

Abstract
IL-1β is critical for TH17 cell survival, expansion, and effector function in vivo during autoimmune responses, including EAE. However, the spatiotemporal role and cellular source of IL-1β during EAE pathogenesis is poorly defined. In the present study, we uncovered a novel T cell-intrinsic inflammasome that drives IL-1β production during TH17-mediated EAE pathogenesis. TCR activation induced pro-IL-1β expression, while ATP stimulation triggered T cell production of IL-1β via ASC-NLRP3-dependent caspase-8 activation. IL-1R was detected on TH17 but not TH1 cells, and ATP-treated TH17 cells showed enhanced survival compared to ATP-treated TH1 cells, suggesting autocrine action of TH17-derived IL-1β. Together, these data reveal a critical role for IL-1β produced by a TH17 cell-intrinsic ASC-NLRP3-Caspase-8 inflammasome during CNS inflammation.
INTRODUCTION
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system (CNS) that affects an estimated 350,000 people in the United States, and some 2.5 million worldwide1. Experimental autoimmune encephalomyelitis (EAE) is the most commonly utilized animal model of MS. Studies using the EAE model have helped to define the sequence of immunopathogenic events in the development of autoimmune CNS-directed inflammatory disease2. During the initiation stage of EAE, CNS antigen-reactive T cells undergo activation and clonal expansion in the secondary lymphoid organs, while antigen-presenting cells (APCs) simultaneously produce cytokines that regulate the differentiation of effector CD4+ T cells, skewing these cells to classical TH1 (producing IFN-γ) and TH17 (producing IL-17, IL-21, GM-CSF and TNF) T cell lineages. Importantly, recent data demonstrate that both TH1 and TH17 cells are able to independently induce EAE, possibly through different mechanisms3–6.
TH17 cells are generated as a discrete lineage when the peripheral priming microenvironment contains TGFβ and IL-6, and appear to acquire encephalitogenic potential following re-activation and expansion in the presence of IL-1β and IL-237–9. We, along with others, have reported that expression of the IL-1 receptor (IL-1R) is highly induced during TH17 cell differentiation10,11. Mice deficient in IL-1R have been shown to display a significant reduction in EAE disease severity, while mice deficient in IL-1Ra, the endogenous soluble IL-1R antagonist, were shown to have worse disease than wild-type controls10,12,13. IL-1β stimulation of TH17 cells leads to strong and prolonged activation of the mammalian target of rapamycin (mTOR) pathway, which plays a critical role in cell proliferation and survival, and is required for TH17-dependent EAE pathogenesis14,15.
The NLRP3 inflammasome consists of NLRP3 linked via a homotypic pyrin domain interaction to the inflammasome adaptor molecule apoptosis-associated speck-like protein containing a C-terminal caspase-activation and recruitment (CARD) domain (ASC). ASC interacts with pro-caspase-1 via a CARD domain, resulting in caspase 1 activation and maturation and production of IL-1β and IL-18. Several independent studies have recently reported a critical role for the NLRP3 inflammasome in EAE pathogenesis16–18. However, the spatiotemporal role and cellular source of IL-1β during EAE pathogenesis is poorly defined. Although many previous studies have reported that ASC-dependent inflammasome signaling mediates T cell function during both host defense and autoimmune processes, it has remained unclear whether ASC has any T cell-intrinsic role.
Here, we report that the inflammasome adaptor molecule ASC plays a critical T cell-intrinsic role in the pathogenesis of TH17-mediated EAE. T cell-intrinsic ASC is required for the effector stage of EAE, and ASC deficiency in T cells impaired TH17- but not TH1-mediated EAE. Mechanistically, TCR activation induced pro-IL-1β expression and nuclear-to-cytosolic translocation of ASC; polarized TH17 cells expressed IL-1R, and produced mature IL-1β in response to ATP via ASC/NLRP3-dependent caspase-8 activation. ATP-treated TH17 cells showed enhanced survival compared to ATP-treated TH1 cells, which was abrogated by IL-1Ra, suggesting an autocrine action of TH17-derived IL-1β. Together, these data suggest a critical role for IL-1β produced by a novel TH17 cell-intrinsic ASC-NLRP3-Caspase-8 inflammasome during CNS inflammation.
