

Abstract
The solute carrier family 2 facilitated glucose transporter member 4 (GLUT4) plays a key role in the insulin-induced glucose uptake by muscle and adipose tissues. In prediabetes and diabetes, GLUT4 expression/translocation has been detected as reduced, participating in mechanisms that impair glycemic control. Recently, a class of short endogenous noncoding RNAs named microRNAs (miRNAs) has been increasingly described as involved in the posttranscriptional epigenetic regulation of gene expression. The present review focuses on miRNAs potentially involved in the expression of GLUT4 expression, and proteins related to GLUT4 and translocation in skeletal muscle, seeking to correlate them with insulin resistance and diabetes. So far, miR-21a-5p, miR-29a-3p, miR-29c-3p, miR-93-5p, miR-106b-5p, miR-133a-3p, miR-133b-3p, miR-222-3p, and miR-223-3p have been reported to directly and/or indirectly regulate the GLUT4 expression; and their expression is altered under diabetes-related conditions. Besides, some miRNAs that have been linked to the expression of proteins involved in GLUT4 translocation machinery in muscle could also impact glucose uptake. That makes these miRNAs promising targets for preventive and/or therapeutic approaches, which could improve glycemic control, thus deserving future new investigations.
1. Introduction
Diabetes mellitus (DM) is a metabolic disorder with high prevalence in the world [1]. It is characterized by hyperglycemia resulting from defects in insulin secretion and/or action. The most prevalent forms of the disease are type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM). T1DM results primarily from the lack of insulin secretion due to autoimmune destruction of pancreatic β cells, whereas T2DM primarily results from hormone resistance in target tissues, particularly in liver, adipose tissue, and skeletal muscle, which can lead to impaired insulin secretion [2].
Several studies have sought to clarify the main molecular mechanisms involved in the pathophysiology of T2DM. It is known that insulin resistance is important to the development and maintenance of glycemic homeostasis loss [3], a phenomenon in which skeletal muscle plays a key role [4]. The skeletal muscle is the main tissue responsible for insulin-stimulated glucose disposal and the major site of peripheral insulin resistance [4]. In muscles, as in the adipose tissue, the insulin-stimulated glucose uptake is performed through the solute carrier family 2, facilitated glucose transporter member 4 (GLUT4), which is rapidly translocated to the plasma membrane in response to the hormone [5, 6]. Although the skeletal muscle glucose disposal can be reduced by impaired GLUT4 translocation [6, 7], long term established reduction in the glucose uptake has currently been related to a defective glucose transporter gene and/or protein expression [8].
Recently, a new element in the pathophysiology of diabetes emerged, receiving great attention. It is a class of small endogenous noncoding RNAs termed microRNAs. MicroRNAs (miRNAs) are regulatory small noncoding RNAs (~22 nucleotides) that bind to the 3′UTR region of mRNAs, destabilizing them or inhibiting their translation, and thus working as negative posttranscriptional regulators of gene expression [9]. Hence, miRNAs participate as important regulators of biological processes, including cellular differentiation, proliferation, apoptosis, and metabolism, as well as tissue development [9]. Thereafter, alterations in the expression of miRNAs have been observed in numerous cellular dysfunctions, and many studies have been performed to investigate the role of miRNAs in the pathophysiology of diabetes [10–12].
This review summarizes the potential participation of miRNAs in the regulation of GLUT4 protein (codified by the SLC2A4 gene) and proteins related to its translocation, in skeletal muscle, exploring their involvement in the pathophysiology of diabetes, as well as their potential role in preventive or therapeutic approaches for diabetes.
2. Skeletal Muscle, GLUT4, and Glycemic Homeostasis
Skeletal muscle makes up approximately 40% of the total body mass. The main function of skeletal muscle is contraction-related, for which the main energetic substrate is glucose. Skeletal muscle is a great consumer of extracellular glucose, which increases under insulin- and contraction-stimulation. In healthy subjects, skeletal muscle accounts for up to ~80% of glucose disposal under insulin-stimulated conditions, as it occurs in postprandial state, thus displaying a fundamental role in glycemic homeostasis [3].
The glucose transport in mammalian tissues occurs primarily by facilitated diffusion, a process that needs carrier proteins. In the early eighties, it was demonstrated that insulin was able to increase glucose transport in adipocytes [13] and skeletal muscle [14]. Later on, an insulin-regulatable glucose transporter was identified in these tissues [15], and a cDNA encoding this glucose transporter was obtained in rat, mouse, and human [16–20]. This insulin-responsive glucose transporter was ultimately named GLUT4, based on the chronological order in which the various isoforms of GLUTs were characterized.
The GLUT4 protein (codified by the gene solute carrier family 2 member 4, SLC2A4) is the major glucose transporter of brown and white adipose tissues and of skeletal and cardiac muscles [21]. Under basal conditions, that is, without insulin stimulus, a great amount of GLUT4 is restrained in tubule-vesicular structures named GLUT4 storage vesicles (GSVs) [22]. Under insulin stimulus, the GSVs are translocated to the cell surface, rapidly increasing the GLUT4 density at plasma membrane, and, consequently, the glucose uptake [5, 6, 23]. Insulin signaling regulates the several steps of GLUT4 translocation, including GSVs releasing from intracellular retention, trafficking, tethering, and, finally, docking and fusion into the cell membrane. In brief, insulin binds to its receptor, activating intrinsic kinase activity, leading to tyrosine phosphorylation of insulin receptor substrate 1 (IRS1) (the predominant IRS isoform in skeletal muscle) and activating phosphatidylinositide 3-kinase (PI3K). PI3K signaling bifurcates into the Rac1 (a Rho GTPase) and the PDK1 (phosphoinositide-dependent protein kinase) pathways, which converge to GLUT4 translocation, both of which ultimately stimulate small GTPases [6, 24, 25].
Rac1 pathway involves rearrangement of actin-filaments and cortical actin branching, mediated by actin-related proteins 2/3 (ARP2/3) complexes [26], which provides a path along which GSVs are transported, and is crucial for retention and fusion of GSV beneath the plasma membrane [27]. PDK1 pathway leads to activation of AKT (particularly isoform AKT2) and of the atypical protein kinases C zeta and lambda (PKCζ/λ) [28], all involved in the GSV translocation. AKT-mediated signal involves downstream participation of the RAB-GTPase-activating proteins named TBC1D4 (also known as AS160) and TBC1D1. Several RAB-GTPases are targets for TBC1D4 in muscle cells [29–33] and are involved in intracellular vesicle traffic through engagement of mechanical effectors responsible for vesicle budding, mobilization, and fusion [34]. Finally, unconventional myosin-Ic (MYO1C) and SNAP-associated receptor (SNARE) proteins such as vesicle-associated membrane protein 2 (VAMP2), syntaxin-4 (STX4), synaptosomal-associated protein 23 (SNAP23), syntaxin-binding protein 3 (STXBP3), and syntaxin-binding protein 4 (STXBP4) were described as essential elements for insulin-mediated tethering and fusion of GSVs in muscle cells [6, 23, 35, 36].
Besides, in skeletal muscle, contraction-induced GLUT4 translocation also occurs, involving at least two proteins: AMP-activated protein kinase (AMPK) and Ca2+/calmodulin-dependent protein kinase (CaMK2) [37], which can operate in addition to insulin-triggered ones. Thus, GLUT4 is the major mediator of extracellular glucose clearance, playing a key role in the regulation of glycemic homeostasis [3, 6]. In addition to the acute effect of insulin and muscle contraction upon GLUT4 translocation, it has also been demonstrated that both stimuli are able to increase Slc2a4/GLUT4 expression, by, respectively, activating or inhibiting enhancer or repressor transcriptional factors [38–40].
Considering what was described above, it becomes evident that insulin and muscle contraction modulate glycemic homeostasis by regulating both translocation and expression of GLUT4.
3. Skeletal Muscle GLUT4 Expression and Translocation: From Insulin Resistance to T2DM
In prediabetes and T2DM, reduced insulin-stimulated glucose uptake by skeletal muscle is a current feature [3, 41]. Additionally, in T1DM, skeletal muscle glucose disposal can also be reduced because of glucotoxicity and/or hyperinsulinemia [42], pointing out that insulin therapy is obligatory. Thus, under all conditions related to impaired glycemic control, insulin resistance is involved, and, thus, altered skeletal muscle GLUT4 expression and translocation must be involved.
