbs were generated and characterized. Hybridomas were screened initially with gp41-28×3, and then with 15-mer peptides containing epitopes for 2F5 (657EQELLALDKWASLWN671) and 4E10/10E8 (669LWNWFDITNWLWYIK683). Based on the level of reactivity and epitope targeting profiles, three hybridomas (6C10, 9F6 and 21B5) were selected for detailed characterization. Based on the binding to various 15-mer peptides, the epitope for 6C10 was mapped approximately to 657EQELLAL663, just upstream of the 2F5 epitope (Fig. 4A). Since 6C10 epitope is situated within the two 10-mer peptides that were most reactive (653QEKNEQELLA and 656NEQELLALDK; Fig. 2), 6C10 could represent the most predominant antibody induced.
Fig. 4. Epitope mapping analyses of mAbs.
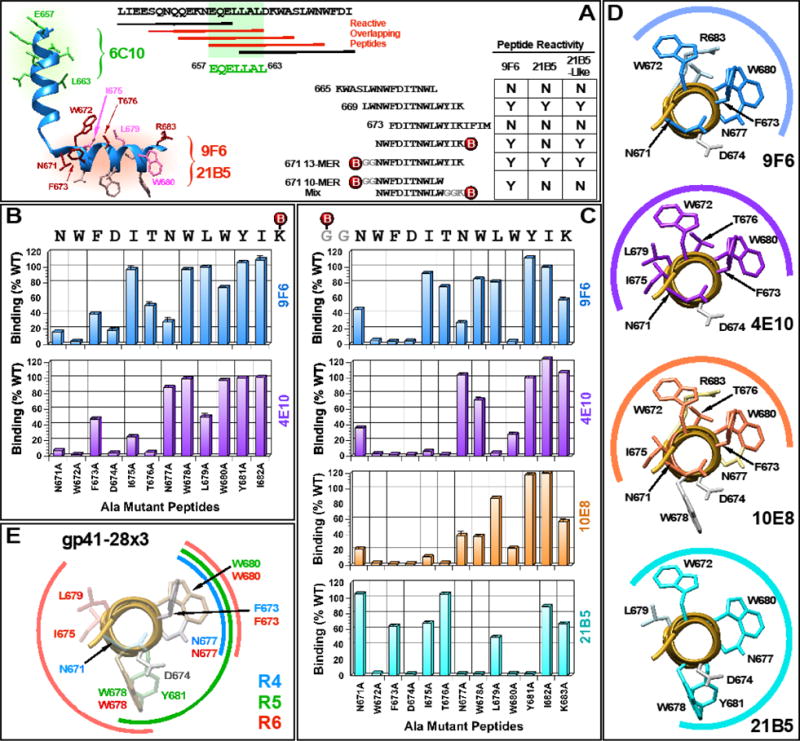
(A) Linear epitope mapping of 6C10, 9F6, 21B5 and twelve 21B5-like mAbs using overlapping peptides. A structure of a 28-mer peptide co-crystallized with 10E8 (PDB: 4G6F) is shown to illustrate general locations of the epitopes. (B and C) Fine-epitope mapping analyses using C- or N-terminally biotinylated alanine mutant peptides. (D) Graphic presentations of residues important for 9F6 and 21B5 binding. 4E10 and 10E8 are included for comparison. (E) A summary of all residues targeted by antibodies induced by immunization with gp41-28×3 alone. Data from all three rabbits (R4, R5 and R6) are compiled.
Although 9F6 and 21B5 strongly bound 669LWNWFDITNWLWYIK683, both failed to bind upstream (665KWASLWNWFDITNWL679) and downstream (673FDITNWLWYIKIFIM686) peptides (Fig. 4A). This result suggested that the critical residues are also present within the four terminal residues at either ends of the 669-peptide (i.e. LWNW and WYIK). Alternatively, but not exclusively, the presence of four residues on 665- and 673-peptides at the N- or C-terminal ends, respectively (i.e. KWAS and IFIM), could be interfering with antibody binding. Additional mapping analyses with shorter peptides indicated that 9F6 epitope resided within 671NWFDITNWLW680, whereas 21B5 epitope extended further downstream. 21B5 bound the N-terminally biotinylated GGNWFDITNWLWYIK683 peptide, but not the 671NWFDITNWLWYIK683 peptide biotinylated at the C-terminal lysine, suggesting that the antibody binds near K683 and that biotin sterically interferes with access. Interestingly, we isolated twelve 21B5-like hybridomas that bound both the N- and C-terminally biotinylated 13-mer peptide.
Detailed epitope mapping of 9F6 and 21B5 mAbs
To better define the 9F6 and 21B5 epitopes, ELISAs were performed using the two panels of scanning alanine mutant peptides as described in Fig. 3. For comparison, 4E10 and 10E8 were also analyzed. As shown in Fig. 4B, mutating N671, W672, F673, T676 and N677 residues to alanine markedly diminished 9F6 binding when tested with C-terminally biotinylated peptides. To a lesser degree, mutating W680 also reduced binding. This target profile was remarkably similar to that of 4E10, except for I675 and L679, which were recognized by 4E10 but not by 9F6. Conversely, 9F6 targeted N677 whereas 4E10 did not. A similar targeting profile was observed when N-terminally biotinylated peptides were used (Fig. 4C). However, one major difference was the W680A mutation, which showed a severe binding defect for both 9F6 and 4E10 using the N-terminally biotinylated peptide. It should be noted that while mutating W680 only moderately reduced peptide binding (Brunel et al., 2006), it is important for neutralization by 4E10 (Brunel et al., 2006; Zwick et al., 2005). Interestingly, the K683A mutation reduced 9F6 binding to 60% of the wild type even though the 671 10-mer peptide (671NWFDITNWLW680) was sufficient to bind the mAb efficiently. The results are graphically summarized in Fig. 4D, which show significant overlap between 9F6 and 4E10/10E8 binding.
Overall, the antibody targeting profile of 10E8 was fairly similar to that of 4E10, although there were some notable differences (e.g. L679). Mutated residues that most severely diminished 10E8 binding were N671, W672, F673, I675, T676 and W680. To a lesser extent, mutating N677 and W678 also reduced recognition. The reduced binding by the W678A mutation was somewhat unexpected since it resides on the opposite side of the neutralization face, although Huang et al. (Huang et al., 2012) reported that mutating this residue reduced peptide inhibition of 10E8 neutralization by ~20%. Also unexpected was the reduction of only 40% for the K683A mutation, when R683 was critical (Huang et al., 2012). Similarly, the L679A mutation resulted in only a slight reduction when it had previously shown to exhibit a moderate effect (~50% reduction in neutralization inhibition) (Huang et al., 2012).
The five most critical residues for 21B5 binding were W672, N677, W678, W680 and Y681. Thus, in contrast to 9F6, 21B5 targeted W678 and Y681, both which are located directly on the opposite side of the neutralization face (Fig. 4D). Conversely, 21B5 failed to target N671 and only weakly bound F673, both of which are targeted by 9F6, as well as 4E10 and 10E8. Overall, 9F6 better resembled 4E10/10E8 than 21B5.
Sequence analysis of MPER-binding mAbs
To determine the origin of MPER-specific mAbs, antibody genes were PCR-amplified from hybridomas, cloned and sequenced. Analyses indicated that 6C10 heavy chain was derived from IGHV1S45*01 germline, while 9F6 and 21B5 originated from IGHV1S40*01(Fig. S2A–C). 6C10 and 21B5 kappa chains were derived from the IGKV1S32*01 and IGKV1S44*01 germlines, respectively. Unfortunately, we have not been able to recover a productive light chain from the 9F6 hybridoma using known primer sets for either kappa or lambda chains, despite exhaustive efforts. We suspect our primers may not bind the 9F6 light chain, due to either somatic mutations in the primer-binding site or suboptimal design based on incomplete information about the rabbit germline repertoire. Among the three mAbs, the heavy chain of 9F6 was the most conserved to germline, sharing 96.53% nucleotide identity, followed by 6C10 and 21B5 (91.32% and 87.85%, respectively). Similarly, the kappa chain of 6C10 was less divergent than that of 21B5 (90.32% and 89.25%, respectively). Surprisingly, high levels of mutations were observed in FR1 for 6C10 and 21B5. The importance of these mutations in antibody function has not been determined.
