

Abstract
Primary hyperparathyroidism (PHPT) is a common disorder in which parathyroid hormone (PTH) is excessively secreted from one or more of the four parathyroid glands. A single benign parathyroid adenoma is the cause in most people. However, multiglandular disease is not rare and is typically seen in familial PHPT syndromes. The genetics of PHPT is usually monoclonal when a single gland is involved and polyclonal when multiglandular disease is present. The genes that have been implicated in PHPT include proto-oncogenes and tumour-suppressor genes. Hypercalcaemia is the biochemical hallmark of PHPT. Usually, the concentration of PTH is frankly increased but can remain within the normal range, which is abnormal in the setting of hypercalcaemia. Normocalcaemic PHPT, a variant in which the serum calcium level is persistently normal but PTH levels are increased in the absence of an obvious inciting stimulus, is now recognized. The clinical presentation of PHPT varies from asymptomatic disease (seen in countries where biochemical screening is routine) to classic symptomatic disease in which renal and/or skeletal complications are observed. Management guidelines have recently been revised to help the clinician to decide on the merits of a parathyroidectomy or a non-surgical course. This Primer covers these areas with particular attention to the epidemiology, clinical presentations, genetics, evaluation and guidelines for the management of PHPT.
Primary hyperparathyroidism (PHPT) is a common disorder characterized by an excessive secretion of parathyroid hormone (PTH) from one or more of the four parathyroid glands. It is typically due to a single benign parathyroid adenoma. The classic disorder is associated with hypercalcaemia, but a normocalcaemic variant is now recognized. The incidence of PHPT increased markedly in countries where biochemical screening tests have come into use. With the increase in incidence of PHPT, its clinical presentation has changed from a symptomatic disease with skeletal (for example, fragility fractures) and renal (for example, kidney stones) manifestations to one in which patients are not overtly symptomatic. Asymptomatic PHPT has been the subject of four decades of discussion about how best to deal with a disorder that can be cured by surgery but which presents often as an incidental discovery without symptoms. It is important to note that individuals who are ‘asymptomatic’ can still show evidence of complications and target organ involvement. Conversely, symptomatic PHPT still occurs and can be the most prominent form of the disease in countries where biochemical screening tests are not routinely used. This Primer provides, within a historical framework, the evolution of this disease clinically, advances in our knowledge of its epidemiology, pathogenesis, presentations, evaluation and management.
Epidemiology
When the first several hundred individuals with PHPT were described in the 1930s at the Massachusetts General Hospital, 57% had kidney stones, 8% had peptic ulcer disease and 23% had bone complications1. Over the past five decades, the clinical presentation of PHPT has changed in several regions of the world2. Since the 1970s, PHPT has become more routinely diagnosed at an asymptomatic stage owing to increased screening2. The clinical evolution of PHPT from symptomatic to asymptomatic has occurred primarily in the United States and Europe, although other countries have more recently appreciated this change as well3. One of the vexing issues that clinicians deal with is whether the asymptomatic patient has evidence of target organ involvement. This is the reason why this group of patients has been the subject of such intense interest over the past four decades.
Comparing prevalence and clinical aspects of the disease within and between countries is problematical owing to varying levels of screening availability and sources of information (for example, referral centres versus country demographics). Nevertheless, summarizing the varying presentations of PHPT globally is instructive.
North America
PHPT presents primarily as an asymptomatic disorder in North America4. The prevalence of kidney stones and skeletal manifestations has markedly decreased in the past 40 years. In addition, prevalence data show considerable sex and ethnic differences. The Kaiser Permanente Health Care database in California, USA, showed a significantly higher incidence of PHPT among black individuals (92 per 100,000 in women and 46 per 100,000 in men) than in white individuals (81 per 100,000 in women and 29 per 100,000 in men)5. Other races demonstrated a lower incidence. In Asian-American populations, for example, the incidence was 52 per 100,000 in women and 28 per 100,000 in men. In the Hispanic population, the incidence was 49 per 100,000 in women and 17 per 100,000 in men5. During the 15-year period of this study, the Kaiser Permanente Health Care database showed an overall threefold increase in incidence, which is perhaps a reflection of increased screening5 (FIG. 1). Another survey conducted in the United States estimated a PHPT prevalence of 0.86% for the general population6. The prevalence of normocalcaemic and hypercalcaemic PHPT in Canada was evaluated in the Canadian Multicenter Osteoporosis Study7 and was found to be 3.3% and 1.4%, respectively.
Figure 1. Incidence of PHPT.
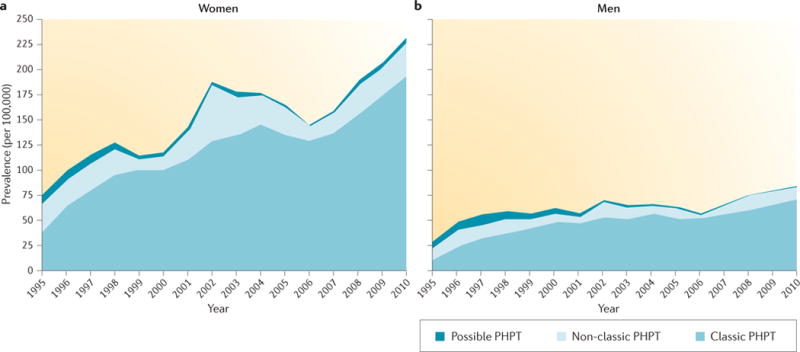
The incidence of primary hyperparathyroidism (PHPT) in women (part a) and men (part b) as determined from a large epidemiological database from the United States. Over a 15-year period, the incidence tripled even in the setting of routine use of biochemical screening tests5. Republished with permission of Endocrine Society, from Incidence and prevalence of primary hyperparathyroidism in a racially mixed population, Yeh, M. W. et al., 98, 3, 1122–1129, 2013; permission conveyed through Copyright Clearance Center, Inc.
In the United States, a 60% increase in the incidence of parathyroid cancer, a very rare presentation of PHPT, was reported between 1988 and 2003 (REF. 8). This apparent increase could be owing to greater awareness of the entity and uncertainty, at times, in the pathological characteristics of parathyroid cancer versus parathyroid adenoma and the tendency, over the years, to favour a cancer diagnosis when uncertain.
Europe
In Europe, PHPT is also identified most commonly as a relatively common, asymptomatic disorder. A Swedish study identified PHPT in 1.6% of women and 0.3% of men9. The incidence of PHPT also increased in Denmark between 1999 and 2010 (REF. 10), probably reflecting increased screening. Monitoring in Scotland, UK, over a 20-year period (1997–2006) showed, as has been shown everywhere, that PHPT predominantly affects women11.
Latin America
In Latin America, 47% of patients presented with asymptomatic disease in a case series of 124 patients, whereas 25% had overt skeletal manifestations12. Approximately 44% of people with PHPT had kidney stones at the time of presentation in a study from Argentina13. In Brazil, 81.8% of patients were asymptomatic at the time of presentation14. However, kidney stones and osteitis fibrosa cystica (a distinct PHPT-associated complication that is associated with bone loss, excessive fibrosis of the collagen matrix and the formation of bone cysts) were present in 18.2% and 6.1% of patients, respectively. The mean age was 61.1 years (±16 years).
Asia
Although epidemiological studies on the prevalence of PHPT have not been conducted in Asian countries, it seems that, in countries such as China and India, patients with PHPT still present with classic PHPT, namely, with organ manifestations and relatively high serum levels of calcium15,16. However, a clear trend for PHPT to present more commonly as an asymptomatic disorder in China has been emerging over the past 5–10 years17. In the 35-year period between 1958 and 1993, 97% of PHPT in China was characterized by kidney stones, classic skeletal lesions (for example, osteitis fibrosa cystica, generalized skeletal demineralization and pathological fractures) and other features of symptomatic PHPT16. Recently, the clinical profile of 249 patients with PHPT who were diagnosed and treated from 2000 to 2010 in one clinical centre in China was compared to American patients17,18. Chinese patients were younger (51.3 years of age) than American patients (66.4 years of age) at diagnosis and were less likely to be women (female/male ratio of 2.07/1) than patients in New York City, USA (female/male ratio of 4.5/1). The average calcium levels (11.7 ± 1.4 mg per dl in China versus 10.6 ± 0.6 mg per dl in the United States), PTH levels (402 pg per ml (range: 103–2,700 pg per ml) in China versus 68 pg per ml (range: 48–95 pg per ml) for the United States) and creatinine levels (1.03 mg per dl (range: 0.55–2.75 mg per dl) for China and 0.79 mg per dl (range: 0.59–1.23 mg per dl) for the United States) in serum were all significantly higher, whereas the average 25-hydroxyvitamin D concentration (13 ng per ml (range: 5–30 ng per ml) in China versus 37 ± 16 ng per ml in the United States) was much lower. Among all Chinese patients, 60% manifested classic symptoms related to PHPT, including polydipsia and polyuria (79.7%), kidney stones (78.4%), bone pain (69.9%) and fatigue (62.8%).
However, the clinical profile of Chinese patients is changing. The proportion of asymptomatic patients with PHPT rose from <20% before 2006 to ~50% in the period 2007–2010. This change has largely been driven by the availability of routine serum calcium testing and the incidental discovery of a parathyroid gland on neck ultrasonography17. A survey in Hong Kong, China, also demonstrated a steady increase in the prevalence of asymptomatic PHPT from 5% between 1973 and 1982 to 39% between 1983 and 1992 and to 59% between 1993 and 2002 (REF. 19).
Although China seems to be witnessing a change in the clinical presentation of PHPT, other Asian countries including India, Iran, Pakistan, Saudi Arabia and Thailand still mostly report symptomatic PHPT; asymptomatic PHPT remains rare in these regions (0–2.2% of all PHPT diagnoses)15,20–24. The number of patients diagnosed in a single centre in north India steadily increased between 1990 and 2010, with 28 cases diagnosed during 1990–1999 and 174 cases during 2000–2010. PHPT was predominantly diagnosed in women (70.3%), and virtually all 202 patients (99.1%) were symptomatic with kidney and skeletal involvement25. A similar presentation was described in Western India20.
Parathyroid carcinoma has been reported in 6% of patients with PHPT in China (a total of 235 patients) and was even as high as 11.5% (a total of 87 patients) in Japan17,26. These figures are much higher than in countries in the west. However, a decreasing trend for the incidence of parathyroid carcinoma over the past 10 years was noted in Chinese patients with PTHT17.
Africa and Australia
Information about PHPT in Africa is limited. A retrospective analysis of 28 patients with PHPT in a referral hospital in Durban, South Africa, between 2003 and 2009 reported that PHPT was still presenting with symptoms27.
In a recent study from Australia, 561 patients with PHPT were followed up for >30 years. Mortality figures were higher than the general Australian population28.
Summary
In summary, the breakdown of individuals with symptomatic versus asymptomatic PHPT has regional specificity. In countries for which screening biochemistries are not routinely part of the health care system, symptomatic PHPT seems to predominate. In countries where screening biochemistries are routine, asymptomatic PHPT takes centre stage. In short, symptomatic disease tends to be more common in countries where the prevalence is lower. All studies, independent of the breakdown of symptomatic versus asymptomatic PHPT in a given country, have shown that postmenopausal women are affected much more commonly than men. The incidence rates between premenopausal women and men are simi-lar5. European studies suggest increased mortality29, but mortality rates are not higher in North America30.
Mechanisms/pathophysiology
The pathophysiology of PHPT relates to the loss of the homeostatic control of PTH synthesis and secretion (FIG. 2), leading to increased PTH secretion by individual cells or increased parathyroid cell proliferation, but with each cell secreting a normal level of PTH.
Figure 2. Control of parathyroid hormone synthesis and secretion.
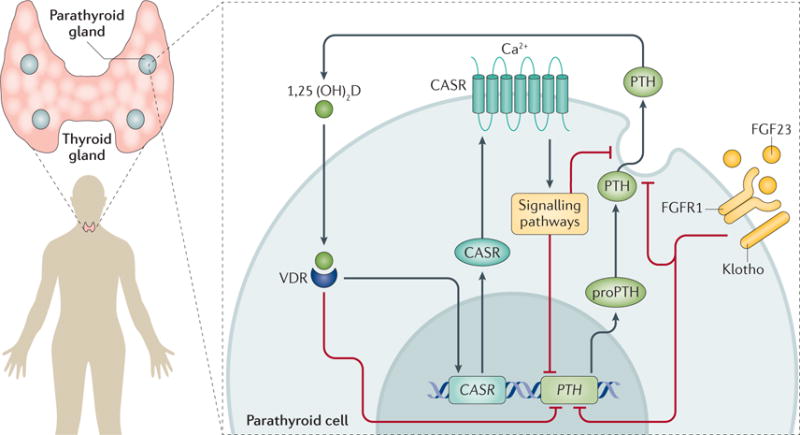
In humans, the parathyroid hormone gene (PTH) is located on chromosome 11. The formation of mature PTH requires cleavage of the pre-sequences and pro-sequences (25 and 6 amino acid residues, respectively); the mature PTH peptide (84 amino acids) is packaged into secretory vesicles in the parathyroid chief cells. PTH can either be secreted by exocytosis, degraded within the secretory vesicles (producing fragments that are released in the circulation) or sequestered in a stored pool169. The principal regulators of PTH secretion are extracellular ionized calcium (Ca2+) and 1,25-dihydroxyvitamin D (1,25(OH)2D). Other potentially important regulators include serum phosphate and fibroblast growth factor 23 (FGF23). A rise in extracellular ionized calcium levels activates the calcium-sensing receptor (CASR), which suppresses PTH expression. In addition, PTH expression is suppressed by 1,25(OH)2D and FGF23. FGFR1, fibroblast growth factor receptor 1; VDR, vitamin D receptor.