RESULTS
T cell-specific ASC deficiency delayed and greatly attenuated EAE
To investigate whether Asc has any T cell-intrinsic role, we bred a mouse strain in which all three exons of the Pycard gene (that encodes Asc) are flanked by lox(p) sites (Ascf/f mice) onto the Lck-cre transgenic mouse strain, which expresses Cre under the control of the Lck proximal promoter, generating Ascf/+Lck-Cre mice (Fig. 1a and Supplementary. Fig. 1). We generated mice with T cell-specific deletion of Asc by further crossbreeding to obtain Ascf/−Lck-Cre and littermate control Ascf/+Lck-Cre mice (Supplementary. Fig. 2). We tested the impact of T cell-specific Asc deletion on neuroinflammation and demyelination by subjecting Ascf/−Lck-Cre and littermate control Ascf/+Lck-Cre mice to immunization with neuroantigen, MOG35–55 peptide. Mice with T cell-specific ASC deficiency had attenuated disease severity compared to controls (Fig. 1b). Asc+/+ and Asc+/− mice exhibited similar EAE phenotype upon immunization (data not shown), indicating haploinsufficiency of Asc has no impact on EAE phenotype. While Ascf/+Lck-Cre developed severe EAE, Ascf/fLck-Cre mice were protected, mirroring the data shown in Fig. 1b (data not shown). Inflammatory mononuclear cell infiltration in the brain, including CD4+ T cells, B cells, neutrophils and macrophages, was similarly reduced in mice with T cell-specific Asc ablation (Fig. 1c–d) and the expression of inflammatory cytokine and chemokine expression in the spinal cord was also significantly decreased (Fig. 1e). Histopathological analysis showed reduced infiltrating immune cell accumulation and resultant demyelination in spinal cord of Ascf/−Lck-Cre mice compared to controls (Fig. 1f). Together, these data indicate that deletion of Asc from T cells greatly protects mice from the pathogenesis of EAE, with a marked attenuation of disease severity.
Figure 1.
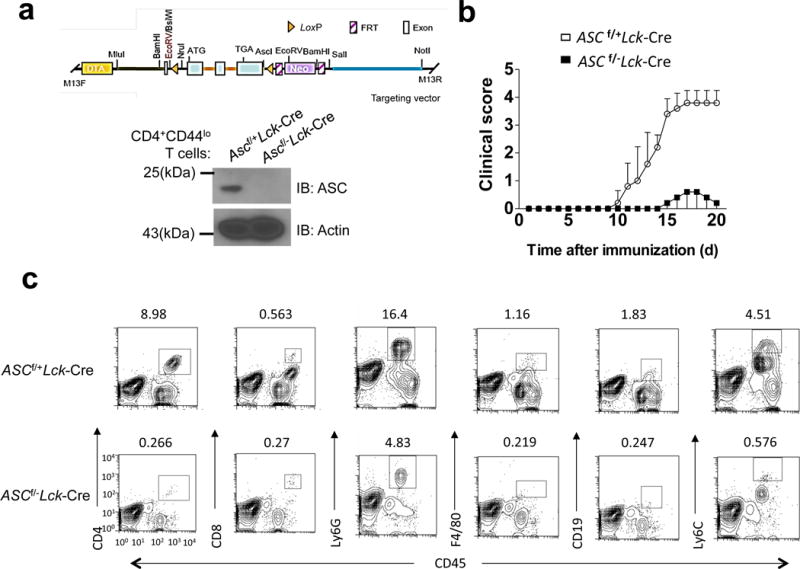
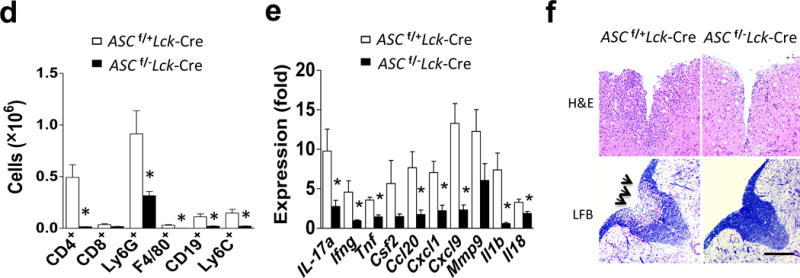
Genetic deletion of the inflammasome adaptor ASC in T cells protects from EAE. (a) Targeting vector design for generation of a novel mouse strain with all three Pycard exons flanked by lox(p) sites (upper panel), and Western blot analysis of ASC expression in FACS-sorted naïve CD4+ T cells from Ascf/+Lck-Cre and Ascf/−Lck-Cre mice (lower panel). (b) Mean clinical score of EAE in Ascf/+Lck-Cre and Ascf/−Lck-Cre mice (n=5 mice per group) induced by active immunization with MOG35–55. (c–d) Immune cell infiltration in the brains of MOG35–55-immunized Ascf/+Lck-Cre and Ascf/−Lck-Cre mice (n = 5, 7 days after disease onset) was analyzed by flow cytometry with the antibodies as indicated. (d) Absolute numbers of CNS-infiltrating cells were determined at the peak of disease. Brains and spinal cords were harvested together and mononuclear infiltrating cells were stained with the indicated antibodies, followed by flow cytometric analysis. (e) Real-time PCR analysis of inflammatory gene expression as indicated in the spinal cords of Ascf/+Lck-Cre and Ascf/+Lck-Cre mice (n=5) at the peak of disease. (f) Hematoxylin and eosin (H&E) staining (upper panels) and Luxol fast blue staining (lower panels) of lumbar spinal cords from Ascf/+Lck-Cre and Ascf/−Lck-Cre mice harvested at the peak of disease. Scale bar, 200μm. *P < 0.05 (Unpaired t test, d and e). *P < 0.05 (Two-way ANOVA,b). Data are representative of three independent experiments. Error bars represent s.d (b). Error bars represent s.e.m (d,e). 5 mice per group in each experiment.