Of the twelve 21B5-like mAbs that could bind C-terminally biotinylated 671-13mer peptide, five representative hybridomas (21B6, 6F9, 31A4, 27A1 and 17-4H2) were selected for further characterization. Sequence analyses showed that all five mAbs belonged to the same clonal family as 21B5, as determined by predicted germline, junction, and CDR3 identity (Fig. S2D). It should be noted, however, that IMGT predicted 21B6 heavy chain might be derived from the IGHV1S45*01, which is highly related to IGHV1S40*01 and differ only at the very 5′ end of the V-gene. Although convergent affinity maturation of antibodies derived from different germlines is a possibility, we suspect a more likely scenario is a gene conversion event between the two V genes following V-D-J recombination. The heavy chains of 21B5-like mAbs were less divergent from the germline than 21B5 (91.93%–97.22% identity). Differences from germline were most highly concentrated in the CDR2. The kappa chains were more divergent than the heavy chains, with germline identity ranging from 87.81% to 89.96%. Of note, when comparing similarities versus differences of 21B5 to the other clonal family members (Fig. S2D, cyan highlights versus yellow), mutations in the kappa chain were much more conserved across the family than the heavy chain, where 21B5 represents a more distinctive sequence.
Bioinformatics analyses of B cell repertoire and antibody maturation
To gain insights into possible maturation pathways of the MPER-specific mAbs, the antibody repertoire was analyzed by NGS of PBMC collected four days after the first three immunizations (A1, A2, A3), as well as terminal PBMC (TP) and spleen (TS). A total of 13.3M raw reads were obtained, from which 290,848 unique productive heavy chain and 89,760 kappa chain sequences were derived (Table S1). The lambda chain was not sequenced, as it is underutilized in rabbits (Appella et al., 1974). Clonally related heavy chain sequences were inferred, and the full-length sequences related to MPER-specific mAbs were determined (Table S2). A total of 19,133 clonally related sequence clusters were found. Of these, 10,924 were represented in more than one sample (Fig. S3). Heavy chains used primarily two V-genes (IGHV1S40 and IGHV1S45) and J-gene IGHJ4 (Fig. S4). D-gene usage was more diverse. The most commonly used V-gene for the kappa chain was IGKV1S32 (Fig. S5). Note that the apparent predominant usage of kappa J-gene IGKJ1-2 is an artifact from amplification with a 3′ primer that closely resembles the IGKJ1-2*01 sequence.
The interrelationships between all sequenced antibody heavy chains (“heavy chain antibodyome”) are graphically shown in Fig. 5A, and the clonal families of mAbs 6C10, 9F6 and 21B5 are highlighted. CDR3 spectratypes of the heavy and kappa chains are plotted in Fig. 5B. The non-Gaussian distribution of the CDR3 length suggests active antibody developments. While κCDR3 length remained fairly constant, peaking at 12 a.a., significant variation in HCDR3 length was observed after each immunization, indicating fluctuations in antibody repertoires generated in response to different antigenic stimulations. After the second immunization, when a strong anamnestic response against gp41-28×3 was observed (Fig. 2B), the peak HCDR3 length changed from 13 to 14 a.a. (Fig. 5B). It is worth noting that, 6C10 has a HCDR3 length of 14 a.a. (ARDLDDVIGWNFGW), suggesting that the shift in the peak HCDR3 length could be due to a strong antibody response against the cluster II immunodominant region (peptides 653 and 656; Fig. 3). Consistent with this notion, IGHV1S45 and IGKV1S32, from which 6C10 heavy and light chains were derived, became the most dominant V-genes used after the second immunization (Fig. S4, S5). After the third immunization, there was a notable shift from 14 back to 13 a.a, which could be due to antibody responses directed against new antigens (e.g. vaccinia virus antigens). The dual peaks in the terminal PBMC sample (TP) may be the result of the fourth immunization with gp41-28×3 and rVV-gp160DH12.
Fig. 5. Clonal and phylogenetic analysis of the heavy chain repertoire.
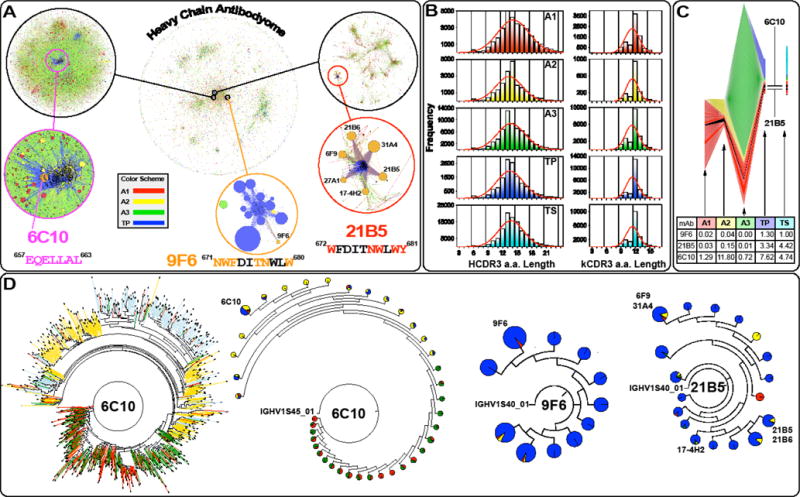
(A) Heavy chain antibodyome: Each unique CDR3 sequence is represented as a point, colored by its sampling time (A1-A3: PBMC after the first, second and third immunizations; TP: terminal PBMC). Due to large amount of data, the analyses was limited to antibodies recovered from PBMC only. The same color-coding is used throughout the figure. Sequences forming clonal families are joined by lines. The approximate locations of 6C10, 9F6 and 21B5 mAb families are shown in expanded detail (Note: they are zoomed at different levels). The presence of multiple large clonal families indicates diverse responses to immunizations. Families remote from the isolated mAbs are dominated by reads from the A3 sample. (B) Heavy and kappa chain CDR3 spectratypes are shown for each sample. The distribution shows distinct and different skews in samples A2 and A3, and a composite of the two in sample TP, suggesting that clonal populations stimulated at the earlier timepoints have been integrated into longer term memory. (C) Antibody clone dynamics colored by sampling time and earliest identification of clonotypes. The overall band height at each sample point is proportional to the number of novel CDR3 nucleotide sequences in that sample, normalized to account for variation in sequencing depth. The height of each individual band (representing a single clonotype) is proportional to the number of novel CDR3 nucleotide sequences identified in that clonotype. Black bands indicate the 6C10 and 21B5 clonal families. The underlying table shows the percentage of novel CDR3s that are clonally related to the indicated mAb. In both cases, CDR3s found in multiple timepoints are counted only in the earliest (or leftmost) sample and clonal relationship is inferred by CDR3 sequence identity alone. (D) Dendrograms of mAb clonal families (inferred by CDR3 sequence identity and V-/J-gene origin), in which pie charts indicate by size the number of descendants at each node, and by color their timepoint origin (only PBMC samples are included). The dendrogram for 6C10 is shown for comparison in both the conventional and accumulated (pie chart) form. The limited representation of earlier timepoints in the 21B5 and 9F6 charts suggests that development of these clonal families occurred almost exclusively after the A3 sampling time. By contrast, the dendrogram for the Ab 6C10 clonal family shows development at each timepoint, suggesting that refinements have been introduced by successive immunizations. Dendrograms for the three mAbs are shown at different zoom levels. Thus, their size should not be compared between different mAbs.
The development of the B cell repertoire after the third immunization was dominated by novel clonal families (Fig. 5C). This dominance could also be seen in Fig. 5A, where a large number of distinct clusters are predominantly associated with the A3 sample. The vast majority of the novel clonal families are expected to be antibodies against vaccinia virus antigens. Consequently, the proportions of clonal family sizes of the MPER-specific mAbs all contracted. Evaluation of the size of clonal family sizes showed clear immunodominance of 6C10 over both 21B5 and 9F6 epitopes (Fig. 5C). Importantly, the development of the 6C10 lineage had already begun after the first immunization, whereas significant levels of 9F6 and 21B5 clonal families were observed only in terminal samples (TP and TS; Figs. 5C and 5D). Alternatively, 9F6 and 21B5 could have begun to expand after the fourth immunization with gp41-28×3 and rVV-gp160DH12, a sample we were not able to analyze.
Since 9F6 best targeted the 4E10/10E8 binding site, its maturation process was examined in greater detail (Fig. 6). A total of 12 substitutions were observed compared to the inferred naive germline sequence, of which only six were within the CDRs (Fig. 6A). Two of these mutations are potentially the result of a gene conversion event with IGHV1S45 (G14E and A15G; Fig. 6B). Phylogenetic lineage analyses indicated that there was another mutation at an early stage of antibody evolution (T102A), which reverted back to the germline sequence (A102T). All substitutions except for V106L and T108I are observed in reads from the A1 sample point onwards. V106L and T108I are observed only in the terminal sample. The sequence identity/divergence plot of 9F6 heavy chain illustrates good coverage of the development history of the mAb (Fig. 6C).