Since the characterization of GLUT4, and considering its central role in glycemic homeostasis, several studies have been performed attempting to understand the role of the skeletal muscle SLC2A4/GLUT4 expression and translocation in the pathophysiology of insulin resistance and/or diabetes. However, the interpretation of the results must be carefully performed, because muscle tissues show multiple fiber types, displaying different rates of insulin sensitivity and GLUT4 expression. Red (type I, slow-twitch, oxidative) fibers are significantly more insulin-responsive than white (type IIa/b, fast-twitch oxidative/glycolytic) muscle fibers and have higher levels of GLUT4 protein [43, 44]. So, the skeletal muscle participation upon glycemic homeostasis must take the whole-body proportion between white and red muscles into account.
Skeletal muscle SLC2A4/GLUT4 expression under conditions of impaired glycemic homeostasis has been investigated since the early nineties. In prediabetes, T2DM and T1DM experimental models, GLUT4 expression in skeletal muscle has been regularly detected to be reduced [42, 45–49]. Besides, transgenic animals with muscle-specific inactivation of GLUT4 show a diabetic phenotype [50, 51], and the muscle-specific GLUT4 overexpression in diabetic animals improves glycemic control [52, 53], pointing out the fundamental role of GLUT4 in glycemic homeostasis [7]. In humans, although some pioneering studies reported that SLC2A4/GLUT4 expression was unchanged in skeletal muscles of prediabetes and T2DM subjects [54, 55], other studies reported a reduction of GLUT4 [56], which was further confirmed in more sensitive quantitative analyses of GLUT4 [57, 58]. Therefore, repressed Slc2a4/GLUT4 expression in skeletal muscle becomes a focus in the pathophysiology and treatment of impaired glycemic homeostasis conditions [8, 59–61]. Finally, in insulin-deficient animals [42, 62] and T1DM humans [63, 64] under insulin therapy, the Slc2a4/GLUT4 expression seems to be unaltered, as compared to nondiabetic state, reinforcing the important role of the glycemic homeostasis in the regulation of this gene.
Insulin signaling damage also plays a role in insulin resistance of skeletal muscle, by impairing GLUT4 storage vesicles (GSV) translocation. In T2DM humans, reduced activation of early steps of insulin signaling, such as IRS1/PI3K [24, 28], and downstream steps such as activation AKT, PKC-zeta, and TBC1D4 [28, 65, 66] have been observed to be impaired, thus compromising insulin-stimulated GSV translocation. On the other hand, in T2DM, muscle contraction-stimulated glucose transport is preserved, and the activity of proteins related to contraction-induced GSV translocation, such as AMPK and CAMKII, has been described as unaltered [37]. Importantly, despite reduced activation of several proteins related to the GSV translocation machinery, the expression (cellular content) of these proteins is unaltered in T2DM, except by the PKC-zeta that was found reduced [28].
Behavioral (diet and exercise) and pharmacological (metformin, thiazolidinediones, glimepiride, insulin, resveratrol, and quercetin) interventions that improve glycemic control have been described as able to increase Slc2a4/GLUT4 expression and translocation [8, 39, 42, 49, 67–72]. Besides, although some insulin sensitizers were developed without the knowledge of their effects on the machinery of the GSV translocation [24], some of them such as exercise, metformin, and thiazolidinediones have revealed improvement of some steps of the insulin signaling cascade [24, 28].
In both T1DM and T2DM, muscle glucose disposal is variably impaired, and, because of that, skeletal muscle was emphasized by Coleman et al. as an important target for delaying diabetic complications [73]. The present data highlight the importance of increasing muscle SLC2A4 gene expression to improve glycemic control, since the GLUT4 density in the GSV is primordial for an effective GLUT4 translocation. In fact, Slc2a4 gene becomes a promising target for pharmacogenomics of insulin resistance [8], and, for that, it is mandatory to unravel mechanisms related to the SLC2A4 gene expression, such as its epigenetic regulation by microRNAs.
4. MicroRNAs (miRNAs)
MicroRNAs (miRNAs) are a class of short (~22 nt) endogenous noncoding RNAs that participate in the epigenetic regulation of gene expression [9]. Since the discovery of the first miRNA (named lin-4) in nematode Caenorhabditis elegans [74, 75] and the identification of the first miRNA in humans (named let-7) [76], hundreds of miRNAs have been found in both animals and plants and have revolutionized the understanding of the regulation of gene expression.
The vast majority of miRNAs are initially transcribed by RNA polymerase II resulting in a long primary transcript called pri-miRNA. The pri-miRNA is subsequently processed: firstly in the nucleus, by the Drosha–DGCR8 complex, to a ~60–70 nt precursor miRNA (pre-miRNA) hairpin-loop structure, which is exported to the cytoplasm where the Dicer–TRBP complex will generate a ~22 nt double-stranded miRNA [77]. One strand of this duplex, representing a mature miRNA, is then incorporated into the miRNA-induced silencing complex (miRISC). As part of miRISC, miRNAs imperfectly base-pair with sequences in the 3′ untranslated region (UTR) of target mRNAs and inhibit protein synthesis by either repressing translation or promoting mRNA deadenylation and decay [77]. This interaction is nucleated by perfect Watson-Crick base pairing of nucleotides 2–7 at the 5′ end of the miRNA (termed “seed sequence”) with a complementary seed match site in the 3′-UTR of the target mRNA [78]. However, it is important to note that the base pairing may not occur only in the 3′-UTR, but also in the open reading frame (ORF) or 5′-UTR of the mRNA target [79]. Importantly, a single miRNA can regulate the expression of hundreds of genes, and the expression of a single gene can be regulated by multiple miRNAs [80]. Moreover, computational predictions suggest that more than 60% of all mammalian protein-coding genes may be regulated my miRNAs [81]. Indeed, miRNAs have been implicated in the regulation of many key biological processes, and, miRNA deregulation is a common feature in several pathophysiological conditions, including metabolic disorders such as diabetes [11, 12, 82–84], suggesting that miRNAs could serve as targets for preventive or therapeutic interventions.
5. miRNAs and Skeletal Muscle
Many miRNAs are expressed in a tissue-specific manner. This concept was confirmed in a study by Lagos-Quintana et al. [85] showing that miR-1, miR-122-5p, and miR-124-3p expression was restricted to striated muscle, liver, and brain, respectively. Currently, a tissue-specific miRNA is defined as a miRNA that is expressed in a specific tissue at a level that is at least 20-fold higher than it is expressed in all other tissues [86]. Some miRNAs, including miR-1, miR-133a/b-3p, and miR-206-3p, are highly expressed in skeletal muscle, corresponding to nearly 25% of total miRNA expression in skeletal muscle, and, thus, they are habitually referred to as muscle-specific miRNAs or myomiRs [87]. Other myomiRs described include miR-208a-3p, miR-208b-3p, and miR-499-5p [88]. Most myomiR family members are expressed in both the heart and the skeletal muscle, except for miR-208a-3p, which is cardiac-specific [88], and miR-206 which is only found in skeletal muscle, mainly in slow-twitch muscles such as the soleus [89]. In addition, miR-208b-3p and miR-499-5p, encoded by myosin genes, may be useful markers of the muscle fiber type [88].
Several of these miRNAs are organized under bicistronic clusters on the same chromosome (i.e., miR-1-1/133a-2, miR-1-2/133a-1, and miR-206/133b) and are transcribed together [90]. These myomiRs are controlled by fundamental myogenic regulatory factors, including myoblast determination protein 1 (MYOD1), myogenic factor 5 (MYF5), myogenic factor 6 (MYF6), and myogenin (MYOG), as well as serum response factor (SRF) and myocyte enhancer factor 2 A/B/C/D (MEF2A/B/C/D) [91, 92]. Together, they regulate the key processes of myogenesis, including myoblast/satellite cell proliferation and differentiation [91]. Furthermore, myomiRs are modulated during multiple biological processes involved in skeletal muscle growth, development, and maintenance, including hypertrophy and atrophy [87, 93].
Identifying the role and regulation of skeletal muscle miRNAs expression during several phases of muscle development, as well as under health impaired conditions, will significantly enhance the understanding of skeletal muscle biology and may result in new therapies to target muscle diseases or chronic diseases associated with impaired muscle function [94].