In parathyroid hyperplasia, the increased number of parathyroid cells maintain their normal sensitivity to calcium, whereas in parathyroid adenomas, the parathyroid cells show a lower than normal sensitivity to the inhibitory action of calcium (REFS 31,32). Both conditions give rise to PHPT and can cause hypercalcaemia.
PTH endocrine axis and target tissues
Calcium is the main regulator of PTH secretion, with an inverse sigmoidal relationship between calcium concentration and PTH release that is mediated by the interaction of calcium with the calcium-sensing receptor (CASR) present on the surface of parathyroid cells33 (FIG. 3). The active form of vitamin D — 1,25(OH)2D — suppresses PTH transcription and parathyroid cell proliferation. These effects are not only due to circulating 1,25(OH)2D synthesized in the kidney but also due to local synthesis by the 1α-25-hydroxyvitamin D hydroxylase enzyme present in parathyroid cells34. An increase in the serum levels of phosphate indirectly stimulates PTH synthesis and secretion as well as parathyroid cell proliferation by binding to calcium, thereby lowering the serum calcium concentration35. Some studies have suggested that serum phosphate can directly affect parathyroid cell function by increasing PTH mRNA stability36. Finally, studies in rats and rat parathyroid cell cultures have shown that fibroblast growth factor 23 (FGF23), released from osteocytes (bone cells involved in mechanosensing and mineral homeostasis), inhibits PTH synthesis and secretion and, perhaps, parathyroid cell proliferation by binding to its cognate FGF receptor 1 (FGFR1) and the coreceptor Klotho on the membrane of parathyroid cells37. The relevance of this observation to the pathogenesis of PHPT in humans remains uncertain. Indeed, patients with PHPT have increased levels of FGF23 in association with high serum levels of PTH, suggesting that the increased FGF23 levels might be an adaptive response to counteract increased 1,25(OH)2D levels as a consequence of high PTH levels38.
Figure 3. Relationship between calcium and parathyroid hormone levels in normal conditions and in PHPT.
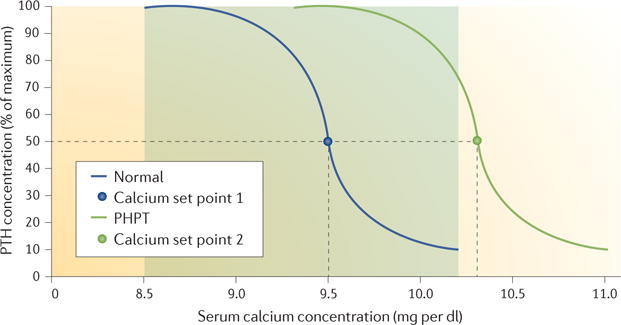
In normal conditions, the level of parathyroid hormone (PTH) secretion depends on the level of calcium, with a high level of PTH secreted when serum calcium is at the lower limit of the normal range and a low level when serum calcium is at the upper limit of the normal range. The range of normal serum calcium levels is indicated by the shaded area. The calcium set point corresponds to the concentration of calcium, which reduces PTH secretion by 50%. In primary hyperparathyroidism (PHPT), the parathyroid adenoma is relatively insensitive to the feedback suppression by calcium, and the curve is shifted to the right with an increase in the set point for serum calcium. As a consequence, PHPT is often associated with hypercalcaemia.
The main target organs of PTH are the bone and the kidney. PTH is a central regulator of bone homeostasis, through its action on bone-forming osteoblasts, osteocytes and bone-resorbing osteoclasts39. The final effect of PTH on bone mass is either anabolic or catabolic and will depend on the dose and periodicity of PTH signalling40. In healthy individuals, PTH is released both with circadian dynamics and in a pulsatile manner, such that its dual anabolic and catabolic actions are well balanced. Conversely, in patients with PHPT, the chronically increased PTH levels lead to bone loss mediated by the receptor activator of nuclear factor-кβ ligand (RANKL, also known as TNFSF11), osteoporosis (particularly at sites that are more rich in cortical bone) and fragility fractures. It is not clear to what extent changes in these secretory dynamics are associated with bone loss and fragility fractures in PHPT41.
In the kidney, PTH stimulates tubular calcium reabsorption and phosphate excretion and stimulates the activity of 1α-25-hydroxyvitamin D hydroxylase. When patients with PHPT develop hypercalciuria, the filtered calcium load is greater than the capacity of the kidney to reabsorb calcium efficiently, even under the influence of PTH42.
Aetiological factors of tumorigenesis
PHPT is usually (>90%) sporadic and caused by a solitary benign adenoma (85–90%), less frequently by multiglandular involvement consisting of either multiple adenomas or hyperplasia of all four glands (5–10%), and very rarely by parathyroid carcinoma (<1%)43. Multiglandular involvement may present in an asynchronous manner and, therefore, can be confused with single-gland adenoma when only one abnormal gland is found at the time of initial surgery44. Over time, another parathyroid adenoma can become clinically evident. Conversely, multiple adenomas, presenting simultaneously, can also occur45. Very rarely, PHPT may be caused by ectopic secretion of PTH by a non-parathyroid tumour44.
The aetiology of PHPT remains elusive in the majority of patients. A history of external neck irradiation in childhood46, exposure to a nuclear incident in adults47 and long-term lithium therapy (as opposed to short-term lithium therapy that may cause secondary hyperparathyroidism)48 are present in a few cases. PHPT may also be part of hereditary endocrine syndromes, including multiple endocrine neoplasia type 1 (MEN1) and MEN2A, MEN4 (which is a MENl-like condition (associated with one or more of the main MEN1-associated tumours, but without MEN1 gene mutations), hereditary hyperparathyroidism-jaw tumour syndrome, familial isolated hyperparathyroidism, familial hypocalciuric hypercalcaemia (FHH) and neonatal severe hyperparathyroidism (TABLE 1). The identification of hereditary cases can sometimes be difficult in the absence of a family history and, in these settings, a de novo germline mutation should be considered.
Table 1.
Gene abnormalities associated with syndromic and non-syndromic forms of PHPT
| Disorder | OMIM entry | Gene | Chromosomal locus | Pattern of inheritance |
|---|---|---|---|---|
| Syndromic forms of PHPT | ||||
| MEN1 | 131100 | MEN1 | 11q13.1 | Autosomal dominant |
| MEN2A | 171400 | RET | 10q11.21 | Autosomal dominant |
| MEN4 | 610755 | CDKN1B | 12p13.1 | Autosomal dominant |
| Hereditary hyperparathyroidism-jaw tumour | 145001 | CDC73 (also known as HRPT2) | 1q31.2 | Autosomal dominant |
| Non-syndromic forms of PHPT | ||||
| Familial isolated hyperparathyroidism | 145000 | MEN1, CDC73 and CASR, among others* | 11q13.1,1q31.2 and 3q13.3–q21.1 | Autosomal dominant |
| FHH type 1 | 145980 | CASR | 3q13.3–q21.1 | Autosomal dominant |
| FHH type 2 | 145981 | GNA11 | 19p13.3 | Autosomal dominant |
| FHH type 3 | 600740 | AP2S1 | 19q13.32 | Autosomal dominant |
| Neonatal severe hyperparathyroidism | 239200 | CASR | 3q13.3–q21.1 | Autosomal recessive |
FHH, familial hypocalciuric hypercalcaemia; MEN, multiple endocrine neoplasia; OMIM, Online Mendelian Inheritance in Man; PHPT, primary hyperparathyroidism. *Not yet identified.
Molecular mechanisms of tumorigenesis
Evaluation of the clonal status of a parathyroid tumour (including adenomas and carcinomas), has shown that most are monoclonal, which implies that the tumours derive from a single abnormal cell49,50. By contrast, hyperplastic parathyroid glands probably originate from a stimulus for generalized (polyclonal) parathyroid cell proliferation, even though monoclonal tumours may also occur in this context51. Thus, in some cases, apparently hyperplastic glands are in fact adenomas. The clonality of parathyroid tumours has also been shown in MEN1 (REFS 52,53).
The clonality ofmost parathyroid tumours suggests the involvement of genes that control parathyroid cell growth and/or PTH synthesis or secretion. Two broad categories of genes might be involved: proto-oncogenes and tumour-suppressor genes (FIG. 4). Notably, tumorigenesis is often a multistep process during which cells acquire a series of mutations or deletions in multiple genes, which can in some instances coexist. Moreover, epigenetic changes could contribute, in certain specific ways, to tumour development. Several genes have been implicated in sporadic and hereditary forms of PHPT. Although syndromic forms account for <10% of PHPT, the identification of genes involved in the pathogenesis of inherited forms has contributed to the understanding of parathyroid tumorigenesis.
Figure 4. Oncogenes and tumour-suppressor genes in parathyroid tumours.
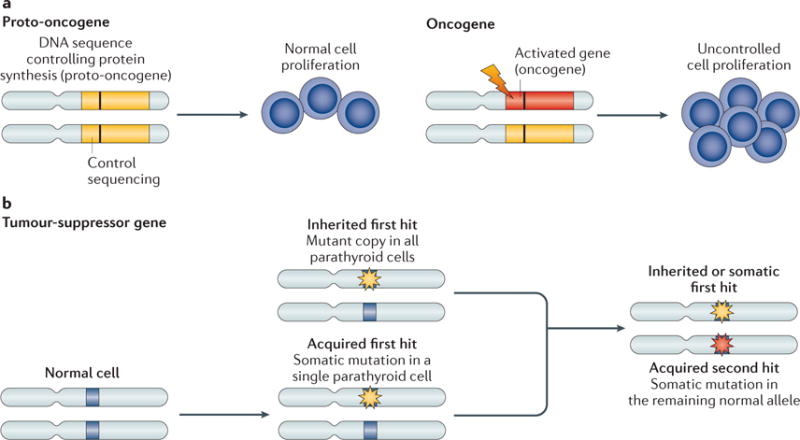
a | Proto-oncogenes have a physiological role in the control of cell proliferation, differentiation and cell death. The conversion of proto-oncogenes to oncogenes (due to chromosome inversion, translocation or point mutation) causes an increased expression of its encoded protein or the synthesis of a new protein with increased function. Most oncogenes are dominant mutations; a single copy of the gene is sufficient to promote tumorigenesis. The conversion of a proto-oncogene mainly occurs in somatic cells. Its presence in a germline cell does not necessarily result in neoplastic transformation, but can increase the risk of it in the offspring. b | Tumour-suppressor genes encode proteins that normally inhibit cell proliferation and, once mutated, contribute to tumour development through their functional inactivation. Loss-of-function mutations in tumour-suppressor genes act recessively; one copy of the gene suffices to control cell proliferation. Inactivation of a tumour-suppressor gene is usually caused by point mutations or deletions, which may either be inherited in all parathyroid cells or occur somatically in a single parathyroid cell (first hit). An acquired somatic deletion or mutation of the other copy of the gene (second hit) will lead to loss of function of the related gene product.
FIGURE 5 summarizes the main molecular mechanisms involved in parathyroid tumorigenesis.
Figure 5. Parathyroid tumorigenesis.
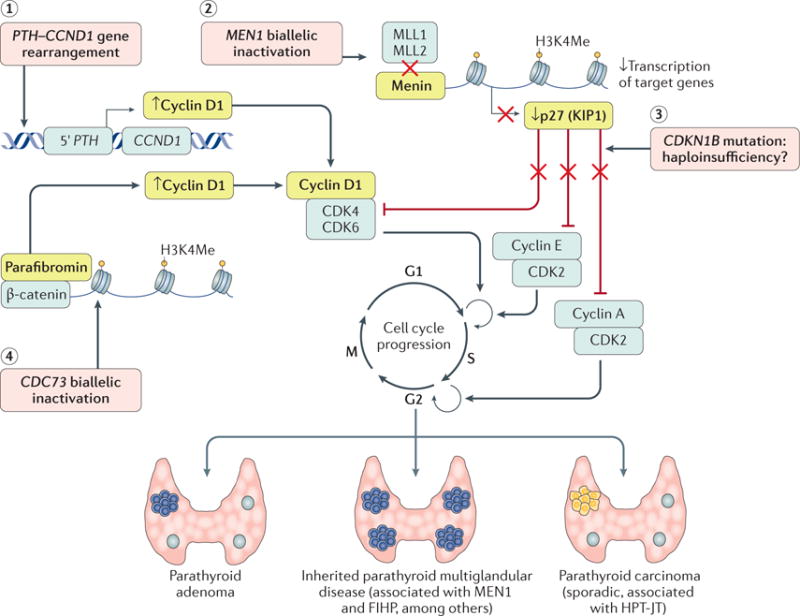
(1) Cyclin D1 (encoded by CCND1), a regulatory subunit of cyclin-dependent kinase 4 (CDK4) and CDK6 required for progression through the G1 phase of the cell cycle, is frequently (20–40%) overexpressed in parathyroid adenomas. In a subset of these adenomas, a pericentromeric inversion of chromosome 11 places the parathyroid hormone gene (PTH) promoter sequences on 11p15 immediately upstream of CCND1. This rearrangement results in the upregulated expression of cyclin D1 upon activation of CDKs. (2) Loss-of-function mutations of MEN1 (which encodes menin) represent the main cause of multiple endocrine neoplasia type 1 (MEN1) syndrome (in up to 80% of cases), especially in the familial setting, but are also found in a subset of familial isolated hyperparathyroidism (FIHP) and in 12–35% of sporadic parathyroid adenomas. MEN1 functions as a classic tumour-suppressor gene and is frequently associated with somatic loss of heterozygosity. Menin is a component of the histone methyltransferase complex, including the histone-lysine N-methyltransferases MLL1 (encoded by KMT2A) and MLL2 (encoded by KMT2D), required for histone H3 lysine 4 trimethylation (H3K4Me) of the target genes. In physiological conditions, activation of this complex leads to the basal transcription of CDKN1B, encoding the CDK inhibitor p27 (KIP1), which stops progression of the cell cycle by inhibiting CDK complexes acting at multiple phases of the cell cycle, especially at G1-S and S-G2 checkpoints. MEN1 inactivation results in a reduction of nuclear expression levels of p27 (KIP1) and the loss of negative control of cell cycle progression. (3) CDKN1B germline mutations are the molecular cause of the MEN4 syndrome. CDKN1B is an atypical tumour-suppressor gene because biallelic inactivation is an uncommon event, suggesting that only a single inactivated copy of the gene is sufficient to lead to a diseased state (haploinsufficiency). (4) Somatic and germline inactivating CDC73 (which encodes parafibromin) mutations have been frequently identified in patients with parathyroid carcinoma. Germline CDC73 mutations are also responsible for hyperparathyroidism-jaw tumour syndrome (HPT-JT). Parafibromin binds to DNA and leads to repression of cyclin D1 and inhibition of proliferation. In case of CDC73 inactivation, parafibromin seems to associate with β-catenin, a central mediator of WNT signalling, and activates target genes (for example, MYC and CCDN1) through H3K4Me.