Fig. 6. Detailed analyses of 9F6 maturation.
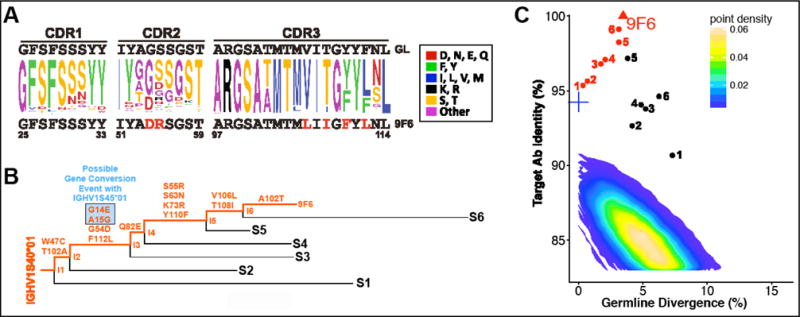
(A) Sequence variation of the HCDRs of the 9F6 clonal family. The germline sequence is shown at the top. The 9F6 sequence is shown at the bottom, with mutated residues indicated in red. (B) Lineage tree derived from representative sequences from the sample, selected to illustrate the inferred development of the antibody. Amino acid changes as a result of a possible gene conversion event are highlighted in blue. (C) Sequence identity/divergence plot of heavy chain sequences sharing the 9F6 germline. Clones shown in panel B are indicated, with inferred intermediates marked in red. The intermediates indicate good coverage of the development history. The contour plot encompasses all full-length productive sequences of the IGHV1S40 germline extracted from PBMC samples. The inferred 9F6 germline is marked by a blue cross.
DISCUSSION
The antigenic repertoire of HIV-1 envelope proteins in virus-infected patients is vast, largely due to chronic virus replication continuously generating antigens with different amino acid sequences and variable glycosylation patterns. In addition, a large number of protein fragments is likely generated by proteolytic degradation. This antigenic complexity is further increased by the drastic conformational changes that occur when the protein mediates fusion between viral and cellular membranes. The transient nature of the gp41 structure makes targeting the MPER neutralizing epitopes even more challenging. Despite this unfavorable antigenic environment, the isolation of bnAbs against the MPER from HIV-1-infected patients, albeit rare, demonstrates the possibility of inducing such antibodies through vaccination. An important question is whether similar bnAbs can be induced using a single immunogen or whether it requires multiple immunogens used in concert. If the latter, then, how many and which immunogens? Would a single cocktail of all of the immunogens together work, or would administering them in a particular, sequential order be better?
During the past three decades, the vast majority of HIV-1 vaccine efforts focused on designing immunogens that can induce bnAbs. The failures to elicit bnAbs using immunogens that are deemed antigenically native dictate that more efforts should be made in exploring novel vaccine strategies, regimens, and/or formulations. Typically, vaccines consist of the same immunogen(s) administered multiple times (i.e. “homologous prime-boost” immunization; Fig. 7A). However, as discussed earlier, it might be difficult, if not impossible, to induce MPER bnAbs using this strategy with a single immunogen. While a trimeric envelope complex may present the MPER in the native conformation, specifically targeting 4E10/10E8 epitopes may be like looking for the proverbial needle in a haystack due to the presence of multiple highly immunogenic epitopes on gp120. Even if naive B cells that encode precursor B cell receptors against the neutralizing epitopes can be stimulated, they may fail to expand efficiently due to the competitive environment of germinal centers. Successive immunizations with the same immunogen would preferentially amplify antibodies targeting the immunodominant epitopes, resulting in the induction of non- or type-specific-neutralizing antibodies.
Fig. 7. Hypothetical comparison of two vaccine strategies.
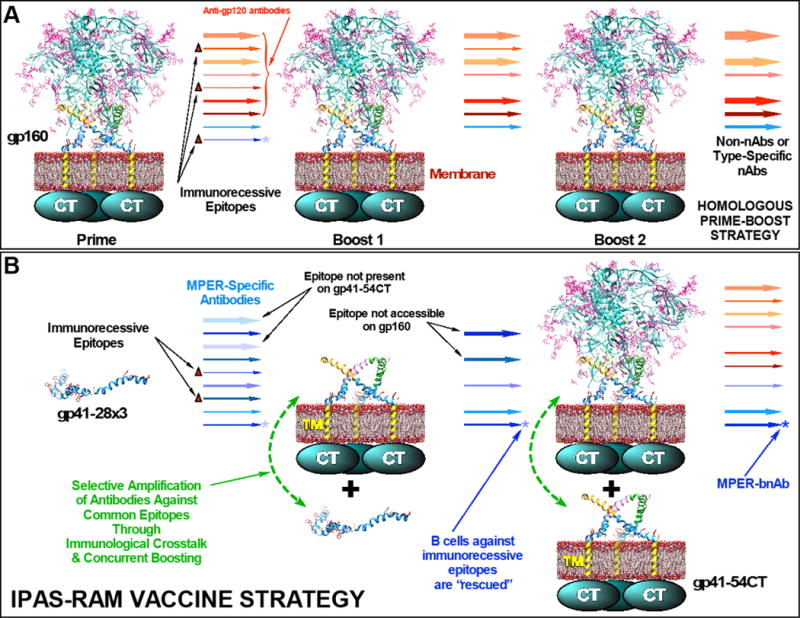
(A) A homologous prime-boost vaccine strategy using a whole, trimeric gp120/gp41 complex. Because MPER neutralizing epitopes (indicated by asterisks) are immunorecessive, B cells that target them are not stimulated sufficiently and are eventually eliminated. (B) In addition to sequential immunization with different immunogens (heterologous prime-boost), the unique and a novel feature of the IPAS-RAM vaccine strategy is co-immunization in a phased manner, which we expect will allow selective amplification of antibodies against common epitopes through immunological crosstalk and concurrent boosting. It is hypothesized that this could potentially rescue B cells against immunorecessive neutralizing epitopes.
In this report, we described our initial effort to explore a novel IPAS-RAM vaccine strategy designed to enhance antibody responses against non-immunogenic target epitopes and expedite the antibody maturation process. The strategy is different from typical vaccinations in that the immune system is exposed to different immunogens sequentially, starting with a small immunogen to focus immune responses towards the desired region (e.g. the MPER) and induce a large antibody repertoire against all possible epitope conformations (Fig. 7B). Subsequent immunizations are carried out with antigenically distinct, but related, immunogens that are progressively more “native” to guide the antibody maturation process. The final immunization is done with a whole envelope protein complex (i.e. trimeric gp120/gp41 on membrane) to increase the likelihood that mature antibodies can bind MPER epitopes in the context of native envelope spikes. During the course of the immunization, B cell clones that encode antibodies against epitopes that are absent or inaccessible on boosting immunogens are not expected to expand and are filtered out. The major hallmark of the IPAS-RAM vaccine strategy that is uniquely different from a simple “heterologous prime-boost” immunization is that immunogens are administered in a phased manner. Because the antigens are phased, anamnestic immune responses against previously administered immunogens could rapidly boost antibody responses against related epitopes on new immunogens (“concurrent boosting”). We hypothesized this strategy of “antigenic relays” and “antigenic filters” would expedite clonal expansion of target-epitope-specific B cells and accelerate antibody maturation, while minimizing antibody responses against non-conserved epitopes. We further hypothesized that the presence of different, but related immunogens simultaneously would allow “immunological crosstalks” (i.e. B cells stimulated by one immunogen engaging common/similar epitopes on different immunogens) that could facilitate antibody maturation.
To demonstrate proof-of-concept, we examined the IPAS-RAM vaccine strategy using three immunogens, gp41-28×3, gp41-54CT, and rVV-gp160DH12. Although we failed to induce neutralizing antibodies, we identified one mAb (9F6) with an epitope targeting profile that closely resembled those of 4E10 and 10E8. As typical for any exploratory study, we acknowledge that our study raised more questions than it answered. They include (1) what led to the induction of 9F6 and why does it fail to neutralize HIV-1; (2) did we use optimal immunogens; (3) is the complex IPAS-RAM vaccine regimen any better than traditional vaccine strategies with simpler regimens; and (4) can the IPAS-RAM vaccine strategy be improved?