6. miRNAs and Proteins of the GLUT4 Translocation Machinery in Insulin Resistance
Over the last few years, several known miRNAs were described as involved in the pathophysiology of diabetes [11, 95], especially in the impaired glycemic homeostasis [10, 80, 96]. Some studies related changes in miRNAs expression in skeletal muscle of diabetic Goto-Kakizaki rats (decreased miR-10b-5p, miR-24-3p) and T2D humans (decreased miR-133a-3p and miR-206), but they did not relate these miRNAs to any target mRNA [97–99]. In a step by step review of the protein related to the GLUT4 translocation machinery in skeletal muscle, several miRNAs have been potentially correlated to some target genes.
Regarding the early steps of insulin signaling, the INSR mRNA was suggested to be target for miR-let-7f-5p in T2DM mice [100] and for miR-15b-5p in T2DM humans [101]. Besides, it was reported that INSR gene expression was downregulated in miR-135a-5p transfected C2C12 cells [102]. IRS1 and/or IRS2 mRNAs were correlated with (1) let-7f-5p in T2DM mice [100]; (2) miR-15b-5p in T2DM humans [101]; (3) miR-29a-3p in IR-obese mice [103]; (4) miR-135a-5p in T2DM mice [104]; and (5) miR-144-3p in T2DM rats [105].
PI3KR1 mRNA was proposed to be regulated only by miR-15b-5p in T2DM humans [101]. AKT2 mRNA reduction was correlated to reduced expression of miR-23a and miR-107 after loss of weight in obese dogs [106], suggesting an unexpected and incomprehensible increasing of AKT2 in the obese insulin resistant dogs before the weight loss. Besides, miR-29/a/b/c-3p regulation of AKT2 was also suggested in GK rats [107]. Finally, regulation of some proteins downstream the PI3K has also been related to miRNAs: (1) MTOR and P70S6K1 were proposed to be regulated by reduction of miR-16-5p in obese rats and mice [108]; (2) MAPK14 expression was related to miR-24-3p in GK rats [99]; and (3) RAC1 decrease was correlated with miR-23a and miR-107 decreasing after weight loss of obese dogs [106]. Additionally, phosphorylation of AKT and TBC1D4 was related to decreased miR-194-5p [109] and increased miR-494-3p [110], respectively; however, this effect cannot be attributed to a direct miRNA epigenetic regulation. Finally, there is no report relating miRNAs and posttranscriptional regulation of AMPK and CAMK2 genes in skeletal muscle of insulin resistant animals or humans.
Table 1 summarizes the miRNAs potentially involved in the regulation of proteins that are related to GLUT4 translocation machinery in skeletal muscle tissue/cells under insulin resistant conditions.
Table 1.
Insulin resistance-induced regulation of miRNAs related to proteins of the GLUT4 translocation machinery in skeletal muscle.
| miRNA∗ | Condition | miRNA expression |
Proposed target∗∗ and its regulation | Reference |
|---|---|---|---|---|
| let-7f-5p | T2DM human and mouse | ↑ | ↓ INSR, IRS2 | Zhu et al. [100] |
| miR-10b-5p | GK rat | ↓ | — | Herrera et al. [97] |
| miR-15b-5p | T2DM human | ↓ | ↓ INSR, IRS1, ↑ PIK3R1 |
Bork-Jensen et al. [101] |
| miR-16-5p | Obese rat and mouse | ↓ | ↓ MTOR, RPS6KB1 | Lee et al. [108] |
| miR-23a | Loss of weight in obese dog | ↓ | ↓ RAC1, AKT2 | Uribe et al. [106] |
| miR-24-3p | GK rat | ↓ | ↑ MAPK14 | Huang et al. [99] |
| miR-29a/b/c-3p | GK rat, IR-obese mouse | ↑ | ↓ AKT2, IRS1 | He et al. [107]; Yang et al., [103] |
| miR-107 | Loss of weight in obese dog | ↓ | ↓ RAC1, AKT2 | Uribe et al. [106] |
| miR-133a-3p | T2DM human | ↓ | — | Gallagher et al. [98] |
| miR-135a-5p | T2DM mouse, IR-C2C12 cell | ↑ | ↓ INSR, IRS2 | Agarwal et al. [104]; Honardoost et al. [102] |
| miR-144-3p | T2DM rat | ↑ | ↓ IRS1 | Karolina et al. [105] |
| miR-206 | T2DM human | ↓ | — | Gallagher et al. [98] |
∗The nomenclature of miRNAs is in accordance with the “miRBase Sequence Database-Release 21.” ∗∗Gene names are based on HGNC (HUGO Gene Nomenclature Committee), and despite some date being related to mouse/rat gene, description was related to human gene (capital letters). IR, insulin resistance/resistant; T2DM, type 2 diabetes mellitus; GK, Goto-Kakizaki; INSR, insulin receptor; IRS1, insulin receptor substrate 1; IRS2, insulin receptor substrate 2; MAPK14, mitogen-activated protein kinase 14; AKT2, AKT serine/threonine kinase 2; PI3KR1, phosphoinositide-3-kinase regulatory subunit 1; MTOR, mechanistic target of rapamycin; RPS6KB1, ribosomal protein S6 kinase B1; RAC1, ras-related C3 botulinum toxin substrate 1 (rho family, small GTP binding protein Rac1).
Finally, it is important to highlight that most epigenetic regulation described here were not validated, but just reported as correlated regulation. So, future studies must be conducted to validate that miRNAs participate in the regulation of proteins of the GLUT4 translocation machinery in the insulin resistance. It is also important to emphasize that these recent studies on miRNA regulation of proteins of the insulin signaling cascade have described alterations in mRNA/protein content in insulin resistant conditions, which do not match the paradigm that insulin resistance alters only the activation of these proteins, but not their content [28].
7. miRNAs Related to the SLC2A4/GLUT4 Expression in Insulin Resistance
To our knowledge, under conditions related to insulin resistance and/or diabetes, just a few studies have investigated the regulation of GLUT4 expression by miRNAs. These studies were not performed uniquely in skeletal muscle, but some of them were conducted in adipose and cardiac tissues or cells, and we are including them in the present analysis. The GLUT4 regulation by miRNAs include (1) confirmed direct effect upon Slc2a4 mRNA expression; (2) confirmed direct effect upon a target that regulates Slc2a4 mRNA expression; and (3) predictable direct effect by in silico analysis and correlation between expression of GLUT4 and the miRNA.
Confirmed direct modulations of Slc2a4/GLUT4 expression were demonstrated for miR-93-5p, miR-223-3p, and miR-106b-5p [111–114].
An elegant study has shown that miR-93-5p was upregulated in subcutaneous adipose tissue from polycystic ovary syndrome (PCOS) and non-PCOS insulin resistant women, and that miR-93-5p expression correlated negatively with GLUT4 and positively with HOMA-IR in these women [111]. Besides, overexpression of miR-93-5p in both human and 3T3-L1 adipocytes decreased GLUT4 protein expression; and, conversely, the inhibition of miR-93-5p increased GLUT4 expression [111]. Besides, by means of luciferase assay, this study confirmed the direct regulation of Slc2a4 by miR-93-5p [111].
Another miRNA confirmed as a direct regulator of Slc2a4/GLUT4 is the miR-223-3p, which has been described as upregulated in adipose tissue of women with insulin resistance [112]. In fact, overexpression of miR-223-3p in human adipocytes was reported to inhibit insulin-stimulated glucose uptake and to decrease GLUT4 protein content [112]. Similarly, upregulation of miR-223-3p was observed in heart of T2DM patients [113]. However, the overexpression of miR-223-3p in cardiomyocyte from neonatal rats was accompanied by increased GLUT4 expression [113], revealing a paradoxical effect as compared to that observed in adipocyte [112].
Recently, a direct negative effect of miR-106b-5p upon Slc2a4 was demonstrated by luciferase assay, in muscle L6 cells. In these cells, overexpression of miR-106b-5p downregulated the GLUT4 content and decreased the glucose consumption and uptake; and, conversely, knockdown of miR-106b-5p increased the levels of GLUT4 and glucose consumption in this cell [114].
Indirect regulation of Slc2a4 mRNA expression may be related to miR-222-3p, miR-133a/b-3p, miR-21a-5p, and miR-29a-3p [115–118].
In omental adipose tissue from women with gestational diabetes mellitus miR-222-3p expression was found upregulated; besides that, miR-222-3p expression was negatively correlated with estrogen receptor 1 (ESR1) and GLUT4 contents and positively correlated with serum estradiol levels [115]. In addition, Esr1 mRNA was shown to be a target for miR-222-3p in 3T3-L1 cells, in which estradiol increased miR-222-3p expression and reduced ESR1 and GLUT4 proteins [115]. Besides, miR-222-3p silencing in 3T3-L1 adipocytes was seen to increase ESR1 and GLUT4 expression, as well as the insulin-stimulated glucose uptake [115]. Regarding that, our group demonstrated that ESR1 is a potent enhancer of Slc2a4/GLUT4 expression in 3T3-L1 adipocytes [119], supporting the proposal of an ESR1-mediated effect of miR-222-3p upon GLUT4.