Syndromic forms of PHPT
Multiple endocrine neoplasia type 1
MEN1 shows an autosomal dominant pattern of inheritance, and the genetic basis responsible for the predisposition to tumour development is a germline mutation in MEN1 (encoding menin) (REF. 54) (FIG. 5; TABLE 1). More than 1,000 mutations have been identified in ~80% of probands and families with MEN1. The mutations are distributed across the translated region of the gene without hotspots; the majority of mutations predict truncation or the absence of the encoded protein (menin)55. Individuals with MEN1 typically inherit the inactivating germline mutation from an affected parent, but up to 10% of affected individuals have a de novo germline mutation. An acquired somatic inactivation of the other copy of MEN1 in one parathyroid cell, leading to the absence of functional menin, contributes to the clonal origin of the parathyroid tumour and other tumours in the patient54 (FIG. 5).
Menin is a 610 amino acid protein that interacts in vitro with various proteins, including the transcription factor AP-1 (encoded by JUN), nuclear factor-кB and the SMAD transcription factors, as well as other cell cycle regulators and cytoskeletal proteins, all suggesting a role in multiple biological pathways56. The interaction of menin with a histone methyltransferase complex, including the histone-lysine N-methyltransferases MLL1 (encoded by KMT2A) and MLL2 (encoded by KMT2D), can be involved in MEN1-associated tissue-selective tumorigenesis57, mediated by the transcriptional activation of CDKN1B (encoding cyclin-dependent kinase inhibitor 1B) and CDKN2C58,59. Recently, a germline mutation of the aryl hydrocarbon receptor-interacting protein gene (AIP) has been reported in a patient with sporadic MEN1 syndrome60. Mutations of other genes, encoding cyclin-dependent kinase inhibitors, including CDKN1A, CDKN1B (MEN4, see below), CDKN2B and CDKN2C, have been identified in MEN1 mutation-negative kindreds with MEN1 disease61.
Multiple endocrine neoplasia type 2A
The inheritance of MEN2A is autosomal dominant and due to gain-of-function mutations in the RET proto-oncogene (TABLE 1). RET is a tyrosine kinase receptor that is involved in the control of growth and differentiation in developing tissues, including those derived from the neural crest. A RET mutation at codon 634 is highly associated with the development of PHPT62. Parathyroid tumours occur in 10–25% of patients with MEN2A; in ~50% of patients that develop tumours, no hypercalcaemia is observed62.
Multiple endocrine neoplasia type 4
In 2006, Pellegata et al.63 described a rat model that spontaneously developed multiple neuroendocrine tumours, with a spectrum similar to that of MEN1 and MEN2 syndromes. The same authors showed that a homozygous germline mutation in Cdkn1b, encoding p27, was responsible for this condition, which was named MENX63. Heterozygous germline mutations of the human homologue CDKN1B were identified in kindreds with classic MENl-associated tumours (for example, pituitary, parathyroid and pancreatic tumours) occurring in association with uncommon MENl-associated or non-associated tumours (for example, adrenal and thyroid tumours, and gonadal and renal tumours, respectively) without a mutation in MEN1 (REF. 64). These kindreds with MENl-associated tumours and CDKN1B mutations are now designated as a new syndrome called MEN4 (REFS 65,66) (TABLE 1). In Gl phase cells (resting), p27 (KIP1) is found in the nucleus, where it controls cell cycle progression from G1 to S phase, the mechanism for which involves inhibiting the cyclin E-CDK2 and cyclin A–CDK2 complexes. In proliferating cells, p27 (KIP1) is also localized to the cytoplasm, where it seems to have a pro-oncogenic function67.
Hereditary hyperparathyroidism-jaw tumour syndrome
Mutations in CDC73 are responsible for hereditary hyperparathyroidism-jaw tumour syndrome68,69 (TABLE 1). This is a rare autosomal disorder consisting of parathyroid tumours, with a high prevalence of parathyroid carcinomas (l5%), ossifying fibromas of the mandible and maxilla and, occasionally, renal and uterine lesions. CDC73 is a tumour-suppressor gene that encodes parafibromin, a 53l amino acid nuclear protein that is also expressed in the kidney, heart, adrenal glands and skeletal muscle. Parafibromin is a member of the human polymerase-associated factor 1 (PAF1) complex, which regulates transcription and chromatin modification70 (FIG. 5).
Non-syndromic forms of hyperparathyroidism Familial isolated hyperparathyroidism
Familial isolated hyperparathyroidism is a familial form of PHPT in which no other manifestations typical of a syndromic form of PHPT are present, suggesting incomplete penetrance. The disorder is genetically heterogeneous, but mutations of MEN1, CDC73, CASR and CDKN1B have been reported in a minority of cases71,72.
FHH and neonatal severe hyperparathyroidism
FHH and neonatal severe hyperparathyroidism are due to inactivating mutations in CASR. FHH is genetically heterogeneous and three different variants have been identified73. Most cases of FHH are caused by heterozygous inactivating mutations of CASR on 3q13.3–q21.1 (FHH type 1)74. The mutations are distributed along the entire gene, but are most predominant in the regions encoding the extracellular and transmembrane domains33. Approximately 30% of patients with FHH type 1 lack mutations in CASR. Recent studies have identified two additional forms of FHH linked to chromosome 19, which are due to mutations of G protein subunit all (GNA11; FHH type 2)75 and adaptor-related protein complex 2σ 1 subunit (AP2S1; FFH type 3)75. Neonatal severe hyperparathyroidism results from inheritance of two abnormal CASR alleles in either the homozygous or the compound heterozygous state73.
The main sites where the CASR is mainly expressed are parathyroid cells and those in the thick ascending loop of the renal tubule. Reduction or loss of function of CASR results in impaired calcium inhibition of PTH secretion in parathyroid cells and in reduced calcium excretion at any serum calcium concentration in the kidney33. Gene inactivation studies in mice support the pathogenetic role of CASR inactivation in FHH type 1 and neonatal severe hyperparathyroidism76.
Sporadic form of PHPT
Parathyroid adenoma
Overexpression of cyclin D1 (encoded by CCND1) and mutations of MEN1 are well-known factors in the pathogenesis of sporadic parathyroid adenomas44. CCND1 was discovered in parathyroid adenomas and mapped on 11q13. In these tumours, a pericentromeric inversion causes a tumour-specific DNA rearrangement of CCND1 with PTH and is responsible for transcriptional activation and overexpression of cyclin D1 (REFS 77,78) (FIG. 5). This genetic abnormality has been detected in ~8% of sporadic parathyroid adenomas, although an increased expression of CCND1 has been reported in ~20–40% of cases. Cyclin D1 plays an important part in the regulation of cell cycle progression. Its effects on proliferation are mediated by the phosphorylation of retinoblastoma protein (RB). Biallelic-acquired inactivating MEN1 mutations have been described in 12–35% of sporadic parathyroid adenomas79–81.
Other genetic abnormalities have been detected in a small proportion of parathyroid adenomas, including mutations in CDC73 (REFS 82,83), CDKN1B84 and AIP85. Interestingly, mutations were germline in two patients with CDKN1B mutations and in one carrying a mutation in AIP Contradictory results have been reported on the presence of CTNNB1 (encoding β-catenin) mutations involving serine/threonine phosphorylation86,87. Up to 30 proteins, involved in various cellular pathways (for example, response to biotic stimuli, cell organization and signal transduction), are differentially expressed in normal parathyroid and parathyroid adenomas88. Of these various proteins, 14-3-3ζ/δ may be one of the key proteins in the pathogenesis of parathyroid adenoma.
Parathyroid carcinoma
Loss of RB1 expression in parathyroid carcinoma was initially reported by Cryns et al.89 and was proposed as a diagnostic tool, but contradictory results were shown by others90 and no abnormalities in the coding or promoter regions of RB1 have been identified91. CDC73 is the most common mutated gene (up to 70%) in sporadic parathyroid carcinoma69,90,91. Interestingly, mutations are germline in about one-third of patients, suggesting that they may be affected by hereditary hyperparathyroidism-jaw tumour syndrome or a variant instead of sporadic tumours90,91. Another mechanism of CDC73 inactivation — methylation of the promoter — has been reported in a small subset of parathyroid carcinoma92. Recently, using whole-exome sequencing, germline and somatic mutations have been detected in 4 out of 22 (18%) patients with parathyroid carcinoma. Occasional mutations of MEN1 have also been reported93. Two recent studies suggest a potential role of microRNA-296 as a novel oncosuppressor gene in parathyroid carcinoma94,95.
Diagnosis, screening and prevention
Clinical manifestations
Osteitis fibrosa cystica describes the skeletal manifestations of PHPT when the disease is advanced. It is typically characterized by bone pain, skeletal deformities and pathological fractures12 (FIG. 6). Conversely, in asymptomatic PHPT, skeletal involvement is typically seen only by bone mineral density (BMD) testing, which shows a reduction, especially of the cortical bone compartment best seen at the distal one-third of the radius site. Recent data using high-resolution peripheral quantitative CT (HRpQCT) have demonstrated that both cortical and trabecular bone compartments are affected in PHPT96,97. Involvement of the trabecular compartment is further documented by the trabecular bone score, which is also reduced in the lumbar spine98,99, and by fractures that are increased at both vertebral and non-vertebral sites100,101. These observations support recommendations to include vertebral imaging either by radiography, CT or vertebral fracture assessment to evaluate skeletal manifestations of PHPT (BOX 1). After parathyroidectomy, BMD increases even in patients with mild disease. When PHPT is associated with osteitis fibrosa cystica, the improvement in BMD can be so marked as to increase into the normal range42,102.
Figure 6. Overt bone disease in PHPT.
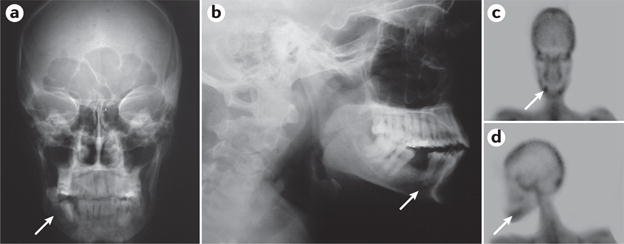
Osteolytic lesion due to a brow n tumour of the jaw from an individual with severe primary hyperparathyroidism (PHPT; arrow; part a and part b). In addition, note the increased uptake of 99m-technetium-methyl diphosphonate on the bone scan (arrow; part c and part d).
Box 1. Parameters for the evaluation of patients with PHPT.
Laboratory tests
General markers: biochemical screening panel including measuring the levels of creatinine and blood urea nitrogen
Mineral homeostasis: serum calcium (corrected for albumin) or ionized, PTH (second-generation or third-generation assay), phosphate and 25-hydroxyvitamin D levels
Urine (24-hour): calcium and creatinine levels. If the urine calcium level is >400 mg per day, a stone risk analysis profile (including urinary phosphate, oxalate, sodium, uric acid, sulfate and citrate levels) should be performed
Bone resorption marker: serum C-telopeptide crosslinked type I collagen or urine N-telopeptide of type I collagen levels*
Bone formation marker: serum bone-specific alkaline phosphatase, procollagen type I N-propeptide or osteocalcin levels*
If clinically indicated: genetic testing
Imaging
Bone mineral density analysis by DXA of the lumbar spine, hip (total and femoral neck) and distal one-third of the radius
Vertebral fracture evaluation by X-ray or vertebral fracture assessment by DXA
Abdominal imaging for kidney stones or nephrocalcinosis (ultrasonography, CT or abdominal X-ray)
Optional, if available: high-resolution peripheral quantitative CT or trabecular bone score evaluation using DXA
DXA, dual-energy X-ray absorptiometry. *Many clinicians find bone formation and bone resorption markers to be helpful as they indicate the level of bone turnover. All the available bone turnover markers are listed, but if the clinician elects to obtain this information, only a single bone formation and a single bone resorption marker would be necessary.
The prevalence of kidney stones in PHPT has varied over the years and, to a certain extent, is dependent on the use of imaging modalities to detect them. Overt kidney stone disease (symptomatic kidney stones) has declined from 60% when series of PHPT were first reported about 50 years ago to more-current estimates of ~20%103–105. By contrast, abdominal imaging of asymptomatic patients with PHPT is yielding a higher prevalence of silent stones and nephrocalcinosis than was thought to be the case in asymptomatic PHPT106,107.