Based on NGS analyses (Fig. 5C), we did not see significant levels of either 9F6 or 21B5-related antibodies until the terminal samples (TP and TS). While we cannot be certain as to what exactly led to the induction of 9F6, bioinformatics analyses provided some insights. In the A1 sample, % identities of the heavy chain V-gene of 9F6 clonal family members to germline ranged from 76.7% to 97.22%, which suggests that repertoire diversification had started. Since we did not have pre-immune samples to examine, it is possible that they existed prior to immunization with gp41-28×3. However, further expansion of all three mAb clonal families after the second immunization (A2) suggests that they were induced by gp41-28×3. The relative expansion of the 9F6 clonal family, as well as that of 21B5 and 6C10, shrank in A3, most likely due to dominant immune responses against vaccinia virus antigens. TP and TS samples were taken at the conclusion of the study. Thus, the marked expansion of the 9F6 and 21B5 clonal families could be due to the co-immunization with rVV-gp160DH12 and gp41-28×3 (fourth immunization) and/or the final immunization with gp41-28×3 alone (administered to increase efficiency of generating MPER-specific hybridomas). However, three lines of evidence suggest that co-immunization might be responsible: (1) We did not see many 9F6 clonal family members in the A2 sample when a strong anamnestic response against gp41-28×3 was induced. (2) In phylogenetic analyses, the closest sequences to the evolutionary development of 9F6 (shown as intermediate sequences in Figs. 6B and 6C) all came from the TP sample. Further, the substitutions V106L and T108I were observed only in the terminal samples. (3) Epitope targeting profiles of antibodies induced by homologous prime-boost immunization using gp41-28×3 only, did not resemble that of 9F6 (compiled in Fig. 4E). Notably, none of the three animals mounted significant antibody responses towards W672 and R683. Thus, while gp41-28×3 may have initiated repertoire diversification, subsequent exposure to both gp41-28×3 and rVV-gp160DH12 simultaneously likely have guided the evolution of an 9F6 intermediate to its mature form. Several studies have reported the potential benefits of co-immunizations by combining different vaccine modalities (e.g. protein and DNA vaccination; (Jaworski et al., 2012; Krebs et al., 2014; J. Li et al., 2013; Patel et al., 2013)). However, potential benefits could also arise from synergy between different immunogens, regardless of the vaccine modality. At the present time, the contribution of gp41-54CT/rVV-gp160DH12 co-immunization is unclear.
The major difference in epitope targeting between 21B5 and 4E10/10E8 is that 21B5 recognizes W678 and Y681, which lie on the opposite side of the neutralizing face. Thus, the lack of neutralizing activity is not unexpected. In contrast, all of the residues critical for 9F6 binding lie within the footprint of 4E10/10E8 (with a possible exception of N677). Therefore, it is not readily apparent why 9F6 fails to exhibit neutralizing activity. It is possible there are steric clashes between 9F6 and other parts of the trimeric envelope complex and/or the membrane surface that prevent 9F6 binding. Comparing a co-crystal structure of 9F6 bound to an MPER peptide to those of 4E10 and 10E8 could provide critical insights.
Linear epitope mapping analyses identified two peptides highly reactive to immune sera (653QEKNEQELLA662 and 656NEQELLALDK665; Fig. 2). This region corresponds to the cluster II immunodominant domain of gp41 (Xu et al., 1991), suggesting that our vaccine regimen tested in rabbits recapitulated antibody responses mounted against gp41 in humans. A large fraction of the antibodies that bind these peptides likely represents clonal family members of 6C10 mAb, which targets 657EQELLAL663 (Fig. 4A). Immunodominance of the 6C10 epitope was also evident from the antibodyome data. After the first immunization, the repertoire of antibodies clonally related to 6C10 was about 25-fold greater than those of 9F6 and 21B5 combined (Fig. 5C). That increased to about 62-fold after the second immunization. Despite partial epitope overlap with 2F5, 6C10 lacked neutralizing activity. Interestingly, the llama mAb 2H10 that recognizes 657EQELLELDK665 has neutralizing activity (Lutje Hulsik et al., 2013), suggesting the importance of targeting further downstream of the 6C10 epitope for inhibiting gp41 function. Other factors such as antibody footprint size, angle of approach, affinity, etc. could also play important roles. Detailed mapping analyses and structural studies of 6C10 could provide additional insights.
All of the MPER bnAbs (2F5, Z13e1, 4E10 and 10E8) bound gp41-28×3 (Fig. 1D). However, 2F5 binding was significantly better than 10E8 (~50-fold) and 4E10/Z13e1 (~3- to 5-fold). This might explain why the nearby 6C10 epitope was so immunogenic. Thus, retrospectively, gp41-28×3 may not have been an ideal priming immunogen to use for eliciting 4E10/10E8-like antibodies. An immunogen that has better exposure and/or affinity to 10E8, as well as one devoid of the cluster II region, might have elicited stronger antibody responses towards 4E10/10E8 epitopes. In this regard, a 4E10 scaffold-based immunogen, T88, which induced non-neutralizing antibodies with a target epitope profile similar to 4E10 (Correia et al., 2010), could have been a better priming immunogen for our IPAS-RAM vaccine strategy.
Our NGS analyses of antibody genes largely focused on heavy chain sequences related to three mAbs against a few select linear epitopes. These sequences likely represent a tiny fraction of the vast range of clonal families and superfamilies whose development were stimulated by immunizations. We were unable to employ single-cell PCR amplification of antibody genes from FACS-sorted antigen-specific B cells due to the lack of suitable markers on rabbit plasmablasts and non-specific binding of MPER immunogens to cells. Isolation of additional mAbs could facilitate and expand the scope of analyses. Alternatively, the NGS analyses could be conducted in conjunction with proteomics analyses of antibodies purified using immunogens (Cheung et al., 2012), although we would not be able to pair authentic heavy and light chains. While further work is needed to dissect the specific developmental stimuli and mechanisms, these analyses have clearly demonstrated the utilities of repertoire sequencing towards better understanding of antibody responses in vaccine settings. Importantly, we have demonstrated that rabbits can be a useful animal model to evaluate antigen-specific B cell population dynamics using NGS data despite the fact that they use gene conversion, in addition to somatic hypermutation, to generate antibody repertoire.
CONCLUSIONS
In this study, we evaluated a novel IPAS-RAM vaccine strategy to induce 4E10/10E8-like bnAbs. Despite our unsuccessful efforts, which we attribute largely to the use of a suboptimal priming immunogen, results of the study demonstrate potential benefits of sequential immunization with multiple immunogens in a phased manner. Thus, further evaluation of the IPAS-RAM vaccine strategy is warranted, perhaps using a better priming immunogen. The target epitope profile of the 9F6 mAb we isolated closely resembled those of 4E10 and 10E8. To our knowledge, 9F6 may be the best vaccine-induced mAb that mimics 10E8. Comparative structural analyses of 9F6 with 10E8 could provide insights into why 9F6 lacks neutralizing activity and how we might be able to improve our vaccine strategy.
MATERIALS AND METHODS
Immunogen generation
The priming immunogen, gp41-28×3, was produced from a pHR1-HR2-28×3 construct. This plasmid was constructed from pGEX-2T gp41(30) (Penn-Nicholson et al., 2008) through multiple subcloning steps and site-directed mutagenesis, details of which will be described elsewhere. The pHR1-HR2-28×3 construct is based on a pET-21a vector. HR1-HR2-28×3 was expressed in E. coli, solubilized with 8M urea, and purified using Ni-NTA as we previously described (Banerjee et al., 2016; Habte et al., 2015; Qin et al., 2014). The fusion protein was cleaved with thrombin (GE Healthcare; cat#27-0846-01) and the gp41-28×3 fragment was purified using Ni-NTA, dialyzed into PBS (pH 8.0) and stored at −80°C. To clone gp41-54CT construct, a region containing the gene (630EWEREISN QGLERALL856; numbering based on HXB2) was PCR amplified from pcDNA-MCON6gp160 (kindly provided by Dr. Beatrice Hahn (Gao et al., 2005)) using a sense primer 5′-cgcggatcc GAG TGG GAG CGC GAG ATC-3′ and an antisense primer 5′-cggaagc TTA atg gtg atg atg gtg atg CAG CAG GGC GCG CTC CAG-3′. Six-histidine tag was incorporated into the primer. The resulting PCR product was digested with BamHI and HindIII (underlined) and inserted into the corresponding sites on a vector based on pcDNA*MCON6-gp120-OD described previously (Qin et al., 2014).