Regulation of GLUT4 expression has also been related to the typical myomiRs miR-133a-3p and miR-133b-3p, which were reported to act indirectly upon Slc2a4/GLUT4 expression. In fact, miR-133 was observed to target the Klf15 mRNA (Krüeppel-like factor 15), inhibiting this transcriptional factor, which is an enhancer of Slc2a4 expression; thus miR-133 leads to the reduction in GLUT4 expression and in insulin-stimulated glucose uptake in rat cardiomyocytes [116].
Ling and colleagues have suggested that miR-21a-5p targets the Pten mRNA (phosphatase tensin homologue), since overexpression of miR-21a-5p in 3T3-L1 adipocytes decreases PTEN protein, despite unchanged Pten mRNA, and considering that Pten 3′UTR has a highly conserved miR-21a-5p recognition element [117]. PTEN is a negative regulator of PI3K/AKT activity [117], which has been demonstrated to be involved in the insulin-induced enhancer effect on Slc2a4 transcription [40]. Thus, miR-21a-5p by decreasing PTEN increases PI3K/AKT activity, which would enhance Slc2a4/GLUT4 expression. Unfortunately, the study by Ling and colleagues [117] did not evaluate Slc2a4 mRNA or total cellular GLUT4 protein expression to reinforce this hypothesis.
Another indirect regulation of GLUT4 is attributed to miR-29a-3p [118], which was seen to target the Ppard mRNA (peroxisome proliferator-activated receptor delta) suppressing PPARD protein, which in turn reduced the PGC1A (peroxisome proliferator-activated receptor gamma coactivator 1-alpha); the latter is an important enhancer of Slc2a4 transcription [120]. In fact, overexpression of miR-29a-3p in skeletal muscle cell induced a GLUT4 reduction, bearing in mind that the miR-29a-3p can also play a direct effect on Slc2a4 mRNA, as commented on above.
Predictable direct regulation of Slc2a4, indicated by in silico analysis and/or correlation between expression of GLUT4 and miRNA, have been proposed to miR-29c-3p, miR-29a-3p, and miR-222-3p [115, 118, 121].
Increased levels of miR-29c-3p with reduced GLUT4 content were observed in cardiomyocytes of obese animals, suggesting an inverse relationship between GLUT4 and miRNA-29c-3p; however, this hypothesis needs to be validated [121]. In addition, upregulation of miR-29a-3p was observed in skeletal muscle of rats with intrauterine growth restriction; and overexpression of miR-29a-3p in muscle C2C12 cells induced a reduction in Slc2a4 mRNA and GLUT4 protein [118]. However, the direct effect of both miR-29c-3p and miR-29a-3p upon GLUT4 expression remains to be demonstrated. In silico analysis points out Slc2a4 mRNA as target to miR-222-3p; however, whether the Slc2a4/GLUT4 regulation observed in response to miR-222-3p involves a direct effect of this microRNA upon Slc2a4 mRNA, or it is just secondary to the ESR1 regulation [115], as commented on above, remains to be determined.
Finally, in obese dogs that have been subjected to weight loss, the expression of both miR-23a and miR-107 was downregulated, and that was accompanied by decreased Slc2a4 mRNA expression; however, in this experimental condition AKT2 and RAC1 were also repressed, and thus the Slc2a4 regulation might be direct and/or indirect [106].
Table 2 shows the miRNAs that have been related to GLUT4 expression, and Figure 1 shows a schematic summary of direct and indirect regulation of GLUT4.
Table 2.
miRNAs related to the Slc2a4/GLUT4 expression in insulin resistance.
| miRNA∗ | Species/tissue | Description | Reference |
|---|---|---|---|
| miR-23a | Dog/skeletal muscle | Downregulated after loss of weight, together with decreased Slc2a4 mRNA | Uribe et al. [106] |
|
| |||
| miR-29c-3p | Mouse/heart | Upregulated in heart of obese mice, together with decreased GLUT4 | Guedes et al. [121] |
|
| |||
| miR-29a-3p | Rat and mouse/ skeletal muscle |
Upregulated in muscle of rats with intrauterine growth restriction; overexpression in C2C12 myocyte decreased GLUT4 | Zhou et al. [118] |
|
| |||
| miR-93-5p | Human/adipose | Upregulated in adipose tissue of women with PCOS and/or IR; overexpression in adipocyte decreased GLUT4 | Chen et al. [111] |
|
| |||
| miR-106b-5p | Rat/skeletal muscle | Upregulated in muscle of rats with T2DM; overexpression in L6 myocyte decreased GLUT4 and glucose uptake | Zhou et al. [114] |
|
| |||
| miR-107 | Dog/skeletal muscle | Downregulated after loss of weight, together with decreased Slc2a4 mRNA | Uribe et al. [106] |
|
| |||
| miR-133a/b-3p | Rat/heart | Overexpression in cardiomyocyte decreased GLUT4 and insulin-stimulated glucose uptake | Horie et al. [116] |
|
| |||
| miR-222-3p | Human/adipose | Upregulated in adipose tissue of women with GDM; overexpression in adipocyte decreased Slc2a4 mRNA | Shi et al. [115] |
|
| |||
| miR-223-3p | Human/adipose | Upregulated in adipose tissue of women with IR; cell overexpression decreased GLUT4 and glucose uptake | Chuang et al. [112] |
|
| |||
| miR-223-3p | Human and rat/heart | Upregulated in heart of humans with T2DM; overexpression in rat cardiomyocyte increased GLUT4 | Lu et al. [113] |
∗The nomenclature of miRNAs is in accordance with the “miRBase Sequence Database-Release 21.” IR, insulin resistance; GDM, gestational diabetes mellitus; PCOS, polycystic ovary syndrome; T2DM: type 2 diabetes mellitus.
Figure 1.
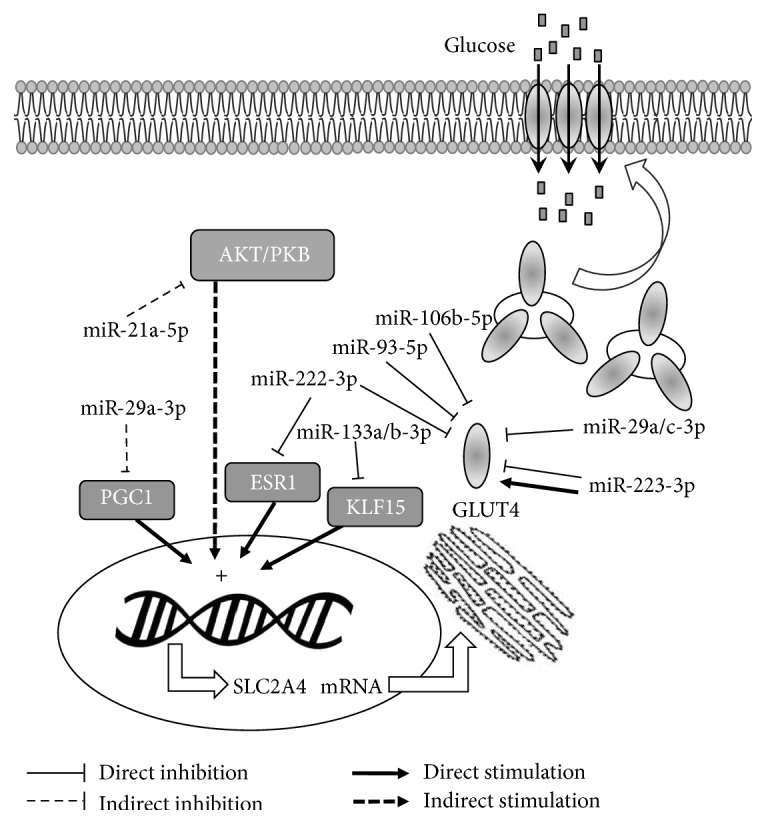
SLC2A4/GLUT4 expression regulation by miRNAs in muscle and adipose tissues. Regulation have been proposed to occur directly in GLUT4 translation and/or in transcription factors that regulate SLC2A4 transcription, and, consequently, GLUT4 expression. Several mechanisms are involved in translocation of GLUT4 storage vesicles to the plasma membrane, increasing GLUT4 density and glucose transport. PI3K, phosphatidylinositol 3-kinase; AKT, RAC-alpha serine/threonine-protein kinase; PGC1A, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; ESR1, estrogen receptor 1; KLF15, Krüeppel-like factor 15; GLUT4, solute carrier family 2, facilitated glucose transporter member 4; SLC2A4, solute carrier family 2 member 4 gene; miR, microRNA.