Weakness and fatigue were initially thought to be associated with a neuromuscular syndrome, characterized by atrophy of type II muscle fibres of the proximal musculature108. However, over time, as the disease has evolved into a more asymptomatic phenotype, these objective findings on neurological examination are no longer seen109. Among asymptomatic patients with PHPT, a detailed examination using electroneurography is required to disclose subtle abnomalities of peripheral nerves110. General symptoms — in particular, fatigue, weakness, anxiety and mood alterations — along with impairment in quality of life (QOL) may or may not improve after surgical cure (see below)111.
Peptic ulcer disease, which used to be considered a frequent complication of PHPT, is now rarely seen and is almost exclusively detected in patients with MEN1 or MEN4 syndromes, who can develop gastrin-producing tumours112. Similarly, the association between PHPT and acute pancreatitis, apart from that related to hypercalcaemia per se, remains to be established113.
With regard to cardiovascular health, hypertension, premature atherosclerosis, valve calcification, left ventricular hypertrophy and arrhythmias have been reported in patients with PHPT114–116, but the data on outcome as well as the mechanisms involved are inconclusive. The Nurses’ Health study114 showed an increased incidence of PHPT in women with hypertension compared with those with normal blood pressure values. In addition, no evidence of increased left ventricular mass or diastolic dysfunction in patients with biochemically mild PHPT was found, but the finding of higher serum calcium and PTH levels in patients with diastolic dysfunction suggests that disease severity of PHPT may be associated with cardiac manifestations115. Iwata et al.116 observed that mild PHPT was associated with subclinical aortic valve calcification. Serum PTH levels, but not serum calcium levels, were a more important predictor of aortic valve calcification than traditional cardiovascular risk factors.
Laboratory diagnosis
Confirmed hypercalcaemia is a major indicator of PHPT117 (BOX 1). Either total calcium (albumin corrected) or ionized calcium can be measured. As ~45% of total serum calcium is bound to albumin, calculating the corrected calcium level involves adjusting the level of total serum calcium by the level of serum albumin. The many technical issues related to the direct measurement of ionized calcium lead many experts to recommend the adjusted total calcium as the best way to report the serum calcium concentration. It is helpful to repeat the serum calcium measurements several times over the course of a 3–6–month period and to retrieve historical serum calcium values that are available from the patient’s record.
A major diagnostic linchpin in PHPT is the measurement of circulating PTH. Second-generation assays that measure the so-called intact PTH molecule [PTH(1–84)] show crossreactivity with the large, inactive, amino-truncated fragment [PTH(7–84)]. This fragment can also be detected in individuals without PHPT and can accumulate in patients with renal failure. The third-generation or bio-intact assay uses a labelled antibody that recognizes an extreme N-terminal PTH fragment [PTH(1–4)], preventing crossreactivity with PTH(7–84). However, this assay also detects a post-translational form of PTH in the region 5–20 that is not truncated. This post-translationally modified form of PTH (N-PTH) represents 10% of PTH in healthy individuals and 15% in patients with chronic kidney disease, and is not thought to be active. In parathyroid cancer, it seems to be overrepresented118. There are few studies comparing the diagnostic sensitivity between the second-generation and third-generation assays, but both perform generally well119.
In PHPT, the serum PTH levels may be frankly increased or in the normal range. However, a ‘normal’ PTH value in someone with hypercalcaemia is clearly not a normal physiological value. For example, in a large epidemiological study in Scotland, UK11, patients with hypercalcaemia on two or more occasions and an intact serum PTH concentration of >28 pg per ml (reference range: 19–65 pg per ml) were considered to have PHPT. Indeed, in individuals without PHPT who become hyper-calcaemic, PTH should be completely suppressed and undetectable by immunoassay.
Normocalcaemic PHPT
Normocalcaemic PHPT has been increasingly detected in patients who are evaluated for low BMD117. Other causes of hyperparathyroidism due to a normal physiological stimulus to increased PTH levels — secondary hyperparathyroidism states — should be excluded (BOX 2). Bone resorption inhibitors, such as bisphosphonates and denosumab, usually cause increases in serum PTH concentrations, rendering a diagnosis of normocalcaemic PHPT under these conditions uncertain120,121. The diagnosis of parathyroid gland autonomy in the setting of chronic kidney disease is more challenging. In patients with advanced chronic kidney disease, the presence of hypercalcaemia along with a progressive rise in serum PTH levels is usually referred to as tertiary hyperparathyroidism. In advanced-stage chronic kidney disease, intact PTH levels in serum should be maintained between 150 and 550 pg per ml to avoid low-turnover or high-turnover bone disease122.
Box 2. Causes of secondary hyperparathyroidism.
Secondary hyperparathyroidism includes conditions in which the parathyroid hormone (PTH) level is increased because of a stimulus that expectedly would increase the levels of PTH.
Decreased intestinal calcium absorption
Vitamin D deficiency: serum 25-hydroxyvitamin D levels of <20 ng per ml
Bariatric surgery
Malabsorption syndromes
Decreased calcium intake
Renal insufficiency (eGFR of <60 ml per minute)
Hypercalciuria
Loop diuretics
Hungry bone syndrome
After successful parathyroid surgery for symptomatic primary hyperparathyroidism with overt skeletal involvement (osteitis fibrosa cystica), a period of time can follow when PTH levels are increased. This is a response to the body’s need to accrue skeletal calcium and, in this setting, endogenous PTH is overproduced temporarily to meet this need
Pseudohypoparathyroidism
This genetic disease of PTH resistance is associated with hypocalcaemia and increased levels of PTH
Drugs
Short-term lithium use
Hydrochlorothiazide
Anticonvulsants, occasionally, if associated with vitamin D deficiency
Antiresorptive agents (for example, bisphosphonates and denosumab)
eGFR, estimated glomerular filration rate.
Evaluation of patients with PHPT
Laboratory evaluation should include measurements of serum phosphate, renal function tests and measurements of serum 25-hydroxyvitamin D (BOX 1). The 24-hour urinary calcium level should also be measured and, if >400 mg per day, a complete stone risk profile analysis should also be measured. A urinary calcium clearance/creatinine clearance ratio of <0.01 suggests, but does not prove, FHH; the age of the patient should also be taken into account as FHH exhibits virtually 100% penetrance by 30 years of age. In addition, in FHH, a family history of hypercalcaemia can often be obtained and should be followed by genetic screening.
Serum phosphate levels are usually low in severe PHPT and in the lower range of normal in milder forms of PHPT123. Bone turnover markers may be increased, the degree to which can be a function of the severity ofthe bone disease (BOX 1). Renal imaging by ultrasound, X-ray or CT should be performed to detect the presence of kidney stones or nephrocalcinosis. Measurement of BMD by dual-energy X-ray absorptiometry should be obtained at the lumbar spine, the hip (total hip and femoral neck) and the distal one-third of the radius in all patients with PHPT. Depending on availability, additional tests such as the trabecular bone score or HRpQCT may be helpful121. In young patients with PHPT (<30 years of age), those with multiglandular disease, a family history of PHPT (affected first-degree relatives) or in those with parathyroid cancer, specific genetic testing (TABLE 1) may be necessary117.
Management
Resection of a parathyroid adenoma (parathyroidectomy) in patients with PHPT is usually curative124,125. Following successful parathyroidectomy, biochemical abnormalities resolve and BMD improves126 with a decreased risk of bone fracture127 and kidney stones128. There may also be improvements in neurocognition and QOL, although randomized controlled trials (RCTs) have failed to show consistent effects129–131. Similarly, cardiovascular end points have not been consistently reached, which might be related to subtle preoperative cardiovascular abnormalities of which the physiological importance is not yet understood. Furthermore, shortterm outcome measures raise questions about long-term improvement and whether the initial abnormalities were clearly related to the disorder132. Patients with symptomatic disease should be referred for parathyroidectomy if there are no medical contraindications. Medical therapy is an option for patients who meet surgical criteria but who cannot undergo surgery. The Conference Proceedings of the Fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism121 provide guidance about which patients with asymptomatic disease should be referred for surgery or those who can be managed expectantly.
Surgical management
A focused approach to remove the single parathyroid adenoma is now the procedure of choice at experienced surgical centres, with use of rapid intraoperative PTH measurements133. This procedure is preceded by successful localization of the parathyroid adenoma. Minimally invasive parathyroidectomy is not generally recommended unless the adenoma is identified by an imaging modality preoperatively. An example of a successful preoperative localization of a parathyroid adenoma is shown in FIG. 7. The minimally invasive parathyroidectomy can be performed under local or regional anaesthesia with same-day discharge. Surgical cure rates with minimally invasive techniques in experienced surgical centres exceed 98% with relatively low risk of surgical complications124,125.
Figure 7. Preoperation location of a parathyroid adenoma.
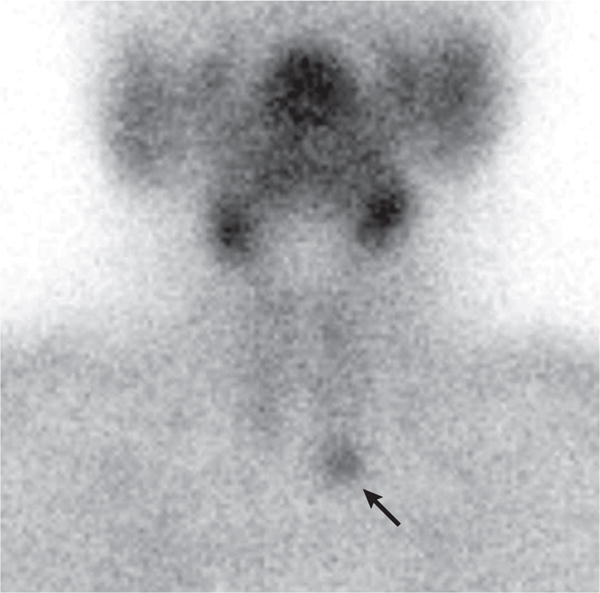
Parathyroid adenoma in the left lower pole of the parathyroid gland is seen by 99m-technetium-sestamibi scanning (arrow). The faint outlines of the thyroid gland are also seen.
Surgery should be considered for any patient who presents with evidence of symptomatic disease, including kidney stones within 10 years of diagnosis or the presence of a fragility fracture. Surgery is also acceptable for asymptomatic patients, provided they do not have medical contraindications121,133. The most recent revision of guidelines in 2014 (REF. 121) is generally consistent with the prior guidelines134 (BOX 3). The revised guidelines take into account the increased risk of vertebral fracture risk and subclinical kidney stones in asymptomatic individuals with PHPT100,101,107. Thus, the new guidelines recommend an evaluation for morphometric vertebral fractures and recommend further evaluation for nephrolithiasis or nephrocalcinosis, or increased risk of kidney stones (biochemical stone risk analysis).
Box 3. Guidelines for parathyroidectomy.
Parathyroidectomy is recommended for symptomatic and asymptomatic patients who meet any of the following criteria168:
Age <50 years
Serum calcium concentration of ≥1 mg per dl above the upper normal limit
T-score at or below −2.5 at the lumbar spine, femoral neck, total hip or distal one-third of the radius, or the presence of vertebral fracture by X-ray or vertebral fracture assessment
Creatinine clearance of <60 ml per minute, increased risk of kidney stones by biochemical stone risk analysis, and kidney stones or nephrocalcinosis by abdominal imaging
The guidelines recommend genetic testing of patients suspected of having a genetic disorder, such as young patients (<30 years of age), patients with syndromic findings and/or a positive family history of syndromic PHPT, and patients with multiglandular disease, parathyroid carcinoma or atypical adenoma117,121. An atypical adenoma is defined histologically as containing features that are not typical of a benign adenoma. Some of these atypical features include an abundance of mitotic figures and cellular atypia. Sometimes, the atypical adenoma will present at the time of surgery as oddly adherent to surrounding structures. The surgical approach for patients with a genetic aetiology may differ from those with a sporadic parathyroid adenoma, in that multiglandular disease is more common when there is a genetic aetiology. Preoperative localization studies are recommended with ultrasound, 99m-technetium-sestamibi nuclear scintigraphy, CT, MRI or PET scans. The choice of preoperative imaging depends on the experience of the surgical centre133.
Non-surgical management
Natural history and monitoring
Patients who do not meet surgical criteria or those who decline surgery should be monitored for signs of disease progression. The longest natural history study prospectively observed patients with primary hyperparathyroidism for 15 years126. Biochemical parameters remained stable for 12 years, with a trend towards increasing levels of serum calcium in years 13–15. BMD remained stable for the first 8–10 years of observation, with subsequent decline in densitometric parameters at the distal radius and hip. Over the 15-year follow-up period, 37% of patients with asymptomatic PHPT ultimately developed one or more criteria for surgery.
The revised guidelines121 recommend annual measurements of serum calcium, serum creatinine and estimated glomerular filtration rate. BMD should be monitored every 1–2 years at the spine, hip and forearm. Follow-up vertebral imaging with X-ray or vertebral fracture analysis should be performed if clinically indicated for a patient with prospective height loss or new back pain. Imaging for new kidney stones should be performed if clinical suspicions emerge. Patients who do not meet criteria for parathyroidectomy on initial evaluation should be considered for surgical management if during the monitoring period serum calcium concentrations rise to >1 mg per d1 above the upper normal limit, creatinine clearance falls to <60 cc per minute, the T-score at the spine, hip or distal one-third of the radius site falls below −2.5 or there is a substantial reduction in BMD, or if a vertebral fracture or kidney stone develops121.