T cell-specific ASC deficiency had no impact on T cell priming
Given the phenotype observed in Ascf/−Lck-Cre mice after immunization with MOG35–55 peptide, we hypothesized that ASC in CD4+ T cells might be important for the priming of MOG35–55 -reactive T effector populations in the secondary lymphoid organs. However, intracellular staining of T cells obtained from lymph nodes of MOG35–55-immunized mice cultured in TH17- and TH1-polarizing conditions in the presence of MOG35–55 indicated that T cell-specific ASC dificiency had no impact on the frequency and total numbers of IL-17A+ and IFN-γ+ CD4+ T cells (Fig. 2a). In addition, supernatants from cultures of MOG35–55 -primed lymph node cells from Ascf/−Lck-Cre− mice showed similar levels of IL-17A and IFN-γ as compared to controls (Fig. 2b). Furthermore, Ascf/−Lck-Cre and Ascf/+Lck-Cre sorted naïve CD4+ T cells (CD4+ CD44lo) had a similar capacity to differentiate to TH1, TH2, TH17 and Treg populations in an antigen-independent manner in vitro (Fig. 2c–g). Together, these findings suggested that T cell specific ASC deficiency does not impact T cell development, ex vivo T cell differentiation, or primary MOG35–55 -specific T cell priming in vivo.
Figure 2.
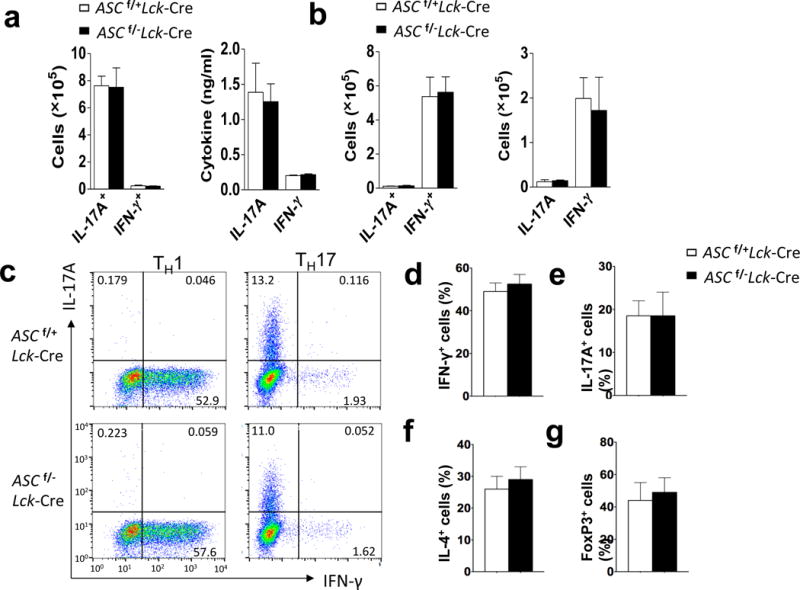
MOG-reactive T effector cell priming in the secondary lymphoid organs is unaffected by ASC deficiency in T cells. (a) Absolute numbers of CD4+ IL-17A+ and CD4+ IFN-γ+ cells (left panel) and concentrations of IL-17A and IFN-γ in the supernatant of cells (right panel) from the draining lymph nodes of mice (n=5 mice per group) 10 days after immunization with MOG35–55, followed by ex vivo re-stimulation in the presence of MOG35–55 and IL-23. (b) Absolute numbers of CD4+IL-17A+ and CD4+IFN-γ+ cells (left panel) and concentrations of IL-17A and IFN-γ in the supernatant of cells (right panel) from the draining lymph nodes of mice (n=5) 10 days after immunization with MOG35–55,, followed by ex vivo restimulation in the presence of MOG35–55 and IL-12. (c) FACS-sorted naïve (CD4+CD44lo) CD4+ T cells were cultured on an anti-CD3/CD28 coated plate in TH17- or TH1-polarizing condition, followed by intracellularly staining for IL-17A and IFN-γ and analyzed by flow cytometry. (d–g) Mean percentage of cells positive for the indicated cytokines/protein following intracellular staining with the indicated antibodies and flow cytometric analysis on day 3 of in vitro differentiation of TH1 (d), TH17 (e), TH2 (f), and Treg (g) cell populations. Data are representative of three independent experiments (a–g). n=5 mice per group in each experiment (a–b) and n=3 mice per group in each experiment (c–g).