8. Conclusions
In adipose and muscle tissues, several miRNAs have already been described as repressors of GLUT4 expression and, consequently, of tissue glucose disposal. In addition, in skeletal muscle, some miRNAs have been linked to the machinery involved in GLUT4 translocation. Moreover, the expression of these miRNAs has been described as altered in humans or experimental models with insulin resistance, thus revealing their potential participation in the pathophysiology of diabetes. Among these miRNAs, miR-29a-3p, miR-29c-3p, miR-93-5p, miR-222-3p, miR-223-3p, and miR-106b-5p are proposed to target the Slc2a4 3′UTR, whereas miR-133a-3p, miR-133b-3p, and miR-222-3p are proposed to target enhancers of Slc2a4 gene transcription. Besides, these and other miRNAs have been related to the expression/activity of proteins involved in the GLUT4 machinery translocation, which might also impact tissue glucose disposal and glycemic homeostasis as well. Thus, the study of miRNAs potentially involved in the regulation of GLUT4 expression and translocation represents an important field that can contribute to establishing preventive and/or therapeutic approaches for insulin resistance and diabetes.
Acknowledgments
The authors acknowledge grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) which has supported referenced studies from our research group in the last decades. J. V. Esteves was recipient of FAPESP Fellowship #2012/20432-0. The authors thank Dr. Adauri Brezolin for English revision of the manuscript.
Conflicts of Interest
The authors declare that there are no conflicts of interest associated with the manuscript.
References
- 1.Guariguata L., Whiting D. R., Hambleton I., Beagley J., Linnenkamp U., Shaw J. E. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Research and Clinical Practice. 2014;103(2):137–149. doi: 10.1016/j.diabres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 2.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2011;34(supplement 1):S62–S69. doi: 10.2337/dc11-s062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeFronzo R. A. Pathogenesis of type 2 diabetes mellitus. Medical Clinics of North America. 2004;88(4):787–835. doi: 10.1016/j.mcna.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Zierath J. R., Krook A., Wallberg-Henriksson H. Insulin action and insulin resistance in human skeletal muscle. Diabetologia. 2000;43(7):821–835. doi: 10.1007/s001250051457. [DOI] [PubMed] [Google Scholar]
- 5.Simpson F., Whitehead J. P., James D. E. GLUT4—at the cross roads between membrane trafficking and signal transduction. Traffic. 2001;2(1):2–11. doi: 10.1034/j.1600-0854.2001.020102.x. [DOI] [PubMed] [Google Scholar]
- 6.Klip A. The many ways to regulate glucose transporter 4. Applied Physiology, Nutrition and Metabolism. 2009;34(3):481–487. doi: 10.1139/H09-047. [DOI] [PubMed] [Google Scholar]
- 7.Herman M. A., Kahn B. B. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. The Journal of Clinical Investigation. 2006;116(7):1767–1775. doi: 10.1172/jci29027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corrêa-Giannella M. L., MacHado U. F. SLC2A4gene: a promising target for pharmacogenomics of insulin resistance. Pharmacogenomics. 2013;14(8):847–850. doi: 10.2217/pgs.13.45. [DOI] [PubMed] [Google Scholar]
- 9.Bartel D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 10.Guay C., Roggli E., Nesca V., Jacovetti C., Regazzi R. Diabetes mellitus, a microRNA-related disease? Translational Research. 2011;157(4):253–264. doi: 10.1016/j.trsl.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Chen H., Lan H.-Y., Roukos D. H., Cho W. C. Application of microRNAs in diabetes mellitus. Journal of Endocrinology. 2014;222(1):R1–R10. doi: 10.1530/joe-13-0544. [DOI] [PubMed] [Google Scholar]
- 12.Raffort J., Hinault C., Dumortier O., Van Obberghen E. Circulating microRNAs and diabetes: potential applications in medical practice. Diabetologia. 2015;58(9):1978–1992. doi: 10.1007/s00125-015-3680-y. [DOI] [PubMed] [Google Scholar]
- 13.Cushman S. W., Wardzala L. J. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. The Journal of Biological Chemistry. 1980;255(10):4758–4762. [PubMed] [Google Scholar]
- 14.Wardzala L. J., Jeanrenaud B. Potential mechanism of insulin action on glucose transport in the isolated rat diaphragm. Apparent translocation of intracellular transport units to the plasma membrane. The Journal of Biological Chemistry. 1981;256(14):7090–7093. [PubMed] [Google Scholar]
- 15.James D. E., Brown R., Navarro J., Pilch P. F. Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature. 1988;333(6169):183–185. doi: 10.1038/333183a0. [DOI] [PubMed] [Google Scholar]
- 16.Birnbaum M. J. Identification of a novel gene encoding an insulin-responsive glucose transporter protein. Cell. 1989;57(2):305–315. doi: 10.1016/0092-8674(89)90968-9. [DOI] [PubMed] [Google Scholar]
- 17.Charron M. J., Brosius F. C., III, Alper S. L., Lodish H. F. A glucose transport protein expressed predominately in insulin-responsive tissues. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(8):2535–2539. doi: 10.1073/pnas.86.8.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukumoto H., Kayano T., Buse J. B., et al. Cloning and characterization of the major insulin-responsive glucose transporter expressed in human skeletal muscle and other insulin-responsive tissues. The Journal of Biological Chemistry. 1989;264(14):7776–7779. [PubMed] [Google Scholar]
- 19.James D. E., Strube M., Muecdler M. Molecular cloning and characterization of an insulin-regulatable glucose transporter. Nature. 1989;338(6210):83–87. doi: 10.1038/338083a0. [DOI] [PubMed] [Google Scholar]
- 20.Kaestner K. H., Christy R. J., McLenithan J. C., et al. Sequence, tissue distribution, and differential expression of mRNA for a putative insulin-responsive glucose transporter in mouse 3T3-L1 adipocytes. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(9):3150–3154. doi: 10.1073/pnas.86.9.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uldry M., Thorens B. The SLC2 family of facilitated hexose and polyol transporters. Pflügers Archiv. 2004;447(5):480–489. doi: 10.1007/s00424-003-1085-0. [DOI] [PubMed] [Google Scholar]
- 22.Leto D., Saltiel A. R. Regulation of glucose transport by insulin: traffic control of GLUT4. Nature Reviews Molecular Cell Biology. 2012;13(6):383–396. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- 23.Foley K., Boguslavsky S., Klip A. Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry. 2011;50(15):3048–3061. doi: 10.1021/bi2000356. [DOI] [PubMed] [Google Scholar]
- 24.Björnholm M., Zierath J. R. Insulin signal transduction in human skeletal muscle: identifying the defects in Type II diabetes. Biochemical Society Transactions. 2005;33(2):354–357. doi: 10.1042/bst0330354. [DOI] [PubMed] [Google Scholar]
- 25.Klip A., Sun Y., Chiu T. T., Foley K. P. Signal transduction meets vesicle traffic: the software and hardware of GLUT4 translocation. American Journal of Physiology—Cell Physiology. 2014;306(10):C879–C886. doi: 10.1152/ajpcell.00069.2014. [DOI] [PubMed] [Google Scholar]
- 26.Chiu T. T., Jensen T. E., Sylow L., Richter E. A., Klip A. Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cellular Signalling. 2011;23(10):1546–1554. doi: 10.1016/j.cellsig.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 27.Chiu T. T., Patel N., Shaw A. E., Bamburg J. R., Klip A. Arp2/3- and cofilin-coordinated actin dynamics is required for insulin-mediated GLUT4 translocation to the surface of muscle cells. Molecular Biology of the Cell. 2010;21(20):3529–3539. doi: 10.1091/mbc.E10-04-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karlsson H. K. R., Zierath J. R. Insulin signaling and glucose transport in insulin resistant human skeletal muscle. Cell Biochemistry and Biophysics. 2007;48(2-3):103–113. doi: 10.1007/s12013-007-0030-9. [DOI] [PubMed] [Google Scholar]