Medical management
Adequate hydration and avoidance of dehydration is always recommended. Calcium intake should not be restricted and should follow national guidelines135. 25-hydroxyvitamin D levels of >20 ng per ml are recommended, although some experts continue to recommend levels of >30 ng per ml. To replete vitamin D when needed, initial doses of 600–1,000 IU daily are recommended, although a recent study showed that doses of 2,800 IU daily were well tolerated136. Serum calcium levels should be monitored during vitamin D supplementation.
Cinacalcet, a calcimimetic, is approved for specific indications in PHPT by the European Medicines Agency (EMA) and the US FDA. The EMA approved the use of cinacalcet for patients with hypercalcaemia who meet surgical criteria but in whom parathyroidectomy is not possible or not clinically ‘appropriate’ (REF. 137). Concurrent medical contraindications to surgery would be an example. Another example is a patient with PHPT who has had several negative operations in search for the elusive adenoma. The FDA approval is for severe hypercalcaemia in patients with PHPT who are unable to undergo parathyroidectomy138. Adverse effects of cinacalcet include nausea, vomiting, diarrhoea and headache, but are uncommon when the single 30 mg daily dose is used. Approval of cinacalcet followed the pivotal randomized, double-blind, placebo-controlled trial in PHPT139. The primary end point — normalization of serum calcium levels — was achieved in 73% of enrolled patients. Serum calcium levels remained in the normal range during the 4.5-year open-label extension140. Subsequent prospective trials have shown similar results141–143. No significant effects on BMD have been observed with cinacalcet therapy.
Alendronate, a bisphosphonate, improved BMD in patients with PHPT144,145. In a 2-year randomized crossover study of alendronate versus placebo in postmenopausal women, the alendronate group had a significant increase in lumbar spine BMD at 2 years compared with baseline and compared with the control group144. The total hip BMD increased at 1 year and subsequently remained stable. Serum and urine concentrations of calcium and PTH did not change. A trial in men showed similar results145. Limited data suggest that oestrogen may also reduce bone resorption in PHPT, with no change in calcium or PTH levels146. In patients who require both a reduction in serum calcium levels and an improvement in BMD, combination therapy with cina-calcet and alendronate is reasonable, but has only been studied in a retrospective manner142,147,148.
Normocalcaemic PHPT
The optimal management strategy for the normocalcaemic variant of PHPT has not been established, but the 2014 guidelines offer recommendations121. Patients should be referred for parathyroidectomy if they have or develop complications of PHPT, even if they remain normocalcaemic. Serum calcium, PTH and creatinine levels should be monitored annually and BMD should be evaluated every 1–2 years. Patients who develop hypercalcaemia can be managed as per the guidelines for asymptomatic PHPT. In one study, BMD improved following parathyroidectomy149 in patients with hypercalcaemic PHPT126,150. There are little data available about the impact of medical therapy on patients with normocalcaemic disease. Alendronate improved BMD in a small cohort151.
Quality of life
Although we define asymptomatic PHPT as a condition in which classic target organs are not affected, many such patients report nonspecific symptoms, including fatigue, irritability, weakness, malaise, somatization, lack of mental clarity, sleep disturbance, anxiety and depression. These non-classic features of PHPT have been vexing and inconclusive to date as far as their association with PHPT and their reversibility after successful parathyroidectomy are concerned. However, these relatively common complaints among many patients with PHPT recall the classic descriptions of the disease of “mental fuzziness” that resolved with parathyroidectomy1.
Various cross-sectional and prospective studies have shown conflicting results with regard to the nonclassic symptoms of PHPT and improvement following parathyroidectomy152–162. The inconsistency of the data among these studies did not permit the expert Workshop Panel of the Fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism to recommend parathyroid surgery if such symptomatology was present in the absence of any clear surgical guidelines121,134.
Walker and colleagues163 published one of the most detailed neuropsychiatric evaluations in individuals with PHPT (TABLE 2). Thirty-nine postmenopausal women with mild, asymptomatic disease were prospectively studied before and after parathyroidectomy compared to 89 healthy age-matched controls. A battery of validated instruments to measure cognition and their relationships to underlying psychological symptoms and biochemical disease severity were used. At baseline, individuals with PHPT had worse symptom scores for depression and anxiety and worse performance on tests of verbal memory and non-verbal abstraction than controls. Six months following parathyroidectomy, these variables improved and were no longer different than controls. Baseline differences in cognition and improvement following parathyroidectomy were not associated with psychological symptoms or serum calcium or PTH concentrations.
Table 2.
Assessment of quality of life in PHPT
| Study | Quality of life and neurocognition | Psychological symptoms |
|---|---|---|
| Prospective study | ||
| Walker et al.163 | Worse scores for immediate and delayed recall of contextually related material (Wechsler Memory Scale Logical Memory Test) in patients with PHPT compared with controls, both of which improved 6 months after parathyroidectomy; non-verbal abstraction (Booklet Category Test) was worse compared with controls and improved after parathyroidectomy | More symptoms of depression (Beck Depression Index), and state and trait anxiety (State-Trait Anxiety Inventory) in patients with PHPT compared with controls; significant improvements in depression and trait anxiety 6 months after parathyroidectomy |
| Randomized controlled trials | ||
| Rao et al.129 | Modest but significant improvements in social functioning and emotional role functioning in individuals 1 year after parathyroidectomy compared with medical management (SF-36) | Modest but significant improvements in anxiety and phobia in individuals 1 year after parathyroidectomy compared with medical management (SCL-90-R) |
| Ambrogini et al.130 | Modest but significant beneficial effects on bodily pain, general health perception, vitality and mental health in individuals 1 year after parathyroidectomy compared with medical management (SF-36) | No between-group differences 1 year after parathyroidectomy (SCL-90-R) |
| Bollerslev et al.131 | Modest but significant improvements in mental health and the mental component summary scores 1 year, but not two years, after parathyroidectomy compared with medical management (SF-36) | No between-group differences (Comprehensive Psychopathological Rating Scale) |
PHPT, primary hyperparathyroidism; SCL-90-R, Symptom Checklist-90-Revised scale; SF-36, 36-Item Short-Form Health Survey.
Various older, observational studies have demonstrated deficits in QOL and neuropsychiatric features in individuals with the mild form of PHPT, with conflicting data regarding symptomatic improvement following parathyroidectomy. Many of these studies are unfortunately limited by methodological flaws, including a failure to apply standardized evaluation tools, small sample sizes, lack of controls or an inappropriate control population, and inclusion of subjects with symptomatic disease. A major problem inherent to this research is the lack of adoption of a validated disease-specific tool to evaluate QOL. Most studies use the 36-Item Short-Form Health Survey (SF-36), one of the most widely used general measures of QOL composed of eight domains on physical and mental health. Pasieka and colleagues164 validated a disease-specific QOL questionnaire, and Webb and colleagues165 state that validation studies are in progress for their disease-specific questionnaire.
The most rigorous data available are from RCTs of parathyroidectomy versus observation129–131. Rao et al. randomized 53 individuals with PHPT to observation versus parathyroidectomy and were followed up for up to 2 years. Five domains of the SF-36 (social functioning, physical functioning, emotional role functioning, vitality and general health perception) significantly worsened during the 2-year follow-up without surgery. Following parathyroidectomy, only modest, but significant, benefits in social functioning and emotional role functioning were observed, with a significant decline in physical functioning. Using the Symptom CheckList-90-Revised scale (SCL-90-R), individuals in the surgical group demonstrated less anxiety and phobia than individuals who were followed without surgery. No significant differences were noted in the other domains and no significant worsening was noted in the individuals who were followed without surgery. Ambrogini et al.130 randomized 50 individuals with PHPT who were followed for up to 1 year. At 12 months, the surgical group had significantly improved scores for bodily pain, general health perception, vitality and mental health compared with medically managed controls, although the absolute differences were modest. No differences were noted in the remaining SF-36 or SCL-90-R domains. Bollerslev et al.131 randomized 191 subjects with PHPT who were followed for up to 2 years. They used the SF-36 questionnaire and the modified Comprehensive Psychopathological Rating Scale (CPRS), using normative Swedish data as a comparison. At baseline, individuals with PHPT had lower SF-36 scores for vitality, social functioning, emotional role functioning, mental health and the mental health component summary than normative controls, as well as a lower CPRS score. A small but significant decrease in physical functioning and the physical component summary score was observed over 2 years in the observation group, whereas there were no changes noted in the surgical group. For the mental health domains, the surgical group had significantly increased values for emotional role functioning and mental health at 1 year compared with baseline, but not at 2 years. At 2 years, the observation group had increased values for mental health from baseline. Using the CPRS questionnaire, both the observation and the surgical groups had significantly more symptoms than controls over the 2 years without significant change over time. There was a trend towards a better outcome in the surgical group at 1 year (P = 0.08) but not at 2 years.
Perrier et al.166 randomized 18 subjects with PHPT to parathyroidectomy or observation and followed subjects for up to 6 months in a pilot study of cognition in addition to sleep and brain function using functional MRI. No between-group differences in cognition, daytime sleepiness or voxel counts by functional MRI were found at 6 months. Total sleep time and left precentral gyrus activity were found to be inversely correlated with PTH levels.
Rolighed and colleagues167 recently reported the results of a randomized, placebo-controlled study, which investigated whether vitamin D supplementation improved QOL in PHPT. Subjects given 2,800 IU of cholecalciferol for 26 weeks before and 26 weeks following parahyroidectomy demonstrated significant improvements in 25-hydroxyvitamin D levels. However, QOL and muscle strength were not improved with cholecalciferol alone, but QOL significantly improved following parathyroidectomy independent of the 25-hydroxyvitamin D level. Aberg et al.152 also showed that, although QOL improved following parathyroidectomy, no additive effect of vitamin D supplementation was evident at doses of 1,600 IU daily.
Outlook
PHPT now often presents without overt target organ involvement. Asymptomatic PHPT becomes more evident as countries adopt biochemical screening methods. Revised guidelines have helped to identify who among these individuals are recommended to undergo parathyroidectomy. However, high-resolution imaging of the skeleton and the kidneys have called attention to the fact that the absence of overt symptomatology does not rule out involvement. Patients with asymptomatic PHPT need renal and skeletal testing before clinical decisions can be made. With a more rigorous evaluative approach, it is likely that a greater proportion of patients will meet one or more guidelines for parathyroid surgery.
This Primer has highlighted several outstanding research issues that remain to be explored in PHPT. Our understanding of normocalcaemic PHPT with regard to diagnosis, natural history, epidemiology, pathophysiology and target organ involvement all remain key questions to be addressed. The optimal steady-state value of 25-hydroxyvitamin D and means by which vitamin D should be replaced in individuals with PHPT who are deficient in vitamin D need to be elucidated. A particularly vexing issue relates to neurocognitive features that often accompany patients with PHPT. Despite several attempts to provide conclusive data on the putative association between neurocognitive function and PHPT as well as possible changes after successful parathyroidectomy, we still lack clear evidence in support of this idea. Microstructural analyses of bone using noninvasive high-resolution imaging technology have given new insights into multiskeletal compartment involvement in PHPT, involving both cortical and trabecular bone. More information is needed to determine how these findings relate to bone strength and fracture risk and the extent to which improvement might follow after successful parathyroid surgery. Although the latest guidelines121 recommend a more vigorous approach to biochemical stone risk factor analysis in individuals with PHPT who have hypercalciuria, the predictive value and reversibility of stone risk after parathyroidectomy are not yet known.
Acknowledgments
This work was supported, in part, by the NIH grant DK 32333.
Footnotes
Competing interests
J.P.B. is a consultant for Merck, Amgen, Shire Pharmaceuticals and Radius, and receives research support from Shire Pharmaceuticals. A.A.K. receives research grants from Amgen and Shire Pharmaceuticals. All other authors declare no competing interests.
Author contributions
Introduction (J.P.B.); Epidemiology (J.-M.L. and A.A.K.); Mechanisms/pathophysiology (C.M.); Diagnosis, screening and prevention (F.B.); Management (J.P.B. and N.E.C.); Quality of life (N.E.C.); Outlook (J.P.B.); Overview of Primer (J.P.B.).