The recombinant vaccinia virus expressing gp160 of the HIV-1DH12 isolate (rVV-gp160DH12) was generated from a single virus variant that was plaque-purified from the Dryvax vaccine obtained from the CDC. First, the plaque-purified virus was attenuated by disrupting the B8R gene by homologous recombination using a pB8R-IFN-γ plasmid that encodes a human IFN-γ gene in between two segments of the B8R gene, resulting in rVVΔB8R-IFN-γ. Next, a second plasmid pNVVDHenv (Cho et al., 2001; 1998), which encodes gp160, was used to insert the gene into a thymidine kinase gene of rVVΔB8R-IFN-γ. The resulting virus was propagated and purified as described previously (Cho et al., 2001; 1998). The details of pB8R-IFN-γ construction, and biological properties of rVVAB8R-IFN-γ and rVV-gp160DH12, will be described elsewhere. The MPER sequence of DH12 strain differs from that of MCON6 at two positions (N674 and S676, instead of D674 and T676).
Rabbit immunization
New Zealand white female rabbits (2.5 to 3 kg) were purchased from Charles River (USA) and housed under specific pathogen free environments. All animals were tested in compliance with the animal protocol approved by IACUC of Iowa State University. For the IPAS-RAM vaccine group, three rabbits were immunized and samples were taken as shown in Fig. 2A. Rabbits were primed with 200 μg of gp41-28×3 subcutaneously with zinc-chitosan as an adjuvant as previously described (Habte et al., 2015; Qin et al., 2014). For the second immunization, rabbits were injected subcutaneously with 50 μg of gp41-28×3. Animals were also injected intradermally with 200 μg of gp41-54CT DNA in PBS, followed by electroporation using the AgilePulse In Vivo System (BTX, Harvard Apparatus). For the third immunization, rabbits were injected with 200 μg of gp41-54CT DNA as described above and with rVV-gp160DH12 (1×108 PFUs) through intradermal injection. For the fourth immunization, both 50 μg of gp41-28×3 and rVV-gp160DH12 were administered. For the homologous prime-boost vaccine group, three rabbits were immunized subcutaneously with 200 μg of gp41-28×3 as shown in Fig. 2C. Serum samples were collected prior to immunization (pre-immune) as well as two weeks post-immunizations. PBMC samples were also collected 4 days after each immunization. Samples were stored at −80 °C until used.
Hybridoma generation
Rabbit R3 was injected intravenously using 200 μg of soluble gp41-28×3 in PBS without any adjuvant on week 35. Four days later, the animal was euthanized and the spleen was harvested. Splenocytes were fused to 240E-1 cells (kindly provided by Dr. Katherine L. Knight (Spieker-Polet et al., 1995)), as previously described (Qin et al., 2015; Spieker-Polet et al., 1995). Hybridoma cell culture media were screened by ELISA for specific binding to gp41-28×3 and individual peptides of interest as described below.
Enzyme-linked immunosorbent assays (ELISA)
All ELISA were performed using a protocol described previously (Banerjee et al., 2016; Habte et al., 2015), except for the use of an alternate blocking buffer containing 2.5% milk and 5% calf sera in PBS (pH 7.5). Various coating antigens were used, including gp41-28×3 (30 ng/ml), a mixture of 10-mer peptides biotinylated either at the N- or C-terminus (Habte et al., 2015), and HIV-1 M group consensus Env peptides (15-mer; 100 ng/well) obtained from the NIH AIDS Reagent Program (Cat# 9487). For the fine epitope mapping analyses, two sets of scanning alanine mutant peptides were used: (1) 13-mer 671 peptide (671NWFDITNWLWYIK683) biotinylated at the C-terminal lysine, as previously described (Banerjee et al., 2016; Habte et al., 2015); and (2) 13-mer 671 peptide biotinylated at the N-terminal amine of two glycine linker (GG671NWFDITNWLWYIK683). For all ELISA testing hybridoma binding, goat anti-rabbit, horseradish peroxidase (HRP)-conjugated antibody (Thermo Scientific; Cat# 31430) was used as secondary antibody.
Neutralization assays
Neutralization assays were performed in TZM-bl cells as previously described (M. Li et al., 2005; Qin et al., 2014; Wei et al., 2002). Viruses tested included SF162 (tier 1A, clade B), MW965.26 (tier 1A, clade C), and MN.3 (tier 1A, clade B). Murine leukemia virus Env-pseudotyped virus was used as a negative control.
Hybridoma gene sequence analysis
Total RNA was extracted from hybridoma cells using the RNeasy Mini kit (Qiagen). RNA samples were treated with DNAse (Invitrogen) according to a manufacturer’s protocol. Samples were subjected to cDNA synthesis using random hexamers and SuperScript III Reverse Transcriptase (Invitrogen). Briefly, 1μL of random hexamers (Roche) and 1μL of 10mM dNTPs were added to 11μL of DNAse treated RNA. The mixture was heated to 65°C for 5min, then cooled briefly on ice.
Subsequently, 4μL of 5× First-Strand Buffer, 1uL of 0.1M DTT, 1uL of RNaseOUT™ (Invitrogen), and 1uL of SuperScript III were added. Reaction was incubated at 25°C for 5min, 45°C for 45mins, and 70°C for 15mins.
Heavy and kappa chain sequences were amplified with Platinum Pfx (Invitrogen) according to manufacturer’s recommendations. Primers were based on a previous publication (Lightwood et al., 2006) and were designed for leader sequences (5′ primer) or the C-terminus of the junction. Primers used for heavy chain amplification were 5′-GATATCAAGCTTACGCTCACCATGGAGACTGGGC-3′ and 5′-CGCGCGCTCGAGACGGTGACSAGGGTSCCYKGGCCCC-3′. Primers used for kappa chain amplification were 5′-GATATCAAGCTTCGAATCGACATGGACACGAGGGCCCCC-3′ and 5′-TCTAGACGTACGTTTGACCACCACCTCGGTCCCTC-3′. Cycling conditions were as follows: Initial denaturation at 94°C for 5mins; followed by 40 cycles of 94°C for 30sec, 68°C for 1.5mins; final extension at 68°C for 7mins; hold at 4°C. PCR products were examined via agarose gel to confirm specific amplification. Products were subcloned for DNA sequencing.
For the 9F6 hybridoma, alternate light chain primers were tested as well. For the kappa chain, 5′ and 3′ primers from a second publication were tested (Rader et al., 2000). The two 5′ primers used bound the beginning of the FR1 region: 5′-GGGCCCAGGCGGCCGAGCTCGTGMTGACCCAGACT-3′ and GGGCCCAGGCGGCCGAGCTCGATMTGACCAGACT-3′. An alternate 3′ primer, 5′-AGATGGTGCAGCCACAGTTCGTAGGATCTCCAGCTCGGTCCC-3′, bound a similar region in the junction as did our original primer. An additional 3′ primer which bound the 3′ untranslated region immediately after the stop codon, 5′-TCACTGGCGGTGCCCTGGCAGGCGTCT-3′, was designed in-house based on NCBI nucleotide sequences. The lambda chain primer set described in Rader et al. (Rader et al., 2000) was also tested.
PBMC isolation
For PBMC isolation, EDTA-treated blood was initially spun at 2000×g for 10 min, with brakes turned off, to remove plasma and RBC. The collected cell layer was diluted 1:3 with PBS. Diluted blood was layered over Ficoll-Paque™ PLUS (GE Healthcare) in a 4:3 (blood:Ficoll) ratio. The layered sample was centrifuged at 400×g for 45mins, with brakes turned off. PBMC layer was removed, and washed twice with 10 mL of PBS, followed by centrifugation at 600×g for 10min, with brakes. PBMC were stored in freezing media (90% fetal bovine serum and 10% DMSO) at −140°C until RNA isolation.
Antibody gene RT-PCR for NGS analysis
Total RNA extraction and cDNA synthesis from PBMCs (~6×106 per timepoint) was done as described for hybridomas. cDNA synthesis was scaled up by 6-fold to accommodate this amount of starting material. Resulting cDNA was split into two aliquots and subjected to separate antibody heavy and light chain gene amplification using Platinum Pfx polymerase (Invitrogen). Reaction mixture (150uL volume, split into 3 tubes) was: 60μL of cDNA template, 15μL of 10× buffer, 4.5μL of 10mM dNTPs, 3μL of MgSO4, 6μL of each primer (10μM concentration), and 1.2μL of polymerase. Primers and cycling conditions were the same as described for hybridoma sequencing. PCR products were examined via agarose gel to confirm specific amplification. Extra primers and nucleotides were removed using QIAquick PCR Purification Kit (Qiagen). Following purification, heavy and light chain concentrations were calculated, and equal amounts of each were pooled for library preparation.