- 29.Ishikura S., Bilan P. J., Klip A. Rabs 8A and 14 are targets of the insulin-regulated Rab-GAP AS160 regulating GLUT4 traffic in muscle cells. Biochemical and Biophysical Research Communications. 2007;353(4):1074–1079. doi: 10.1016/j.bbrc.2006.12.140. [DOI] [PubMed] [Google Scholar]
- 30.Randhawa V. K., Ishikura S., Talior-Volodarsky I., et al. GLUT4 vesicle recruitment and fusion are differentially regulated by Rac, AS160, and Rab8A in muscle cells. The Journal of Biological Chemistry. 2008;283(40):27208–27219. doi: 10.1074/jbc.m804282200. [DOI] [PubMed] [Google Scholar]
- 31.Sun Y., Bilan P. J., Liu Z., Klip A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(46):19909–19914. doi: 10.1073/pnas.1009523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Y., Chiu T. T., Foley K. P., Bilan P. J., Klip A. Myosin Va mediates Rab8A-regulated GLUT4 vesicle exocytosis in insulin-stimulated muscle cells. Molecular Biology of the Cell. 2014;25(7):1159–1170. doi: 10.1091/mbc.E13-08-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y., Jaldin-Fincati J., Liu Z., Bilan P. J., Klip A. A complex of Rab13 with MICAL-L2 and α-actinin-4 is essential for insulin-dependent GLUT4 exocytosis. Molecular Biology of the Cell. 2016;27(1):75–89. doi: 10.1091/mbc.E15-05-0319). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seabra M. C., Wasmeier C. Controlling the location and activation of RabGTPases. Current Opinion in Cell Biology. 2004;16(4):451–457. doi: 10.1016/j.ceb.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 35.Rowland A. F., Fazakerley D. J., James D. E. Mapping insulin/GLUT4 circuitry. Traffic. 2011;12(6):672–681. doi: 10.1111/j.1600-0854.2011.01178.x. [DOI] [PubMed] [Google Scholar]
- 36.Boguslavsky S., Chiu T., Foley K. P., et al. Myo1c binding to submembrane actin mediates insulin-induced tethering of GLUT4 vesicles. Molecular Biology of the Cell. 2012;23(20):4065–4078. doi: 10.1091/mbc.E12-04-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jessen N., Goodyear L. J. Contraction signaling to glucose transport in skeletal muscle. Journal of Applied Physiology. 2005;99(1):330–337. doi: 10.1152/japplphysiol.00175.2005. [DOI] [PubMed] [Google Scholar]
- 38.Silva J. L. T., Giannocco G., Furuya D. T., et al. NF-κB, MEF2A, MEF2D and HIF1-a involvement on insulin- and contraction-induced regulation of GLUT4 gene expression in soleus muscle. Molecular and Cellular Endocrinology. 2005;240(1-2):82–93. doi: 10.1016/j.mce.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 39.Lima G. A., Anhê G. F., Giannocco G., Nunes M. T., Correa-Giannella M. L., Machado U. F. Contractile activity per se induces transcriptional activation of SLC2A4 gene in soleus muscle: involvement of MEF2D, HIF-1a, and TRα transcriptional factors. American Journal of Physiology—Endocrinology and Metabolism. 2009;296(1):E132–E138. doi: 10.1152/ajpendo.90548.2008. [DOI] [PubMed] [Google Scholar]
- 40.Moraes P. A., Yonamine C. Y., Pinto Junior D. C., Esteves J. V. D., Machado U. F., Mori R. C. Insulin acutely triggers transcription of Slc2a4 gene: participation of the AT-rich, E-box and NFKB-binding sites. Life Sciences. 2014;114(1):36–44. doi: 10.1016/j.lfs.2014.07.040. [DOI] [PubMed] [Google Scholar]
- 41.Edwards C. M., Cusi K. Prediabetes: a worldwide epidemic. Endocrinology and Metabolism Clinics of North America. 2016;45(4):751–764. doi: 10.1016/j.ecl.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 42.Okamoto M. M., Anhê G. F., Sabino-Silva R., et al. Intensive insulin treatment induces insulin resistance in diabetic rats by impairing glucose metabolism-related mechanisms in muscle and liver. Journal of Endocrinology. 2011;211(1):55–64. doi: 10.1530/JOE-11-0105. [DOI] [PubMed] [Google Scholar]
- 43.Henriksen E. J., Bourey R. E., Rodnick K. J., Koranyi L., Permutt M. A., Holloszy J. O. Glucose transporter protein content and glucose transport capacity in rat skeletal muscles. American Journal of Physiology—Endocrinology and Metabolism. 1990;259(4):E593–E598. doi: 10.1152/ajpendo.1990.259.4.E593. [DOI] [PubMed] [Google Scholar]
- 44.Kern M., Wells J. A., Stephens J. M., et al. Insulin responsiveness in skeletal muscle is determined by glucose transporter (Glut4) protein level. Biochemical Journal. 1990;270(2):397–400. doi: 10.1042/bj2700397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Camps M., Castello A., Munoz P., et al. Effect of diabetes and fasting on GLUT-4 (muscle/fat) glucose-transporter expression in insulin-sensitive tissues. Heterogeneous response in heart, red and white muscle. Biochemical Journal. 1992;282(3):765–772. doi: 10.1042/bj2820765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hardin D. S., Dominguez J. H., Garvey W. T. Muscle group-specific regulation of GLUT 4 glucose transporters in control, diabetic, and insulin-treated diabetic rats. Metabolism. 1993;42(10):1310–1315. doi: 10.1016/0026-0495(93)90130-G. [DOI] [PubMed] [Google Scholar]
- 47.Machado U. F., Shimizu Y., Saito M. Decreased glucose transporter (GLUT 4) content in insulin-sensitive tissues of obese aurothioglucose- and monosodium glutamate-treated mice. Hormone and Metabolic Research. 1993;25(9):462–465. doi: 10.1055/s-2007-1002149. [DOI] [PubMed] [Google Scholar]
- 48.Klip A., Tsakiridis T., Marette A., Ortiz P. A. Regulation of expression of glucose transporters by glucose: a review of studies in vivo and in cell cultures. The FASEB Journal. 1994;8(1):43–53. doi: 10.1096/fasebj.8.1.8299889. [DOI] [PubMed] [Google Scholar]
- 49.Mori R. C. T., Hirabara S. M., Hirata A. E., Okamoto M. M., Machado U. F. Glimepiride as insulin sensitizer: increased liver and muscle responses to insulin. Diabetes, Obesity and Metabolism. 2008;10(7):596–600. doi: 10.1111/j.1463-1326.2008.00870.x. [DOI] [PubMed] [Google Scholar]
- 50.Zisman A., Peroni O. D., Abel E. D., et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nature Medicine. 2000;6(8):924–928. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]
- 51.Kim J. K., Zisman A., Fillmore J. J., et al. Glucose toxicity and the development of diabetes in mice with muscle-specific inactivation of glut4. The Journal of Clinical Investigation. 2001;108(1):153–160. doi: 10.1172/jci200110294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leturque A., Loizeau M., Vaulont S., Salminen M., Girard J. Improvement of insulin action in diabetic transgenic mice selectively overexpressing GLUT4 in skeletal muscle. Diabetes. 1996;45(1):23–27. doi: 10.2337/diab.45.1.23. [DOI] [PubMed] [Google Scholar]
- 53.Tsao T.-S., Burcelin R., Katz E. B., Huang L., Charron M. J. Enhanced insulin action due to targeted GLUT4 overexpression exclusively in muscle. Diabetes. 1996;45(1):28–36. doi: 10.2337/diab.45.1.28. [DOI] [PubMed] [Google Scholar]
- 54.Pedersen O. J., Bak J. F., Andersen P. H., et al. Evidence against altered expression of GLUT1 or GLUT4 in skeletal muscle of patients with obesity or NIDDM. Diabetes. 1990;39(7):865–870. doi: 10.2337/diab.39.7.865. [DOI] [PubMed] [Google Scholar]
- 55.Garvey W. T., Maianu L., Hancock J. A., Golichowski A. M., Baron A. Gene expression of GLUT4 in skeletal muscle from insulin-resistant patients with obesity, IGT, GDM, and NIDDM. Diabetes. 1992;41(4):465–475. doi: 10.2337/diab.41.4.465. [DOI] [PubMed] [Google Scholar]
- 56.Dohm G. L., Elton C. W., Friedman J. E., et al. Decreased expression of glucose transporter in muscle from insulin-resistant patients. American Journal of Physiology—Endocrinology and Metabolism. 1991;260(3):E459–E463. doi: 10.1152/ajpendo.1991.260.3.E459. [DOI] [PubMed] [Google Scholar]
- 57.Gaster M., Staehr P., Beck-Nielsen H., Schrøder H. D., Handberg A. GLUT4 is reduced in slow muscle fibers of type 2 diabetic patients: is insulin resistance in type 2 diabetes a slow, type 1 fiber disease? Diabetes. 2001;50(6):1324–1329. doi: 10.2337/diabetes.50.6.1324. [DOI] [PubMed] [Google Scholar]