References
- 1.Cope O. The study of hyperparathyroidism at the Massachusetts General Hospital. N Engl J Med. 1966;274:1174–1182. doi: 10.1056/NEJM196605262742105. [DOI] [PubMed] [Google Scholar]
- 2.Pallan S, Rahman MO, Khan AA. Diagnosis and management of primary hyperparathyroidism. BMJ. 2012;344:e1013. doi: 10.1136/bmj.e1013. [DOI] [PubMed] [Google Scholar]
- 3.Bilezikian JP, Silverberg SJ. Clinical practice. Asymptomatic primary hyperparathyroidism. N Engl J Med. 2004;350:1746–1751. doi: 10.1056/NEJMcp032200. This paper reports on the changing clinical presentation of PHPT from symptomatic to asymptomatic. [DOI] [PubMed] [Google Scholar]
- 4.Lowe H, McMahon DJ, Rubin MR, Bilezikian JP, Silverberg SJ. Normocalcemic primary hyperparathyroidism: further characterization of a new clinical phenotype. J Clin Endocrinol Metab. 2007;92:3001–3005. doi: 10.1210/jc.2006-2802. [DOI] [PubMed] [Google Scholar]
- 5.Yeh MW, et al. Incidence and prevalence of primary hyperparathyroidism in a racially mixed population. J Clin Endocrinol Metab. 2013;98:1122–1129. doi: 10.1210/jc.2012-4022. This is a recent survey of the prevalence of PHPT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Press DM, et al. The prevalence of undiagnosed and unrecognized primary hyperparathyroidism: a population-based analysis from the electronic medical record. Surgery. 2013;154:1232–1237. doi: 10.1016/j.surg.2013.06.051. [DOI] [PubMed] [Google Scholar]
- 7.Berger C, et al. Characteristics of hyperparathyroid states in the Canadian multicentre osteoporosis study (CaMos) and relationship to skeletal markers. Clin Endocrinol (Oxf) 2015;82:359–368. doi: 10.1111/cen.12569. [DOI] [PubMed] [Google Scholar]
- 8.Lee PK, Jarosek SL, Virnig BA, Evasovich M, Tuttle TM. Trends in the incidence and treatment of parathyroid cancer in the United States. Cancer. 2007;109:1736–1741. doi: 10.1002/cncr.22599. [DOI] [PubMed] [Google Scholar]
- 9.Palmer M, Jakobsson S, Akerstrom G, Ljunghall S. Prevalence of hypercalcaemia in a health survey: a 14-year follow-up study of serum calcium values. Eur J Clin Invest. 1988;18:39–46. doi: 10.1111/j.1365-2362.1988.tb01163.x. [DOI] [PubMed] [Google Scholar]
- 10.Abood A, Vestergaard P. Increasing incidence of primary hyperparathyroidism in Denmark. Dan Med J. 2013;60:A4567. [PubMed] [Google Scholar]
- 11.Yu N, Donnan PT, Murphy MJ, Leese GP. Epidemiology of primary hyperparathyroidism in Tayside, Scotland, UK. Clin Endocrinol (Oxf) 2009;71:485–493. doi: 10.1111/j.1365-2265.2008.03520.x. [DOI] [PubMed] [Google Scholar]
- 12.Bandeira F, Griz L, Caldas G, Bandeira C, Freese E. From mild to severe primary hyperparathyroidism: the Brazilian experience. Arq Bras Endocrinol Metab. 2006;50:657–663. doi: 10.1590/s0004-27302006000400011. [DOI] [PubMed] [Google Scholar]
- 13.Spivacow FR, Martinez C, Polonsky A. Primary hyperparathyroidism: postoperative long-term evolution. Medicina (B Aires) 2010;70:408–414. (in Spanish) [PubMed] [Google Scholar]
- 14.Eufrazino C, Veras A, Bandeira F. Epidemiology of primary hyperparathyroidism and its non-classical manifestations in the city of Recife, Brazil. Clin Med Insights Endocrinol Diabetes. 2013;6:69–74. doi: 10.4137/CMED.S13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pradeep PV, Jayashree B, Mishra A, Mishra SK. Systematic review of primary hyperparathyroidism in India: the past, present, and the future trends. Int J Endocrinol. 2011;2011:921814. doi: 10.1155/2011/921814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bilezikian JP, Meng X, Shi Y, Silverberg SJ. Primary hyperparathyroidism in women: a tale of two cities — New York and Beijing. Int J Fertil Womens Med. 2000;45:158–165. [PubMed] [Google Scholar]
- 17.Zhao L, et al. The changing clinical patterns of primary hyperparathyroidism in Chinese patients: data from 2000 to 2010 in a single clinical center. J Clin Endocrinol Metab. 2013;98:721–728. doi: 10.1210/jc.2012-2914. [DOI] [PubMed] [Google Scholar]
- 18.Liu JM, et al. Primary hyperparathyroidism: a tale of two cities revisited — New York and Shanghai. Bone Res. 2013;1:162–169. doi: 10.4248/BR201302005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo CY, et al. Surgical treatment for primary hyperparathyroidism in Hong Kong: changes in clinical pattern over 3 decades. Arch Surg. 2004;139:77–82. doi: 10.1001/archsurg.139.1.77. [DOI] [PubMed] [Google Scholar]
- 20.Gopal RA, et al. Clinical profile of primary hyperparathyroidism from western India: a single center experience. J Postgrad Med. 2010;56:79–84. doi: 10.4103/0022-3859.65279. [DOI] [PubMed] [Google Scholar]
- 21.Malabu UH, Founda MA. Primary hyperparathyroidism in Saudi Arabia: a review of 46 cases. Med J Malaysia. 2007;62:394–397. [PubMed] [Google Scholar]
- 22.Prasarttong-Osoth P, Wathanaoran P, Imruetaicharoenchoke W, Rojananin S. Primary hyperparathyroidism: 11-year experience in a single institute in Thailand. Int J Endocrinol. 2012;2012:952426. doi: 10.1155/2012/952426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamidi S, Soltani A, Hedayat A, Kamalian N. Primary hyperparathyroidism: a review of 177 cases. Med Sci Monit. 2006;12:CR86–9. [PubMed] [Google Scholar]
- 24.Biyabani SR, Talati J. Bone and renal stone disease in patients operated for primary hyperparathyroidism in Pakistan: is the pattern of disease different from the west? J Pak Med Assoc. 1999;49:194–198. [PubMed] [Google Scholar]
- 25.Shah VN, Bhadada S, Bhansali A, Behera A, Mittal BR. Changes in clinical and biochemical presentations of primary hyperparathyroidism in India over a period of 20 years. Indian J Med Res. 2014;139:694–699. [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi T, Sugimoto T, Chihara K. Clinical and biochemical presentation of primary hyperparathyroidism in Kansai district of Japan. Endocr J. 1997;44:595–601. doi: 10.1507/endocrj.44.595. [DOI] [PubMed] [Google Scholar]
- 27.Paruk IM, Esterhuizen TM, Maharaj S, Pirie FJ, Motala AA. Characteristics, management and outcome of primary hyperparathyroidism in South Africa: a single-centre experience. Postgrad Med J. 2013;89:626–631. doi: 10.1136/postgradmedj-2012-131707. [DOI] [PubMed] [Google Scholar]
- 28.Clifton-Bligh PB, et al. Mortality associated with primary hyperparathyroidism. Bone. 2015;74:121–124. doi: 10.1016/j.bone.2014.12.067. [DOI] [PubMed] [Google Scholar]
- 29.Lundgren E, et al. Increased cardiovascular mortality and normalized serum calcium in patients with mild hypercalcemia followed up for 25 years. Surgery. 2001;130:978–985. doi: 10.1067/msy.2001.118377. [DOI] [PubMed] [Google Scholar]
- 30.Wermers RA, et al. Survival after the diagnosis of hyperparathyroidism: a population-based study. Am J Med. 1998;104:115–122. doi: 10.1016/s0002-9343(97)00270-2. [DOI] [PubMed] [Google Scholar]
- 31.Brown EM, et al. Dispersed cells prepared from human parathyroid glands: distinct calcium sensitivity of adenomas versus primary hyperplasia. J Clin Endocrinol Metab. 1978;46:267–275. doi: 10.1210/jcem-46-2-267. [DOI] [PubMed] [Google Scholar]
- 32.Silverberg SJ. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Rosen CJ, et al., editors. John Wiley & Sons; 2014. pp. 543–552. [Google Scholar]
- 33.Brown EM. Role of the calcium-sensing receptor in extracellular calcium homeostasis. Best Pract Res Clin Endocrinol Metab. 2013;27:333–343. doi: 10.1016/j.beem.2013.02.006. This is a recent review that highlights the role of the CASR in physiological and pathological conditions. [DOI] [PubMed] [Google Scholar]
- 34.Segersten U, et al. 25-hydroxyvitamin D3-1 α-hydroxylase expression in normal and pathological parathyroid glands. J Clin Endocrinol Metab. 2002;87:2967–2972. doi: 10.1210/jcem.87.6.8604. [DOI] [PubMed] [Google Scholar]
- 35.Naveh-Many T, Rahamimov R, Livni N, Silver J. Parathyroid cell proliferation in normal and chronic renal failure rats. The effects of calcium, phosphate, and vitamin D. J Clin Invest. 1995;96:1786–1793. doi: 10.1172/JCI118224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moallem E, Kilav R, Silver J, Naveh-Many T. RNA-protein binding and post-transcriptional regulation of parathyroid hormone gene expression by calcium and phosphate. J Biol Chem. 1998;273:5253–5259. doi: 10.1074/jbc.273.9.5253. [DOI] [PubMed] [Google Scholar]
- 37.Ben-Dov IZ, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Witteveen JE, van Lierop AH, Papapoulos SE, Hamdy NA. Increased circulating levels of FGF23: an adaptive response in primary hyperparathyroidism? Ear J Endocrinol. 2012;166:55–60. doi: 10.1530/EJE-11-0523. [DOI] [PubMed] [Google Scholar]
- 39.Silva BC, Kousteni S. In: The Parathyroids Basic and Clincial Concepts. Bilezikian JP, et al., editors. Academic Press; 2015. pp. 127–137. [Google Scholar]
- 40.Goltzman D. In: The Parathyroids. Basic and Clincial Concepts. Bilezikian JP, et al., editors. Academic Press; 2015. pp. 139–152. [Google Scholar]
- 41.Carpinteri R, et al. Glucocorticoid-induced osteoporosis and parathyroid hormone. J Endocrinol Invest. 2010;33:16–21. [PMC free article] [PubMed] [Google Scholar]
- 42.Silverberg SJ, Bilezikian JP. In: The Parathyroids Basic and Clinical Concepts. Bilezikian JP, et al., editors. Academic Press; 2015. pp. 317–327. [Google Scholar]
- 43.Marcocci C, Cetani F. Clinical practice. Primary hyperparathyroidism. N Engl J Med. 2011;365:2389–2397. doi: 10.1056/NEJMcp1106636. [DOI] [PubMed] [Google Scholar]
- 44.Arnold A, Levine A. In: The Parathyroids Basic and Clincial Concepts. Bilezikian JP, et al., editors. Academic Press; 2015. pp. 279–296. This is a recent comprehensive chapter on the molecular pathogenesis of parathyroid tumours. [Google Scholar]
- 45.Attie JN, Bock G, Auguste LJ. Multiple parathyroid adenomas: report of thirty-three cases. Surgery. 1990;108:1014–1019. [PubMed] [Google Scholar]
- 46.Cohen J, Gierlowski TC, Schneider AB. A prospective study of hyperparathyroidism in individuals exposed to radiation in childhood. JAMA. 1990;264:581–584. [PubMed] [Google Scholar]
- 47.Boehm BO, Rosinger S, Belyi D, Dietrich JW. The parathyroid as a target for radiation damage. N Engl J Med. 2011;365:676–678. doi: 10.1056/NEJMc1104982. [DOI] [PubMed] [Google Scholar]
- 48.Szalat A, Mazeh H, Freund HR. Lithium-associated hyperparathyroidism: report of four cases and review of the literature. Ear J Endocrinol. 2009;160:317–323. doi: 10.1530/EJE-08-0620. [DOI] [PubMed] [Google Scholar]
- 49.Arnold A, Staunton CE, Kim HG, Gaz RD, Kronenberg HM. Monoclonality and abnormal parathyroid hormone genes in parathyroid adenomas. N Engl J Med. 1988;318:658–662. doi: 10.1056/NEJM198803173181102. [DOI] [PubMed] [Google Scholar]
- 50.Friedman E. In: Molecular Biology of the Parathyroid. Naveh-Many T, editor. Plenum Publisher; New York: 2005. pp. 128–139. [Google Scholar]
- 51.Arnold A, et al. Monoclonality of parathyroid tumors in chronic renal failure and in primary parathyroid hyperplasia. J Clin Invest. 1995;95:2047–2053. doi: 10.1172/JCI117890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Friedman E, et al. Clonality of parathyroid tumors in familial multiple endocrine neoplasia type 1. N Engl J Med. 1989;321:213–218. doi: 10.1056/NEJM198907273210402. [DOI] [PubMed] [Google Scholar]
- 53.Thakker RV, et al. Association of parathyroid tumors in multiple endocrine neoplasia type 1 with loss of alleles on chromosome 11. N Engl J Med. 1989;321:218–224. doi: 10.1056/NEJM198907273210403. [DOI] [PubMed] [Google Scholar]
- 54.Chandrasekharappa SC, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 55.Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Ham Matat. 2008;29:22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- 56.Agarwal SK, et al. Menin molecular interactions: insights into normal functions and tumorigenesis. Horm Metab Res. 2005;37:369–374. doi: 10.1055/s-2005-870139. [DOI] [PubMed] [Google Scholar]
- 57.Gracanin A, Dreijerink KM, van der Luijt RB, Lips CJ, Hoppener JW. Tissue selectivity in multiple endocrine neoplasia type 1-associated tumorigenesis. Cancer Res. 2009;69:6371–6374. doi: 10.1158/0008-5472.CAN-09-0678. [DOI] [PubMed] [Google Scholar]
- 58.Karnik SK, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci USA. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milne TA, et al. MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci USA. 2005;102:14765–14770. doi: 10.1073/pnas.0503630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Belar O, et al. Novel mutations in MEN1, CDKN1B and AIP genes in patients with multiple endocrine neoplasia type 1 syndrome in Spain. Clin Endocrinol (Oxf) 2012;76:719–724. doi: 10.1111/j.1365-2265.2011.04269.x. [DOI] [PubMed] [Google Scholar]