Next-generation sequencing
The TruSeq DNA LT sample prep kit (Illumina) was used to make NGS libraries, consisting of both heavy and kappa chain variable region fragments, for a 2×250bp read. The low sample (LS), gel free method was performed according to manufacturer’s protocol, with minor modifications. Following end repair of the cDNA, indexing adapters were added using DNA adapter tubes (Illumina). The library quality was determined by Bioanalyzer High Sensitivity DNA chip (Agilent), and quantification was performed using a Qubit (Life Technologies). The sample libraries were run on an Illumina MiSeq according to manufacturer’s recommendations.
NGS Read Processing and Quality Control
Reads from the two sequencing runs were demultiplexed by Illumina software, following which they were pre-processed using the Repertoire Sequencing Toolkit (pRESTO) (Vander Heiden et al., 2014). Processing consisted of the following steps: (1) Reads having a mean Phred quality score <20 were removed. (2) Paired reads were identified from Illumina headers, and aligned. Unpaired reads, and reads with a significance threshold calculated by pRESTO of <0.1, or mismatch rate <0.2, were removed. (3) Forward and reverse primers were matched and removed from the reads. Reads not matching primers, or which matched with error rate >0.2, were removed. (4) Duplicate reads were merged. (5) Reads were split into heavy and light chain sets, based on 5′ primer identification. (6) Where the same sample was processed in multiple sequencing runs, the resulting sets were merged to provide a single heavy and light chain set per sample. (7) Resulting read sets were clustered to a minimum identity of 97% using uparse (Edgar, 2010). The NGS data have been deposited to the NCBI Short Reads Archives (SRA) as study SRP094044.
Junction Identification and Germline Assignment
Sequences were parsed with IgBLAST v1.4.0 (Ye et al., 2013) using the IMGT germline library for the rabbit (Oryctolagus cuniculus) (M. P. Lefranc and G. Lefranc, 2001), downloaded 14 March 2015. Subsequent alignments were converted to IMGT output format using IgBlastPlus v1.0(Lees and Shepherd, 2015).
Clonal Inference and Analysis
The cluster diagram (Fig. 5A) was rendered in Gephi v0.9.1 (Bastian et al., 2009). For the Streamgraph view (Fig. 5C), heavy chain reads from all samples were combined, together with the sequences for the isolated mAbs. Clonally related sequences were inferred by clustering junction sequences of the same length and with nucleotide identity ≥ 90% using a single-linkage algorithm. The identity threshold was determined by consideration of the nearest-neighbor distribution (Fig. S6). Where the same CDR3 sequence was observed in multiple samples, it was assigned to the earliest sample (or leftmost in the diagram for terminal samples). Code for the Streamgraph view was as previously described (Laserson et al., 2014). Bands represent clusters of 2 or more CDR3 sequences and the height at each sample point is proportional to the number of CDR3 sequences in the cluster. The overall band height at the sample point reflects the number of unique CDR3 sequences observed in the sample. Because the sequencing depth was uneven, equal samples of 100,000 reads of functional v-regions were drawn from each sample, and the number of unique CDR3 sequences was determined. The average was determined over 100 random draws. Overall band heights are proportional to that average number of unique CDR3 sequences.
Phylogenetic Analysis
Full-length sequences from blood plasma samples whose CDR3s clustered with mAbs of interest were reviewed, and those which did not match the mAb V-and J- germline assignments were rejected. To obtain a computationally tractable data set, random downsampling was used to limit the maximum number of sequences from each sample point to 200. Sequences were codon-aligned using TranslatorX (Abascal et al., 2010) following which phylogenetic trees were inferred by IQ-TREE v1.2.2 (Minh et al., 2013). The selection model GTR+G4 was selected by the program from consideration of the log-likelihoods of the initial parsimony tree for all available models. Trees were rendered using the ETE Toolkit (Huerta-Cepas et al., 2010). Intermediate sequences in the development of mAb 9F6, and its germline junction sequence, were inferred by PHYLIP (Felsenstein and Churchill, 1996) and the logo diagram was rendered with Berkley WebLogo (Crooks et al., 2004). Percentage identity to the mAb and divergence from the germline was determined using in-house software.
In-House Software
Software developed in-house for the analysis (clustering, charting of germline attribution, spectratypes, germline identity) is available as part of TRIgS (http://cimm.ismb.lon.ac.uk/trigs/) (Lees and Shepherd, 2015).
Supplementary Material
Highlights.
Constructed two new MPER-based immunogens (gp41-28×3 and gp41-54CT).
Demonstrated potential benefits of a novel IPAS-RAM vaccine strategy.
Generated MPER-specific mAbs, including 9F6, which closely resembles 10E8.
NGS-based antibody repertoire analyses revealed a maturation pathway of 9F6.
Demonstrated feasibility of doing B cell population dynamics analyses in rabbits.
Acknowledgments
We are grateful to Dr. Beatrice Hahn for providing pcDNA-MCON6gp160. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 Consensus group M Env peptides (Cat# 9487), Z13e1 from Dr. Michael Zwick (Cat# 11557), 2F5 from Dr. Hermann Katinger (Cat# 130220) and 4E10 from Dr. Herman Katinger (Cat# 10091). This work was supported by a Grant from the HHS/NIH/NIAID (P01 AI074286) and funding from Iowa State University. MWC has an equity interest in NeoVaxSyn Inc., and serves as the CEO/President. NeoVaxSyn Inc. did not contribute to this work or the interpretation of the data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38:W7–13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appella E, Chersi A, Rejnek J, Reisfeld R, Mage R. Rabbit immunoglobulin lambda chains: isolation and amino acid sequence of cysteine-containing peptides. Immunochemistry. 1974;11:395–402. doi: 10.1016/0019-2791(74)90072-x. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Shi H, Habte HH, Qin Y, Cho MW. Modulating immunogenic properties of HIV-1 gp41 membrane-proximal external region by destabilizing six-helix bundle structure. Virology. 2016;490:17–26. doi: 10.1016/j.virol.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. ICWSM 2009 [Google Scholar]
- Briney B, Sok D, Jardine JG, Kulp DW, Skog P, Menis S, Jacak R, Kalyuzhniy O, de Val N, Sesterhenn F, Le KM, Ramos A, Jones M, Saye-Francisco KL, Blane TR, Spencer S, Georgeson E, Hu X, Ozorowski G, Adachi Y, Kubitz M, Sarkar A, Wilson IA, Ward AB, Nemazee D, Burton DR, Schief WR. Tailored Immunogens Direct Affinity Maturation toward HIV Neutralizing Antibodies. Cell. 2016;166:1459–1470.e11. doi: 10.1016/j.cell.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunel FM, Zwick MB, Cardoso RMF, Nelson JD, Wilson IA, Burton DR, Dawson PE. Structure-function analysis of the epitope for 4E10, a broadly neutralizing human immunodeficiency virus type 1 antibody. J Virol. 2006;80:1680–1687. doi: 10.1128/JVI.80.4.1680-1687.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso RMF, Brunel FM, Ferguson S, Zwick M, Burton DR, Dawson PE, Wilson IA. Structural basis of enhanced binding of extended and helically constrained peptide epitopes of the broadly neutralizing HIV-1 antibody 4E10. J Mol Biol. 2007;365:1533–1544. doi: 10.1016/j.jmb.2006.10.088. [DOI] [PubMed] [Google Scholar]
- Cheung WC, Beausoleil SA, Zhang X, Sato S, Schieferl SM, Wieler JS, Beaudet JG, Ramenani RK, Popova L, Comb MJ, Rush J, Polakiewicz RD. A proteomics approach for the identification and cloning of monoclonal antibodies from serum. Nat Biotechnol. 2012;30:447–452. doi: 10.1038/nbt.2167. [DOI] [PubMed] [Google Scholar]