- 58.Kampmann U., Christensen B., Nielsen T. S., et al. GLUT4 and UBC9 protein expression is reduced in muscle from type 2 diabetic patients with severe insulin resistance. PLoS ONE. 2011;6(11) doi: 10.1371/journal.pone.0027854.e27854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karnieli E., Armoni M. Transcriptional regulation of the insulin-responsive glucose transporter GLUT4 gene: from physiology to pathology. American Journal of Physiology—Endocrinology and Metabolism. 2008;295(1):E38–E45. doi: 10.1152/ajpendo.90306.2008. [DOI] [PubMed] [Google Scholar]
- 60.Olson A. L. Regulation of GLUT4 and insulin-dependent glucose flux. ISRN Molecular Biology. 2012;2012:12. doi: 10.5402/2012/856987.856987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alam F., Islam M. A., Khalil M. I., Gan S. H. Metabolic control of type 2 diabetes by targeting the GLUT4 glucose transporter: intervention approaches. Current Pharmaceutical Design. 2016;22(20):3034–3049. doi: 10.2174/1381612822666160307145801. [DOI] [PubMed] [Google Scholar]
- 62.Alves-Wagner A. B., Sabino-Silva R., Campello R. S., Mori R. C., MacHado U. F. Decreased diabetes-induced glycemic impairment in WKY and SHR involves enhanced skeletal muscle Slc2a4/GLUT4 expression. Diabetology and Metabolic Syndrome. 2014;6, article 97 doi: 10.1186/1758-5996-6-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kahn B. B., Rosen A. S., Bak J. F., et al. Expression of GLUT1 and GLUT4 glucose transporters in skeletal muscle of humans with insulin-dependent diabetes mellitus: regulatory effects of metabolic factors. Journal of Clinical Endocrinology and Metabolism. 1992;74(5):1101–1109. doi: 10.1210/jc.74.5.1101. [DOI] [PubMed] [Google Scholar]
- 64.Ducluzeau P.-H., Perretti N., Laville M., et al. Regulation by insulin of gene expression in human skeletal muscle and adipose tissue. Diabetes. 2001;50(5):1134–1142. doi: 10.2337/diabetes.50.5.1134. [DOI] [PubMed] [Google Scholar]
- 65.Karlsson H. K. R., Zierath J. R., Kane S., Krook A., Lienhard G. E., Wallberg-Henriksson H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes. 2005;54(6):1692–1697. doi: 10.2337/diabetes.54.6.1692. [DOI] [PubMed] [Google Scholar]
- 66.Vind B. F., Pehmøller C., Treebak J. T., et al. Impaired insulin-induced site-specific phosphorylation of TBC1 domain family, member 4 (TBC1D4) in skeletal muscle of type 2 diabetes patients is restored by endurance exercise-training. Diabetologia. 2011;54(1):157–167. doi: 10.1007/s00125-010-1924-4. [DOI] [PubMed] [Google Scholar]
- 67.Richter E. A., Hargreaves M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiological Reviews. 2013;93(3):993–1017. doi: 10.1152/physrev.00038.2012. [DOI] [PubMed] [Google Scholar]
- 68.Dela F., Ploug T., Handberg A., et al. Physical training increases muscle GLUT4 protein and mRNA in patients with NIDDM. Diabetes. 1994;43(7):862–865. doi: 10.2337/diab.43.7.862. [DOI] [PubMed] [Google Scholar]
- 69.Holten M. K., Zacho M., Gaster M., Juel C., Wojtaszewski J. F. P., Dela F. Strength training increases insulin-mediated glucose uptake, GLUT4 content, and insulin signaling in skeletal muscle in patients with type 2 diabetes. Diabetes. 2004;53(2):294–305. doi: 10.2337/diabetes.53.2.294. [DOI] [PubMed] [Google Scholar]
- 70.Al-Khalili L., Forsgren M., Kannisto K., Zierath J. R., Lönnqvist F., Krook A. Enhanced insulin-stimulated glycogen synthesis in response to insulin, metformin or rosiglitazone is associated with increased mRNA expression of GLUT4 and peroxisomal proliferator activator receptor gamma co-activator 1. Diabetologia. 2005;48(6):1173–1179. doi: 10.1007/s00125-005-1741-3. [DOI] [PubMed] [Google Scholar]
- 71.Burgess T. A., Robich M. P., Chu L. M., Bianchi C., Sellke F. W. Improving glucose metabolism with resveratrol in a swine model of metabolic syndrome through alteration of signaling pathways in the liver and skeletal muscle. Archives of Surgery. 2011;146(5):556–564. doi: 10.1001/archsurg.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anhê G. F., Okamoto M. M., Kinote A., et al. Quercetin decreases inflammatory response and increases insulin action in skeletal muscle of ob/ob mice and in L6 myotubes. European Journal of Pharmacology. 2012;689(1–3):285–293. doi: 10.1016/j.ejphar.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 73.Coleman S. K., Rebalka I. A., D'Souza D. M., et al. Skeletal muscle as a therapeutic target for delaying type 1 diabetic complications. World Journal of Diabetes. 2015;6:1323–1336. doi: 10.4239/wjd.v6.i17.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee R. C., Feinbaum R. L., Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 75.Wightman B., Ha I., Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75(5):855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 76.Pasquinelli A. E., Reinhart B. J., Slack F., et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408(6808):86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- 77.Krol J., Loedige I., Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nature Reviews Genetics. 2010;11(9):597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 78.Bartel D. P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ørom U. A., Nielsen F. C., Lund A. H. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Molecular Cell. 2008;30(4):460–471. doi: 10.1016/j.molcel.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 80.Fernández-Hernando C., Ramírez C. M., Goedeke L., Suárez Y. MicroRNAs in metabolic disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(2):178–185. doi: 10.1161/ATVBAHA.112.300144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Friedman R. C., Farh K. K.-H., Burge C. B., Bartel D. P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Research. 2009;19(1):92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rottiers V., Näär A. M. MicroRNAs in metabolism and metabolic disorders. Nature Reviews Molecular Cell Biology. 2012;13(5):239–250. doi: 10.1038/nrm3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.van Rooij E., Kauppinen S. Development of microRNA therapeutics is coming of age. EMBO Molecular Medicine. 2014;6(7):851–864. doi: 10.15252/emmm.201100899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang Z., Chen H., Si H., et al. Serum miR-23a, a potential biomarker for diagnosis of pre-diabetes and type 2 diabetes. Acta Diabetologica. 2014;51(5):823–831. doi: 10.1007/s00592-014-0617-8. [DOI] [PubMed] [Google Scholar]
- 85.Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., Tuschl T. Identification of tissue-specific microRNAs from mouse. Current Biology. 2002;12(9):735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 86.Lee E. J., Baek M., Gusev Y., Brackett D. J., Nuovo G. J., Schmittgen T. D. Systematic evaluation of microRNA processing patterns in tissues, cell lines, and tumors. RNA. 2008;14(1):35–42. doi: 10.1261/rna.804508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McCarthy J. J., Esser K. A., Peterson C. A., Dupont-Versteegden E. E. Evidence of MyomiR network regulation of β-myosin heavy chain gene expression during skeletal muscle atrophy. Physiological Genomics. 2009;39(3):219–226. doi: 10.1152/physiolgenomics.00042.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Rooij E., Quiat D., Johnson B. A., et al. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Developmental Cell. 2009;17(5):662–673. doi: 10.1016/j.devcel.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McCarthy J. J. MicroRNA-206: the skeletal muscle-specific myomiR. Biochimica et Biophysica Acta (BBA)—Gene Regulatory Mechanisms. 2008;1779(11):682–691. doi: 10.1016/j.bbagrm.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Williams A. H., Liu N., van Rooij E., Olson E. N. MicroRNA control of muscle development and disease. Current Opinion in Cell Biology. 2009;21(3):461–469. doi: 10.1016/j.ceb.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ge Y., Chen J. MicroRNAs in skeletal myogenesis. Cell Cycle. 2011;10(3):441–448. doi: 10.4161/cc.10.3.14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang X. H. MicroRNA in myogenesis and muscle atrophy. Current Opinion in Clinical Nutrition and Metabolic Care. 2013;16(3):258–266. doi: 10.1097/MCO.0b013e32835f81b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zacharewicz E., Lamon S., Russell A. P. MicroRNAs in skeletal muscle and their regulation with exercise, ageing, and disease. Frontiers in Physiology. 2013;4, article 266:11. doi: 10.3389/fphys.2013.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Güller I., Russell A. P. MicroRNAs in skeletal muscle: their role and regulation in development, disease and function. Journal of Physiology. 2010;588(21):4075–4087. doi: 10.1113/jphysiol.2010.194175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kantharidis P., Wang B., Carew R. M., Lan H. Y. Diabetes complications: the microRNA perspective. Diabetes. 2011;60(7):1832–1837. doi: 10.2337/db11-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park S.-Y., Jeong H.-J., Yang W.-M., Lee W. Implications of microRNAs in the pathogenesis of diabetes. Archives of Pharmacal Research. 2013;36(2):154–166. doi: 10.1007/s12272-013-0017-6. [DOI] [PubMed] [Google Scholar]