- 61.Arnold A, Marx SJ. In: Primer on the Metabolic Bone Disease and Disorders of Mineral Metabolism. Rosen CJ, et al., editors. John Wiley & Sons; 2014. pp. 553–561. [Google Scholar]
- 62.Wohllk N, et al. Multiple endocrine neoplasia type 2. Best Pract Res Clin Endocrinol Metab. 2010;24:371–387. doi: 10.1016/j.beem.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 63.Pellegata NS, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA. 2006;103:15558–15563. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thakker RV. In: The Parathyroids. Basic and Clinical Concepts. Bilezikian JP, et al., editors. Academic Press; 2015. pp. 341–364. [Google Scholar]
- 65.Marinoni I, Pellegata NS. p27kip1: a new multiple endocrine neoplasia gene? Neuroendocrinology. 2011;93:19–28. doi: 10.1159/000320366. [DOI] [PubMed] [Google Scholar]
- 66.Molatore S, Pellegata NS. The MENX syndrome and p27: relationships with multiple endocrine neoplasia. Prog Brain Res. 2010;182:295–320. doi: 10.1016/S0079-6123(10)82013-8. [DOI] [PubMed] [Google Scholar]
- 67.Lee J, Kim SS. The function of p27KIPI during tumor development. Exp Mol Med. 2009;41:765–771. doi: 10.3858/emm.2009.41.11.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carpten JD, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002;32:676–680. doi: 10.1038/ng1048. [DOI] [PubMed] [Google Scholar]
- 69.Howell VM, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003;40:657–663. doi: 10.1136/jmg.40.9.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Newey PJ, Bowl MR, Thakker RV. Parafibromin — functional insights. J Intern Med. 2009;266:84–98. doi: 10.1111/j.1365-2796.2009.02107.x. [DOI] [PubMed] [Google Scholar]
- 71.Cetani F, et al. Genetic analyses in familial isolated hyperparathyroidism: implication for clinical assessment and surgical management. Clin Endocrinol (Oxf) 2006;64:146–152. doi: 10.1111/j.1365-2265.2006.02438.x. [DOI] [PubMed] [Google Scholar]
- 72.Miedlich S, Lohmann T, Schneyer U, Lamesch P, Paschke R. Familial isolated primary hyperparathyroidism — a multiple endocrine neoplasia type 1 variant? Eur J Endocrinol. 2001;145:155–160. doi: 10.1530/eje.0.1450155. [DOI] [PubMed] [Google Scholar]
- 73.El-Hajj Fuleihan G, Brown EM. In: The Parathyroids Basic and Clincial Concepts. Bilezikian JP, et al., editors. Academic Press; 2015. pp. 365–387. [Google Scholar]
- 74.Pollak MR, et al. Mutations in the human Ca2+-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell. 1993;75:1297–1303. doi: 10.1016/0092-8674(93)90617-y. [DOI] [PubMed] [Google Scholar]
- 75.Nesbit MA, et al. Mutations in AP2SI cause familial hypocalciuric hypercalcemia type 3. Nat Genet. 2013;45:93–97. doi: 10.1038/ng.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ho C, et al. A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat Genet. 1995;11:389–394. doi: 10.1038/ng1295-389. [DOI] [PubMed] [Google Scholar]
- 77.Arnold A, et al. Molecular cloning and chromosomal mapping of DNA rearranged with the parathyroid hormone gene in a parathyroid adenoma. J Clin Invest. 1989;83:2034–2040. doi: 10.1172/JCI114114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mallya SM, Wu HI, Saria EA, Corrado KR, Arnold A. Tissue-specific regulatory regions of the PTH gene localized by novel chromosome 11 rearrangement breakpoints in a parathyroid adenoma. J Bone Miner Res. 2010;25:2606–2612. doi: 10.1002/jbmr.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cetani F, et al. Six novel MEN1 gene mutations in sporadic parathyroid tumors. Hum Mutat. 2000;16:445. doi: 10.1002/1098-1004(200011)16:5<445::AID-HUMU12>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 80.Farnebo F, et al. Alterations of the MEN1 gene in sporadic parathyroid tumors. J Clin Endocrinol Metab. 1998;83:2627–2630. doi: 10.1210/jcem.83.8.4846. [DOI] [PubMed] [Google Scholar]
- 81.Cromer MK, et al. Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. J Clin Endocrinol Metab. 2012;97:E1774–E1781. doi: 10.1210/jc.2012-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cetani F, et al. Should parafibromin staining replace HRTP2 gene analysis as an additional tool for histologic diagnosis of parathyroid carcinoma? Eur J Endocrinol. 2007;156:547–554. doi: 10.1530/EJE-06-0720. [DOI] [PubMed] [Google Scholar]
- 83.Juhlin CC, Hoog A. Parafibromin as a diagnostic instrument for parathyroid carcinoma-lone ranger or part of the posse? Int J Endocrinol. 2010;2010:324964. doi: 10.1155/2010/324964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Costa-Guda J, Marinoni I, Molatore S, Pellegata NS, Arnold A. Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2011;96:E701–E706. doi: 10.1210/jc.2010-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pardi E, et al. Aryl hydrocarbon receptor interacting protein (AIP) mutations occur rarely in sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2013;98:2800–2810. doi: 10.1210/jc.2012-4029. [DOI] [PubMed] [Google Scholar]
- 86.Bjorklund P, Lindberg D, Akerstrom G, Westin G. Stabilizing mutation of CTNNB1/beta-catenin and protein accumulation analyzed in a large series of parathyroid tumors of Swedish patients. Mol Cancer. 2008;7:53. doi: 10.1186/1476-4598-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cetani F, et al. β-catenin activation is not involved in sporadic parathyroid carcinomas and adenomas. Endocr Relat Cancer. 2010;17:1–6. doi: 10.1677/ERC-09-0147. [DOI] [PubMed] [Google Scholar]
- 88.Giusti L, et al. A proteomic approach to study parathyroid glands. Mol Biosyst. 2011;7:687–699. doi: 10.1039/c0mb00191k. [DOI] [PubMed] [Google Scholar]
- 89.Cryns VL, et al. Loss of the retinoblastoma tumorsuppressor gene in parathyroid carcinoma. N Engl J Med. 1994;330:757–761. doi: 10.1056/NEJM199403173301105. [DOI] [PubMed] [Google Scholar]
- 90.Cetani F, et al. Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. J Clin Endocrinol Metab. 2004;89:5583–5591. doi: 10.1210/jc.2004-0294. [DOI] [PubMed] [Google Scholar]
- 91.Shattuck TM, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003;349:1722–1729. doi: 10.1056/NEJMoa031237. [DOI] [PubMed] [Google Scholar]
- 92.Hahn MA, et al. CDC73/HRPT2 CpG island hypermethylation and mutation of 5′-untranslated sequence are uncommon mechanisms of silencing parafibromin in parathyroid tumors. Endocr Relat Cancer. 2010;17:273–282. doi: 10.1677/ERC-09-0291. [DOI] [PubMed] [Google Scholar]
- 93.Singh Ospina N, Sebo TJ, Thompson GB, Clarke BL, Young WF., Jr Prevalence of parathyroid carcinoma in 348 patients with multiple endocrine neoplasia type 1 — case report and review of the literature. Clin Endocrinol (Oxf) 2014 doi: 10.1111/cen.12714. http://dx.doi.org/10.1111/cen.12714. [DOI] [PubMed]
- 94.Corbetta S, et al. Differential expression of microRNAs in human parathyroid carcinomas compared with normal parathyroid tissue. Endocr Relat Cancer. 2010;17:135–146. doi: 10.1677/ERC-09-0134. [DOI] [PubMed] [Google Scholar]
- 95.Yu W, et al. Whole-exome sequencing studies of parathyroid carcinomas reveal novel PRUNE2 mutations, distinctive mutational spectra related to APOBEC-catalyzed DNA mutagenesis and mutational enrichment in kinases associated with cell migration and invasion. J Clin Endocrinol Metab. 2015;100:E360–E364. doi: 10.1210/jc.2014-3238. [DOI] [PubMed] [Google Scholar]
- 96.Hansen S, Beck Jensen JE, Rasmussen L, Hauge EM, Brixen K. Effects on bone geometry, density, and microarchitecture in the distal radius but not the tibia in women with primary hyperparathyroidism: a case-control study using HR-pQCT. J Bone Miner Res. 2010;25:1941–1947. doi: 10.1002/jbmr.98. [DOI] [PubMed] [Google Scholar]
- 97.Stein EM, et al. Primary hyperparathyroidism is associated with abnormal cortical and trabecular microstructure and reduced bone stiffness in postmenopausal women. J Bone Miner Res. 2013;28:1029–1040. doi: 10.1002/jbmr.1841. This paper reports on the use of high-resolution skeletal imaging to detect trabecular bone disease in PHPT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Romagnoli E, et al. “Trabecular Bone Score” (TBS): an indirect measure of bone micro-architecture in postmenopausal patients with primary hyperparathyroidism. Bone. 2013;53:154–159. doi: 10.1016/j.bone.2012.11.041. [DOI] [PubMed] [Google Scholar]
- 99.Silva BC, et al. Trabecular bone score (TBS) — a novel method to evaluate bone microarchitectural texture in patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2013;98:1963–1970. doi: 10.1210/jc.2012-4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Khosla S, et al. Primary hyperparathyroidism and the risk of fracture: a population-based study. J Bone Miner Res. 1999;14:1700–1707. doi: 10.1359/jbmr.1999.14.10.1700. [DOI] [PubMed] [Google Scholar]
- 101.Vignali E, et al. Morphometric vertebral fractures in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab. 2009;94:2306–2312. doi: 10.1210/jc.2008-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kulak CA, et al. Marked improvement in bone mass after parathyroidectomy in osteitis fibrosa cystica. J Clin Endocrinol Metab. 1998;83:732–735. doi: 10.1210/jcem.83.3.4655. [DOI] [PubMed] [Google Scholar]
- 103.Albright F, Reifenstein EC., Jr . The Parathyroid Glands and Metabolic Bone Diseases: Selected Studies. Williams & Wilkins; 1948. [Google Scholar]
- 104.Mallette LE, Bilezikian JP, Heath DA, Aurbach GD. Primary hyperparathyroidism: clinical and biochemical features. Medicine (Baltimore) 1974;53:127–146. [PubMed] [Google Scholar]
- 105.Starup-Linde J, Waldhauer E, Rolighed L, Mosekilde L, Vestergaard P. Renal stones and calcifications in patients with primary hyperparathyroidism: associations with biochemical variables. Ear J Endocrinol. 2012;166:1093–1100. doi: 10.1530/EJE-12-0032. [DOI] [PubMed] [Google Scholar]
- 106.Cassibba S, et al. Silent renal stones in primary hyperparathyroidism: prevalence and clinical features. Endocr Pract. 2014;20:1137–1142. doi: 10.4158/EP14074.OR. [DOI] [PubMed] [Google Scholar]
- 107.Cipriani C, et al. Prevalence of kidney stones and vertebral fractures in primary hyperparathyroidism using imaging technology. J Clin Endocrinol Metab. 2015;100:1309–1315. doi: 10.1210/jc.2014-3708. This paper re-evaluates the presence of kidney stones in PHPT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Patten BM, et al. Neuromuscular disease in primary hyperparathyroidism. Ann Intern Med. 1974;80:182–193. doi: 10.7326/0003-4819-80-2-182. [DOI] [PubMed] [Google Scholar]
- 109.Turken SA, et al. Neuromuscular involvement in mild, asymptomatic primary hyperparathyroidism. Am J Med. 1989;87:553–557. doi: 10.1016/s0002-9343(89)80613-8. [DOI] [PubMed] [Google Scholar]
- 110.Diniz ET, et al. Primary hyperparathyroidism is associated with subclinical peripheral neural alterations. Endocr Pract. 2013;19:219–225. doi: 10.4158/EP12207.OR. [DOI] [PubMed] [Google Scholar]
- 111.Talpos GB, et al. Randomized trial of parathyroidectomy in mild asymptomatic primary hyperparathyroidism: patient description and effects on the SF-36 health survey. Surgery. 2000;128:1013–1020. doi: 10.1067/msy.2000.110844. [DOI] [PubMed] [Google Scholar]
- 112.Linos DA, van Heerdan JA, Abboud CF, Edis AJ. Primary hyperparathyroidism and peptic ulcer disease. Arch Surg. 1978;113:384–386. doi: 10.1001/archsurg.1978.01370160042005. [DOI] [PubMed] [Google Scholar]
- 113.Bess MA, Edis AJ, van Heerden JA. Hyperparathyroidism and pancreatitis. Chance or a causal association? JAMA. 1980;243:246–247. [PubMed] [Google Scholar]
- 114.Vaidya A, Curhan GC, Paik JM, Kronenberg H, Taylor EN. Hypertension, antihypertensive medications, and risk of incident primary hyperparathyroidism. J Clin Endocrinol Metab. 2015;100:2396–2404. doi: 10.1210/jc.2015-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Walker MD, et al. Cardiac structure and diastolic function in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2010;95:2172–2179. doi: 10.1210/jc.2009-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Iwata S, et al. Aortic valve calcification in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2012;97:132–137. doi: 10.1210/jc.2011-2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Eastell R, et al. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99:3570–3579. doi: 10.1210/jc.2014-1414. Conference proceedings from the Fourth International Workshop on the Management of Asymptomatic PHPT. [DOI] [PubMed] [Google Scholar]
- 118.Rubin MR, et al. An N-terminal molecular form of parathyroid hormone (PTH) distinct from hPTH(1-84) is overproduced in parathyroid carcinoma. Clin Chem. 2007;53:1470–1476. doi: 10.1373/clinchem.2007.085506. [DOI] [PubMed] [Google Scholar]