- Cho MW, Kim YB, Lee MK, Gupta KC, Ross W, Plishka R, Buckler-White A, Igarashi T, Theodore T, Byrum R, Kemp C, Montefiori DC, Martin MA. Polyvalent envelope glycoprotein vaccine elicits a broader neutralizing antibody response but is unable to provide sterilizing protection against heterologous Simian/human immunodeficiency virus infection in pigtailed macaques. J Virol. 2001;75:2224–2234. doi: 10.1128/JVI.75.5.2224-2234.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho MW, Lee MK, Carney MC, Berson JF, Doms RW, Martin MA. Identification of determinants on a dualtropic human immunodeficiency virus type 1 envelope glycoprotein that confer usage of CXCR4. J Virol. 1998;72:2509–2515. doi: 10.1128/jvi.72.3.2509-2515.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia BE, Ban YEA, Holmes MA, Xu H, Ellingson K, Kraft Z, Carrico C, Boni E, Sather DN, Zenobia C, Burke KY, Bradley-Hewitt T, Bruhn-Johannsen JF, Kalyuzhniy O, Baker D, Strong RK, Stamatatos L, Schief WR. Computational Design of Epitope-Scaffolds Allows Induction of Antibodies Specific for a Poorly Immunogenic HIV Vaccine Epitope. Structure. 2010;18:1116–1126. doi: 10.1016/j.str.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Escolano A, Steichen JM, Dosenovic P, Kulp DW, Golijanin J, Sok D, Freund NT, Gitlin AD, Oliveira T, Araki T, Lowe S, Chen ST, Heinemann J, Yao KH, Georgeson E, Saye-Francisco KL, Gazumyan A, Adachi Y, Kubitz M, Burton DR, Schief WR, Nussenzweig MC. Sequential Immunization Elicits Broadly Neutralizing Anti-HIV-1 Antibodies in Ig Knockin Mice. Cell. 2016;166:1445–1458.e12. doi: 10.1016/j.cell.2016.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J, Churchill GA. A Hidden Markov Model approach to variation among sites in rate of evolution. Mol Biol Evol. 1996;13:93–104. doi: 10.1093/oxfordjournals.molbev.a025575. [DOI] [PubMed] [Google Scholar]
- Gao F, Weaver EA, Lu Z, Li Y, Liao HX, Ma B, Alam SM, Scearce RM, Sutherland LL, Yu JS, Decker JM, Shaw GM, Montefiori DC, Korber BT, Hahn BH, Haynes BF. Antigenicity and immunogenicity of a synthetic human immunodeficiency virus type 1 group m consensus envelope glycoprotein. J Virol. 2005;79:1154–1163. doi: 10.1128/JVI.79.2.1154-1163.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev IS, Gordon Joyce M, Zhou T, Kwong PD. Elicitation of HIV-1-neutralizing antibodies against the CD4-binding site. Curr Opin HIV AIDS. 2013;8:382–392. doi: 10.1097/COH.0b013e328363a90e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habte HH, Banerjee S, Shi H, Qin Y, Cho MW. Immunogenic properties of a trimeric gp41-based immunogen containing an exposed membrane-proximal external region. Virology. 2015;486:187–197. doi: 10.1016/j.virol.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes BF, Kelsoe G, Harrison SC, Kepler TB. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol. 2012;30:423–433. doi: 10.1038/nbt.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes BF, Moody MA, Alam M, Bonsignori M, Verkoczy L, Ferrari G, Gao F, Tomaras G, Liao HX, Kelsoe G. Progress in HIV-1 vaccine development. Journal of Allergy and Clinical Immunology. 2014;134:3–10. doi: 10.1016/j.jaci.2014.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kang BH, Pancera M, Lee JH, Tong T, Feng Y, Imamichi H, Georgiev IS, Chuang G-Y, Druz A, Doria-Rose NA, Laub L, Sliepen K, van Gils MJ, de la Peña AT, Derking R, Klasse PJ, Migueles SA, Bailer RT, Alam M, Pugach P, Haynes BF, Wyatt RT, Sanders RW, Binley JM, Ward AB, Mascola JR, Kwong PD, Connors M. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41 -gp120 interface. Nature. 2014;515:138–142. doi: 10.1038/nature13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012;491:406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Dopazo J, Gabaldón T. ETE: a python Environment for Tree Exploration. BMC Bioinformatics. 2010;11:24. doi: 10.1186/1471-2105-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia A, Sarkar A, Stanfield RL, Wilson IA. Crystallographic identification of lipid as an integral component of the epitope of HIV broadly neutralizing antibody 4E10. Immunity. 2016;44(1):21–31. doi: 10.1016/j.immuni.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski JP, Krebs SJ, Trovato M, Kovarik DN, Brower Z, Sutton WF, Waagmeester G, Sartorius R, D’Apice L, Caivano A, Doria-Rose NA, Malherbe D, Montefiori DC, Barnett S, De Berardinis P, Haigwood NL. Co-immunization with multimeric scaffolds and DNA rapidly induces potent autologous HIV-1 neutralizing antibodies and CD8+ T cells. PLoS ONE. 2012;7:e31464. doi: 10.1371/journal.pone.0031464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Torrents de la Peña A, Deller MC, Garces F, Sliepen K, Hua Y, Stanfield RL, Sanders RW, Wilson IA. Complete epitopes for vaccine design derived from a crystal structure of the broadly neutralizing antibodies GT128 and 8ANC195 in complex with an HIV-1 Env trimer. Acta Crystallogr D Biol Crystallogr. 2015;71:2099–2108. doi: 10.1107/S1399004715013917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs SJ, McBurney SP, Kovarik DN, Waddell CD, Jaworski JP, Sutton WF, Gomes MM, Trovato M, Waagmeester G, Barnett SJ, DeBerardinis P, Haigwood NL. Multimeric scaffolds displaying the HIV-1 envelope MPER induce MPER-specific antibodies and cross-neutralizing antibodies when co-immunized with gp160 DNA. PLoS ONE. 2014;9:e113463. doi: 10.1371/journal.pone.0113463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong PD, Mascola JR, Nabel GJ. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat Rev Immunol. 2013;13:693–701. doi: 10.1038/nri3516. [DOI] [PubMed] [Google Scholar]
- Laserson U, Vigneault F, Gadala-Maria D, Yaari G, Uduman M, Vander Heiden JA, Kelton W, Taek Jung S, Liu Y, Laserson J, Chari R, Lee JH, Bachelet I, Hickey B, Lieberman-Aiden E, Hanczaruk B, Simen BB, Egholm M, Koller D, Georgiou G, Kleinstein SH, Church GM. High-resolution antibody dynamics of vaccine-induced immune responses. Proceedings of the National Academy of Sciences. 2014;111:4928–4933. doi: 10.1073/pnas.1323862111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees WD, Shepherd AJ. Utilities for High-Throughput Analysis of B-Cell Clonal Lineages. J Immunol Res. 2015;2015:323506. doi: 10.1155/2015/323506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc MP, Lefranc G. The immunoglobulin factsbook 2001 [Google Scholar]
- Li J, Valentin A, Kulkarni V, Rosati M, Beach RK, Alicea C, Hannaman D, Reed SG, Felber BK, Pavlakis GN. HIV/SIV DNA vaccine combined with protein in a co-immunization protocol elicits highest humoral responses to envelope in mice and macaques. Vaccine. 2013;31:3747–3755. doi: 10.1016/j.vaccine.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79:10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightwood DJ, Carrington B, Henry AJ, McKnight AJ, Crook K, Cromie K, Lawson ADG. Antibody generation through B cell panning on antigen followed by in situ culture and direct RT-PCR on cells harvested en masse from antigen-positive wells. J Immunol Methods. 2006;316:133–143. doi: 10.1016/j.jim.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Lutje Hulsik D, Liu Y-Y, Strokappe NM, Battella S, El Khattabi M, McCoy LE, Sabin C, Hinz A, Hock M, Macheboeuf P, Bonvin AMJJ, Langedijk JPM, Davis D, Forsman Quigley A, Aasa-Chapman MMI, Seaman MS, Ramos A, Poignard P, Favier A, Simorre J-P, Weiss RA, Verrips CT, Weissenhorn W, Rutten L. A gp41 MPER-specific llama VHH requires a hydrophobic CDR3 for neutralization but not for antigen recognition. PLoS Pathog. 2013;9:e1003202. doi: 10.1371/journal.ppat.1003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascola JR, Haynes BF. HIV-1 neutralizing antibodies: understanding nature’s pathways. Immunol Rev. 2013;254:225–244. doi: 10.1111/imr.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascola JR, Montefiori DC. The role of antibodies in HIV vaccines. Annu Rev Immunol. 2010;28:413–444. doi: 10.1146/annurev-immunol-030409-101256. [DOI] [PubMed] [Google Scholar]