T cell-intrinsic ASC is required for TH17-mediated EAE
Given that T cell-specific deficiency attenuated EAE pathogenesis but not T cell priming, we assessed the pathogenic role played by Asc-sufficient and Asc-deficient TH1 and TH17 cells by adoptive transfer into naïve recipients. We found that wild-type recipients receiving Ascf/−Lck-Cre cells re-stimulated under TH1-promoting conditions developed similar disease as those receiving TH1 cells from Ascf/+Lck-Cre mice (Fig. 3a). Analysis of the infiltrating mononuclear cell population in the brains of these mice showed no difference in the accumulation of CD4+ or CD8+ T cells, macrophages, neutrophils, or B-cells, consistent with the clinical scores (Fig. 3b). Wild-type mice receiving Ascf/−Lck-Cre cells re-stimulated under TH17-promoting conditions developed disease with reduced severity than those receiving TH17 cells from Ascf/+Lck-Cre mice (Fig. 3c). Surface staining of the brain mononuclear cell population revealed that the numbers of CD4+ T cells, macrophages, and neutrophils were also reduced in mice receiving Asc-deficient TH17 cells (Fig. 3d). Histopathological analysis also reflected this reduced inflammatory cell infiltration, along with a reduction in demyelination in recipient mice receiving ASC-deficient TH17 cells (Fig. 3e). Taken together, these results suggest that T cell-intrinsic ASC is required for TH17- but not TH1-mediated EAE. We then sorted CD4+ TH17 cells by flow cytometry from MOG-immunized Ascf/−Lck-Cre and Ascf/+Lck-Cre control mice and adoptively transferred into irradiated wild-type recipients. The peripheral priming of MOG-reactive TH17 cells was equivalent in Ascf/−Lck-Cre and Ascf/+Lck-Cre control mice (Fig. 2a, b and Supplementary. Fig. 3a). Recipients (CD45.1 mice) of sorted Ascf/−Lck-Cre CD45.2-CD4+ TH17 cells showed much reduced EAE phenotype as compared to recipients of Ascf/+Lck-Cre CD45.2-CD4+ TH17 cells (Supplementary. Fig. 3), together suggesting that Asc in CD4+ TH17 cells is required for the effector stage of EAE development.
Figure 3.