- 97.Herrera B. M., Lockstone H. E., Taylor J. M., et al. Global microRNA expression profiles in insulin target tissues in a spontaneous rat model of type 2 diabetes. Diabetologia. 2010;53(6):1099–1109. doi: 10.1007/s00125-010-1667-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gallagher I. J., Scheele C., Keller P., et al. Integration of microRNA changes in vivo identifies novel molecular features of muscle insulin resistance in type 2 diabetes. Genome Medicine. 2010;2, article 9 doi: 10.1186/gm130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang B., Qin W., Zhao B., et al. MicroRNA expression profiling in diabetic GK rat model. Acta Biochimica et Biophysica Sinica. 2009;41(6):472–477. doi: 10.1093/abbs/gmp035. [DOI] [PubMed] [Google Scholar]
- 100.Zhu H., Ng S.-C., Segr A. V., et al. The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147(1):81–94. doi: 10.1016/j.cell.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bork-Jensen J., Scheele C., Christophersen D. V., et al. Glucose tolerance is associated with differential expression of microRNAs in skeletal muscle: results from studies of twins with and without type 2 diabetes. Diabetologia. 2015;58(2):363–373. doi: 10.1007/s00125-014-3434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Honardoost M., Arefian E., Soleimani M., Soudi S., Sarookhani M. R. Development of insulin resistance through induction of miRNA-135 in C2C12 cells. Cell Journal. 2016;18(3):353–361. doi: 10.22074/cellj.2016.4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang W.-M., Jeong H.-J., Park S.-Y., Lee W. Induction of miR-29a by saturated fatty acids impairs insulin signaling and glucose uptake through translational repression of IRS-1 in myocytes. FEBS Letters. 2014;588(13):2170–2176. doi: 10.1016/j.febslet.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 104.Agarwal P., Srivastava R., Srivastava A. K., Ali S., Datta M. MiR-135a targets IRS2 and regulates insulin signaling and glucose uptake in the diabetic gastrocnemius skeletal muscle. Biochimica et Biophysica Acta (BBA)—Molecular Basis of Disease. 2013;1832(8):1294–1303. doi: 10.1016/j.bbadis.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 105.Karolina D. S., Armugam A., Tavintharan S., et al. MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS ONE. 2011;6(8) doi: 10.1371/journal.pone.0022839.e22839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Uribe J. H., Vitger A. D., Ritz C., Fredholm M., Bjørnvad C. R., Cirera S. Physical training and weight loss in dogs lead to transcriptional changes in genes involved in the glucose-transport pathway in muscle and adipose tissues. Veterinary Journal. 2016;208:22–27. doi: 10.1016/j.tvjl.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 107.He A., Zhu L., Gupta N., Chang Y., Fang F. Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3T3-L1 adipocytes. Molecular Endocrinology. 2007;21(11):2785–2794. doi: 10.1210/me.2007-0167. [DOI] [PubMed] [Google Scholar]
- 108.Lee D. E., Brown J. L., Rosa M. E., et al. microRNA-16 is downregulated during insulin resistance and controls skeletal muscle protein accretion. Journal of Cellular Biochemistry. 2016;117(8):1775–1787. doi: 10.1002/jcb.25476. [DOI] [PubMed] [Google Scholar]
- 109.Latouche C., Natoli A., Reddy-Luthmoodoo M., Heywood S. E., Armitage J. A., Kingwell B. A. MicroRNA-194 modulates glucose metabolism and its skeletal muscle expression is reduced in diabetes. PLoS ONE. 2016;11(5) doi: 10.1371/journal.pone.0155108.e0155108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee H., Jee Y., Hong K., Hwang G. S., Chun K.-H. MicroRNA-494, upregulated by tumor necrosis factor-α, desensitizes insulin effect in C2C12 muscle cells. PLoS ONE. 2013;8(12) doi: 10.1371/journal.pone.0083471.e83471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen Y.-H., Heneidi S., Lee J.-M., et al. Mirna-93 inhibits glut4 and is overexpressed in adipose tissue of polycystic ovary syndrome patients and women with insulin resistance. Diabetes. 2013;62(7):2278–2286. doi: 10.2337/db12-0963/-/DC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chuang T.-Y., Wu H.-L., Chen C.-C., et al. MicroRNA-223 expression is upregulated in insulin resistant human adipose tissue. Journal of Diabetes Research. 2015;2015:8. doi: 10.1155/2015/943659.943659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu H., Buchan R. J., Cook S. A. MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovascular Research. 2010;86(3):410–420. doi: 10.1093/cvr/cvq010. [DOI] [PubMed] [Google Scholar]
- 114.Zhou T., Meng X., Che H., et al. Regulation of insulin resistance by multiple MiRNAs via targeting the GLUT4 signalling pathway. Cellular Physiology and Biochemistry. 2016;38(5):2063–2078. doi: 10.1159/000445565. [DOI] [PubMed] [Google Scholar]
- 115.Shi Z., Zhao C., Guo X., et al. Differential expression of micrornas in omental adipose tissue from gestational diabetes mellitus subjects reveals miR-222 as a regulator of ERα expression in estrogen-induced insulin resistance. Endocrinology. 2014;155(5):1982–1990. doi: 10.1210/en.2013-2046. [DOI] [PubMed] [Google Scholar]
- 116.Horie T., Ono K., Nishi H., et al. MicroRNA-133 regulates the expression of GLUT4 by targeting KLF15 and is involved in metabolic control in cardiac myocytes. Biochemical and Biophysical Research Communications. 2009;389(2):315–320. doi: 10.1016/j.bbrc.2009.08.136. [DOI] [PubMed] [Google Scholar]
- 117.Ling H.-Y., Hu B., Hu X.-B., et al. MiRNA-21 reverses high glucose and high insulin induced insulin resistance in 3T3-L1 adipocytes through targeting phosphatase and tensin homologue. Experimental and Clinical Endocrinology and Diabetes. 2012;120(9):553–559. doi: 10.1055/s-0032-1311644. [DOI] [PubMed] [Google Scholar]
- 118.Zhou Y., Gu P., Shi W., et al. MicroRNA-29a induces insulin resistance by targeting PPARδ in skeletal muscle cells. International Journal of Molecular Medicine. 2016;37(4):931–938. doi: 10.3892/ijmm.2016.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Campello R. S., Alves-Wagner A. B., Lucas T. F., et al. Estrogen receptor 1 agonist PPT stimulates Slc2a4 gene expression and improves insulin-induced glucose uptake in adipocytes. Current Topics in Medicinal Chemistry. 2012;12(19):2059–2069. doi: 10.2174/156802612804910197. [DOI] [PubMed] [Google Scholar]
- 120.Michael L. F., Wu Z., Cheatham R. B., et al. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(7):3820–3825. doi: 10.1073/pnas.061035098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Guedes E. C., França G. S., Lino C. A., et al. MicroRNA expression signature is altered in the cardiac remodeling induced by high fat diets. Journal of Cellular Physiology. 2016;231(8):1771–1783. doi: 10.1002/jcp.25280. [DOI] [PubMed] [Google Scholar]