- McCoy LE, Weiss RA. Neutralizing antibodies to HIV-1 induced by immunization. J Exp Med. 2013;210:209–223. doi: 10.1084/jem.20121827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB. Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm. Retrovirology. 2008;5:111. doi: 10.1186/1742-4690-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, Nguyen MAT, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30:1188–1195. doi: 10.1093/molbev/mst024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero M, Gulzar N, Klaric KA, Donald JE, Lepik C, Wu S, Tsai S, Julien JP, Hessell AJ, Wang S, Lu S, Burton DR. Neutralizing epitopes in the membrane-proximal external region of HIV-1 gp41 are influenced by the transmembrane domain and the plasma membrane. Journal of Virology. 2012;86:2930–2941. doi: 10.1128/JVI.06349-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Barroso I, Salzwedel K, Hunter E, Blumenthal R. Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J Virol. 1999;73:6089–6092. doi: 10.1128/jvi.73.7.6089-6092.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V, Jalah R, Kulkarni V, Valentin A, Rosati M, Alicea C, von Gegerfelt A, Huang W, Guan Y, Keele BF, Bess JW, Piatak M, Lifson JD, Williams WT, Shen X, Tomaras GD, Amara RR, Robinson HL, Johnson W, Broderick KE, Sardesai NY, Venzon DJ, Hirsch VM, Felber BK, Pavlakis GN. DNA and virus particle vaccination protects against acquisition and confers control of viremia upon heterologous simian immunodeficiency virus challenge. Proceedings of the National Academy of Sciences. 2013;110:2975–2980. doi: 10.1073/pnas.1215393110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejchal R, Doores KJ, Walker LM, Khayat R, Huang PS, Wang SK, Stanfield RL, Julien JP, Ramos A, Crispin M, Depetris R, Katpally U, Marozsan A, Cupo A, Maloveste S, Liu Y, McBride R, Ito Y, Sanders RW, Ogohara C, Paulson JC, Feizi T, Scanlan CN, Wong CH, Moore JP, Olson WC, Ward AB, Poignard P, Schief WR, Burton DR, Wilson IA. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science. 2011;334:1097–1103. doi: 10.1126/science.1213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn-Nicholson A, Han DP, Kim SJ, Park H, Ansari R, Montefiori DC, Cho MW. Assessment of antibody responses against gp41 in HIV-1-infected patients using soluble gp41 fusion proteins and peptides derived from M group consensus envelope. Virology. 2008;372:442–456. doi: 10.1016/j.virol.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Purtscher M, Trkola A, Gruber G, Buchacher A, Predl R, Steindl F, Tauer C, Berger R, Barrett N, Jungbauer A. A broadly neutralizing human monoclonal antibody against gp41 of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 1994;10:1651–1658. doi: 10.1089/aid.1994.10.1651. [DOI] [PubMed] [Google Scholar]
- Qin Y, Banasik M, Kim S, Penn-Nicholson A, Habte HH, LaBranche C, Montefiori DC, Wang C, Cho MW. Eliciting neutralizing antibodies with gp120 outer domain constructs based on M-group consensus sequence. Virology. 2014:462–463. doi: 10.1016/j.virol.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Banerjee S, Agrawal A, Shi H, Banasik M, Lin F, Rohl K, LaBranche C, Montefiori DC, Cho MW. Characterization of a Large Panel of Rabbit Monoclonal Antibodies against HIV-1 gp120 and Isolation of Novel Neutralizing Antibodies against the V3 Loop. PLoS ONE. 2015;10:e0128823. doi: 10.1371/journal.pone.0128823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader C, Ritter G, Nathan S, Elia M, Gout I, Jungbluth AA, Cohen LS, Welt S, Old LJ, Barbas CF. The rabbit antibody repertoire as a novel source for the generation of therapeutic human antibodies. J Biol Chem. 2000;275:13668–13676. doi: 10.1074/jbc.275.18.13668. [DOI] [PubMed] [Google Scholar]
- Reardon PN, Sage H, Dennison SM, Martin JW, Donald BR, Alam SM, Haynes BF, Spicer LD. Structure of an HIV-1-neutralizing antibody target, the lipid-bound gp41 envelope membrane proximal region trimer. Proceedings of the National Academy of Sciences. 2014;111:1391–1396. doi: 10.1073/pnas.1309842111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujas E, Gulzar N, Morante K, Tsumoto K, Scott JK, Nieva JL, Caaviero JMM. Structural and thermodynamic basis of epitope binding by neutralizing and nonneutralizing forms of the anti-HIV-1 antibody 4E10. Journal of Virology. 2015;89:11975–11989. doi: 10.1128/JVI.01793-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzwedel K, West JT, Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol. 1999;73:2469–2480. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TYK, Pietzsch J, Fenyo D, Abadir A, Velinzon K, Hurley A, Myung S, Boulad F, Poignard P, Burton DR, Pereyra F, Ho DD, Walker BD, Seaman MS, Bjorkman PJ, Chait BT, Nussenzweig MC. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science. 2011;333:1633–1637. doi: 10.1126/science.1207227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Bohon J, Han DP, Habte H, Qin Y, Cho MW, Chance MR. Structural characterization of HIV gp41 with the membrane-proximal external region. Journal of Biological Chemistry. 2010;285:24290–24298. doi: 10.1074/jbc.M110.111351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sok D, Briney B, Jardine JG, Kulp DW, Menis S, Pauthner M, Wood A, Lee EC, Le KM, Jones M, Ramos A, Kalyuzhniy O, Adachi Y, Kubitz M, MacPherson S, Bradley A, Friedrich GA, Schief WR, Burton DR. Priming HIV-1 broadly neutralizing antibody precursors in human Ig loci transgenic mice. Science. 2016;353:1557–1560. doi: 10.1126/science.aah3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spieker-Polet H, Sethupathi P, Yam PC, Knight KL. Rabbit monoclonal antibodies: generating a fusion partner to produce rabbit-rabbit hybridomas. Proc Natl Acad Sci USA. 1995;92:9348–9352. doi: 10.1073/pnas.92.20.9348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiegler G, Kunert R, Purtscher M, Wolbank S, Voglauer R, Steindl F, Katinger H. A potent cross-clade neutralizing human monoclonal antibody against a novel epitope on gp41 of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 2001;17:1757–1765. doi: 10.1089/08892220152741450. [DOI] [PubMed] [Google Scholar]
- Tian M, Cheng C, Chen X, Duan H, Cheng HL, Dao M, Sheng Z, Kimble M, Wang L, Lin S, Schmidt SD, Du Z, Joyce MG, Chen Y, DeKosky BJ, Chen Y, Normandin E, Cantor E, Chen RE, Doria-Rose NA, Zhang Y, Shi W, Kong WP, Choe M, Henry AR, Laboune F, Georgiev IS, Huang PY, Jain S, McGuire AT, Georgeson E, Menis S, Douek DC, Schief WR, Stamatatos L, Kwong PD, Shapiro L, Haynes BF, Mascola JR, Alt FW. Induction of HIV Neutralizing Antibody Lineages in Mice with Diverse Precursor Repertoires. Cell. 2016;166:1471–1484.e18. doi: 10.1016/j.cell.2016.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gils MJ, Sanders RW. Broadly neutralizing antibodies against HIV-1: templates for a vaccine. Virology. 2013;435:46–56. doi: 10.1016/j.virol.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Vander Heiden JA, Yaari G, Uduman M, Stern JNH, O’Connor KC, Hafler DA, Vigneault F, Kleinstein SH. pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics. 2014;30:1930–1932. doi: 10.1093/bioinformatics/btu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Protocol G Principal Investigators. Koff WC, Wilson IA, Burton DR, Poignard P. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477:466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Protocol G Principal Investigators. Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002;46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O’Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JY, Gorny MK, Palker T, Karwowska S, Zolla-Pazner S. Epitope mapping of two immunodominant domains of gp41, the transmembrane protein of human immunodeficiency virus type 1, using ten human monoclonal antibodies. J Virol. 1991;65:4832–4838. doi: 10.1128/jvi.65.9.4832-4838.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41:W34–40. doi: 10.1093/nar/gkt382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick MB, Jensen R, Church S, Wang M, Stiegler G, Kunert R, Katinger H, Burton DR. Anti-human immunodeficiency virus type 1 (HIV-1) antibodies 2F5 and 4E10 require surprisingly few crucial residues in the membrane-proximal external region of glycoprotein gp41 to neutralize HIV-1. J Virol. 2005;79:1252–1261. doi: 10.1128/JVI.79.2.1252-1261.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick MB, Labrijn AF, Wang M, Spenlehauer C, Saphire EO, Binley JM, Moore JP, Stiegler G, Katinger H, Burton DR, Parren PW. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J Virol. 2001;75:10892–10905. doi: 10.1128/JVI.75.22.10892-10905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.