- 119.Caron P, et al. Nontruncated amino-terminal parathyroid hormone overproduction in two patients with parathyroid carcinoma: a possible link to HRPT2 gene inactivation. Clin Endocrinol (Oxf) 2011;74:694–698. doi: 10.1111/j.1365-2265.2011.04021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bandeira F, Cassibba S. Hyperparathyroidism and bone health. Curr Rheumatol Rep. 2015;17:48. doi: 10.1007/s11926-015-0523-2. [DOI] [PubMed] [Google Scholar]
- 121.Bilezikian JP, et al. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99:3561–3569. doi: 10.1210/jc.2014-1413. Conference proceedings from the Fourth International Workshop on the Management of Asymptomatic PHPT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ketteler M, et al. Revisiting KDIGO clinical practice guideline on chronic kidney disease-mineral and bone disorder: a commentary from a Kidney Disease: Improving Global Outcomes controversies conference. Kidney Int. 2015;87:502–528. doi: 10.1038/ki.2014.425. [DOI] [PubMed] [Google Scholar]
- 123.Bandeira F, et al. Prevalence of cortical osteoporosis in mild and severe primary hyperparathyroidism and its relationship with bone markers and vitamin D status. J Clin Densitom. 2009;12:195–199. doi: 10.1016/j.jocd.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 124.Udelsman R, Lin Z, Donovan P. The superiority of minimally invasive parathyroidectomy based on 1650 consecutive patients with primary hyperparathyroidism. Ann Surg. 2011;253:585–591. doi: 10.1097/SLA.0b013e318208fed9. [DOI] [PubMed] [Google Scholar]
- 125.Van Udelsman B, Udelsman R. Surgery in primary hyperparathyroidism: extensive personal experience. J Clin Densitom. 2013;16:54–59. doi: 10.1016/j.jocd.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 126.Rubin MR, et al. The natural history of primary hyperparathyroidism with or without parathyroid surgery after 15 years. J Clin Endocrinol Metab. 2008;93:3462–3470. doi: 10.1210/jc.2007-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vestergaard P, Mosekilde L. Parathyroid surgery is associated with a decreased risk of hip and upper arm fractures in primary hyperparathyroidism: a controlled cohort study. J Intern Med. 2004;255:108–114. doi: 10.1046/j.0954-6820.2003.01237.x. [DOI] [PubMed] [Google Scholar]
- 128.Mollerup CL, et al. Risk of renal stone events in primary hyperparathyroidism before and after parathyroid surgery: controlled retrospective follow up study. BMJ. 2002;325:807. doi: 10.1136/bmj.325.7368.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rao DS, Phillips ER, Divine GW, Talpos GB. Randomized controlled clinical trial of surgery versus no surgery in patients with mild asymptomatic primary hyperparathyroidism. J Clin Endocrinol Metab. 2004;89:5415–5422. doi: 10.1210/jc.2004-0028. [DOI] [PubMed] [Google Scholar]
- 130.Ambrogini E, et al. Surgery or surveillance for mild asymptomatic primary hyperparathyroidism: a prospective, randomized clinical trial. J Clin Endocrinol Metab. 2007;92:3114–3121. doi: 10.1210/jc.2007-0219. [DOI] [PubMed] [Google Scholar]
- 131.Bollerslev J, et al. Medical observation, compared with parathyroidectomy, for asymptomatic primary hyperparathyroidism: a prospective, randomized trial. J Clin Endocrinol Metab. 2007;92:1687–1692. doi: 10.1210/jc.2006-1836. This is a RCT of parathyroidectomy on QOL in individuals with PHPT. [DOI] [PubMed] [Google Scholar]
- 132.McMahon DJ, et al. Effect of parathyroidectomy upon left ventricular mass in primary hyperparathyroidism: a meta-analysis. J Clin Endocrinol Metab. 2015;100:4399–4407. doi: 10.1210/jc.2015-3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Udelsman R, et al. The surgical management of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99:3595–3606. doi: 10.1210/jc.2014-2000. [DOI] [PubMed] [Google Scholar]
- 134.Silverberg SJ, et al. Current issues in the presentation of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99:3580–3594. doi: 10.1210/jc.2014-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ross AC, Taylor CL, Yaktine AL, Del Valle HB, editors. Dietary Reference Intakes for Calcium and Vitamin D. The National Academies Press; 2011. [PubMed] [Google Scholar]
- 136.Rolighed L, et al. Vitamin D treatment in primary hyperparathyroidism: a randomized placebo controlled trial. J Clin Endocrinol Metab. 2014;99:1072–1080. doi: 10.1210/jc.2013-3978. [DOI] [PubMed] [Google Scholar]
- 137.European Medicines Agency. Mimpara. EMA; 2009. http://www.ema.europa.eu/docs/en_GB/document_library/_EPAR_-Summary_for_the_public/human/000570/_WC500028901.pdf. [Google Scholar]
- 138.Food and Drug Administration. Sensipar. FDA; 2011. http://www.accessdata.fda.gov/drugsatfda_docs/_label/2011/02_1688s017lbl.pdf. [Google Scholar]
- 139.Peacock M, et al. Cinacalcet hydrochloride maintains long-term normocalcemia in patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2005;90:135–141. doi: 10.1210/jc.2004-0842. [DOI] [PubMed] [Google Scholar]
- 140.Peacock M, et al. Cinacalcet treatment of primary hyperparathyroidism: biochemical and bone densitometric outcomes in a five-year study. J Clin Endocrinol Metab. 2009;94:4860–4867. doi: 10.1210/jc.2009-1472. [DOI] [PubMed] [Google Scholar]
- 141.Iglesias P, et al. Acute and one-year effects of cinacalcet in patients with persistent primary hyperparathyroidism after unsuccessful parathyroidectomy. Am J Med Sci. 2008;335:111–114. doi: 10.1097/MAJ.0b013e3181379f3e. [DOI] [PubMed] [Google Scholar]
- 142.Cetani F, et al. Cinacalcet efficacy in patients with moderately severe primary hyperparathyroidism according to the European Medicine Agency prescription labeling. J Endocrinol Invest. 2012;35:655–660. doi: 10.3275/7970. [DOI] [PubMed] [Google Scholar]
- 143.Luque-Fernandez I, Garcia-Martin A, Luque-Pazos A. Experience with cinacalcet in primary hyperparathyroidism: results after 1 year of treatment. Ther Adv Endocrinol Metab. 2013;4:77–81. doi: 10.1177/2042018813482344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Khan AA, et al. Alendronate in primary hyperparathyroidism: a double-blind, randomized, placebo-controlled trial. J Clin Endocrinol Metab. 2004;89:3319–3325. doi: 10.1210/jc.2003-030908. [DOI] [PubMed] [Google Scholar]
- 145.Khan AA, et al. Alendronate therapy in men with primary hyperparathyroidism. Endocr Pract. 2009;15:705–713. doi: 10.4158/EP08178.ORR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Marcus R, Madvig P, Crim M, Pont A, Kosek J. Conjugated estrogens in the treatment of postmenopausal women with hyperparathyroidism. Ann Intern Med. 1984;100:633–640. doi: 10.7326/0003-4819-100-5-633. [DOI] [PubMed] [Google Scholar]
- 147.Faggiano A, et al. Cinacalcet hydrochloride in combination with alendronate normalizes hypercalcemia and improves bone mineral density in patients with primary hyperparathyroidism. Endocrine. 2011;39:283–287. doi: 10.1007/s12020-011-9459-0. [DOI] [PubMed] [Google Scholar]
- 148.Keutgen XM, et al. Calcimimetics versus parathyroidectomy for treatment of primary hyperparathyroidism: retrospective chart analysis of a prospective database. Ann Surg. 2012;255:981–985. doi: 10.1097/SLA.0b013e31824c5252. [DOI] [PubMed] [Google Scholar]
- 149.Koumakis E, et al. Bone mineral density evolution after successful parathyroidectomy in patients with normocalcemic primary hyperparathyroidism. J Clin Endocrinol Metab. 2013;98:3213–3220. doi: 10.1210/jc.2013-1518. [DOI] [PubMed] [Google Scholar]
- 150.Silverberg SJ, Shane E, Jacobs TP, Siris E, Bilezikian JP. A 10-year prospective study of primary hyperparathyroidism with or without parathyroid surgery. N Engl J Med. 1999;341:1249–1255. doi: 10.1056/NEJM199910213411701. [DOI] [PubMed] [Google Scholar]
- 151.Cesareo R, et al. Effects of alendronate and vitamin D in patients with normocalcemic primary hyperparathyroidism. Osteoporos Int. 2015;26:1295–1302. doi: 10.1007/s00198-014-3000-2. [DOI] [PubMed] [Google Scholar]
- 152.Aberg V, et al. Health-related quality of life after successful surgery for primary hyperparathyroidism: no additive effect from vitamin D supplementation: results of a double-blind randomized study. Eur J Endocrinol. 2015;172:181–187. doi: 10.1530/EJE-14-0757. [DOI] [PubMed] [Google Scholar]
- 153.Amstrup AK, Rejnmark L, Mosekilde L. Patients with surgically cured primary hyperparathyroidism have a reduced quality of life compared with population-based healthy sex-, age-, and season-matched controls. Eur J Endocrinol. 2011;165:753–760. doi: 10.1530/EJE-11-0301. [DOI] [PubMed] [Google Scholar]
- 154.Babinska D, et al. Evaluation of selected cognitive functions before and after surgery for primary hyperparathyroidism. Langenbecks Arch Surg. 2012;397:825–831. doi: 10.1007/s00423-011-0885-5. [DOI] [PubMed] [Google Scholar]
- 155.Benge JF, et al. Cognitive and affective sequelae of primary hyperparathyroidism and early response to parathyroidectomy. J Int Neuropsychol Soc. 2009;15:1002–1011. doi: 10.1017/S1355617709990695. [DOI] [PubMed] [Google Scholar]
- 156.Blanchard C, et al. Quality of life is modestly improved in older patients with mild primary hyperparathyroidism postoperatively: results of a prospective multicenter study. Ann Surg Oncol. 2014;21:3534–3540. doi: 10.1245/s10434-014-3731-5. [DOI] [PubMed] [Google Scholar]
- 157.Espiritu RP, et al. Depression in primary hyperparathyroidism: prevalence and benefit of surgery. J Clin Endocrinol Metab. 2011;96:E1737–E1745. doi: 10.1210/jc.2011-1486. [DOI] [PubMed] [Google Scholar]
- 158.Hermsen A, et al. Perioperative changes in cortical excitability, mood, and quality of life in patients with primary hyperparathyroidism: a pilot study using transcranial magnetic stimulation. Eur J Endocrinol. 2014;170:201–209. doi: 10.1530/EJE-13-0552. [DOI] [PubMed] [Google Scholar]
- 159.Kahal H, et al. The effect of parathyroidectomy on neuropsychological symptoms and biochemical parameters in patients with asymptomatic primary hyperparathyroidism. Clin Endocrinol (Oxf) 2012;76:196–200. doi: 10.1111/j.1365-2265.2011.04197.x. [DOI] [PubMed] [Google Scholar]
- 160.Roman SA, et al. The effects of serum calcium and parathyroid hormone changes on psychological and cognitive function in patients undergoing parathyroidectomy for primary hyperparathyroidism. Ann Surg. 2011;253:131–137. doi: 10.1097/SLA.0b013e3181f66720. [DOI] [PubMed] [Google Scholar]
- 161.Ryhanen EM, et al. Health-related quality of life is impaired in primary hyperparathyroidism and significantly improves after surgery: a prospective study using the 15D instrument. Endocr Connect. 2015;4:179–186. doi: 10.1530//EC-15-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tsukahara K, Sugitani I, Fujimoto Y, Kawabata K. Surgery did not improve the subjective neuropsychological symptoms of patients with incidentally detected mild primary hyperparathyroidism. Eur Arch Otorhinolaryngol. 2008;265:565–569. doi: 10.1007/s00405-007-0523-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Walker MD, et al. Neuropsychological features in primary hyperparathyroidism: a prospective study. J Clin Endocrinol Metab. 2009;94:1951–1958. doi: 10.1210/jc.2008-2574. This is a detailed study of neurocognitive features of PHPT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pasieka JL, et al. Patient-based surgical outcome tool demonstrating alleviation of symptoms following parathyroidectomy in patients with primary hyperparathyroidism. World J Surg. 2002;26:942–949. doi: 10.1007/s00268-002-6623-y. [DOI] [PubMed] [Google Scholar]
- 165.Webb SM, et al. Development of a new tool for assessing health-related quality of life in patients with primary hyperparathyroidism. Health Qual Life Outcomes. 2013;11:97. doi: 10.1186/1477-7525-11-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Perrier ND, et al. Prospective, randomized, controlled trial of parathyroidectomy versus observation in patients with “asymptomatic” primary hyperparathyroidism. Surgery. 2009;146:1116–1122. doi: 10.1016/j.surg.2009.09.034. [DOI] [PubMed] [Google Scholar]
- 167.Rolighed L, et al. No beneficial effects of vitamin D supplementation on muscle function or quality of life in primary hyperparathyroidism: results from a randomized controlled trial. Eur J Endocrinol. 2015;172:609–617. doi: 10.1530/EJE-14-0940. [DOI] [PubMed] [Google Scholar]
- 168.Bilezikian JP, et al. Summary statement from a workshop on asymptomatic primary hyperparathyroidism: a perspective for the 21st century. J Clin Endocrinol Metab. 2002;87:5353–5361. doi: 10.1210/jc.2002-021370. [DOI] [PubMed] [Google Scholar]
- 169.Morrissey JJ, Cohn DV. Regulation of secretion of parathormone and secretory protein-I from separate intracellular pools by calcium, dibutyryl cyclic AMP, and (1)-isoproterenol. J Cell Biol. 1979;82:93–102. doi: 10.1083/jcb.82.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]