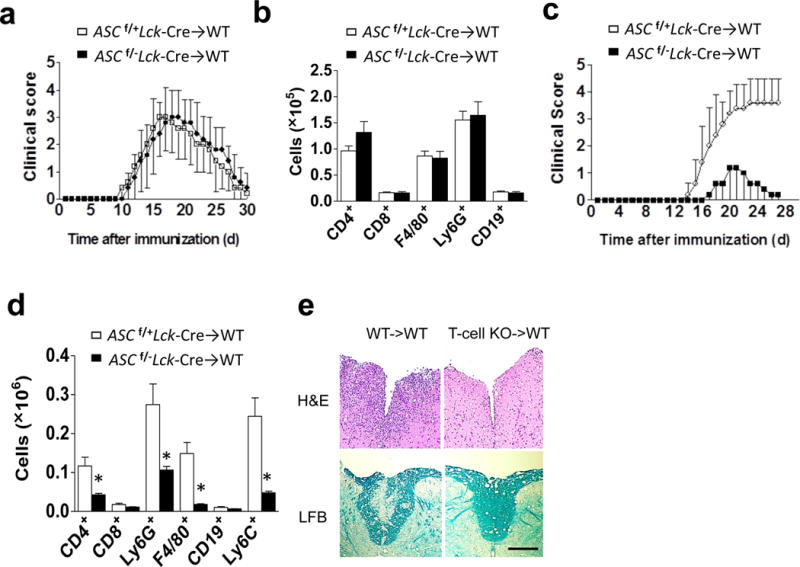
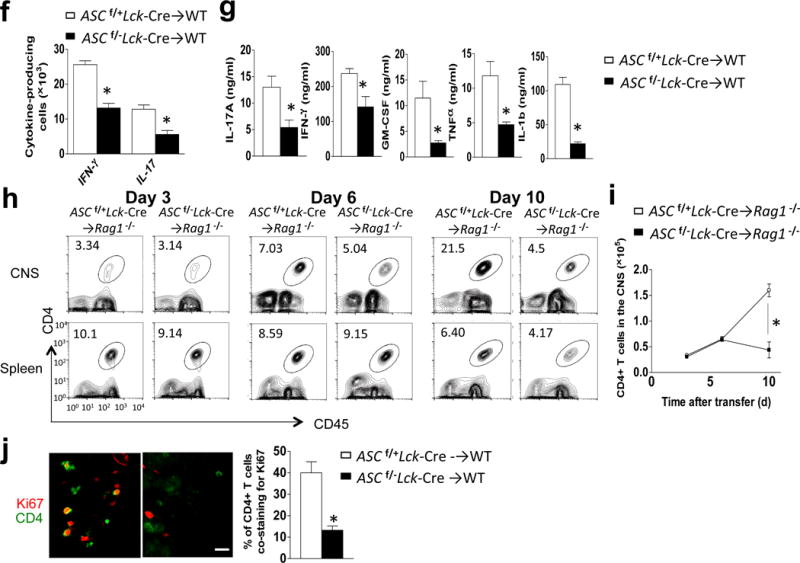
T cell specific ASC deficiency is required for TH17- but not TH1-mediated EAE. (a,c) Mean clinical score of EAE mice (n=5) induced by adoptive transfer of MOG-reactive TH1 (a) and TH17 (c) cells. (b, d) Brains and spinal cords TH1- (b) and TH17- (d) recipient mice were harvested at the peak of disease and stained with the antibodies, followed by flow cytometric analysis. (e) H&E staining (upper panels) and Luxol fast blue staining (lower panels) of lumbar spinal cords at peak of the disease in TH17 recipient mice. Scale bar, 200μm. (f) Flow cytometric analysis of CD4+ cells from the brains of wild-type mice at peak of the disease in TH17 recipient mice. (g) Thy1.2+ CD4+ T cells FACS-sorted from the brains of Thy1.1+ TH17 recipient mice (n=5), were cultured and analyzed by ELISA. (h) Flow cytometric analyses of CD4+ T cells from CNS or spleen from Rag−/− mice (n=5) that received MOG-reactive TH17 cells. (i) Quantification of total CNS-infiltrating CD4+ T cells from (h). (j) Ki-67/CD4 staining of spinal cords from adoptive transfer experiments from (h) (left panels), and percentage of CD4+ T cells co-staining with Ki-67 (right panel). Scale bar, 10μm. Error bars represent s.e.m. *P < 0.05 (Unpaired t test for b, d, f, g, l and j). Error bars represent s.d. *P < 0.05 (Two-way ANOVA for a and c). Data are representative of three independent experiments (a–j). n=5 mice per group in each experiment.
Intracellular staining of brain-infiltrating mononuclear cells revealed that the percentage and absolute number of both IL-17- and IFNγ- secreting cells were decreased in mice receiving TH17 cells from Ascf/−Lck-Cre mice compared to those from Ascf/+Lck-Cre mice (Fig. 3f). Previous studies reported that during the development of EAE, IFN-γ and other pro-inflammatory cytokines (GM-CSF and TNF) in the spinal cord were produced almost exclusively by cells that had produced IL-17 (ex- TH17 cells)19, suggesting that the reduction in IFN-γ-secreting cells in the CNS of mice receiving Ascf/−Lck-Cre TH17 cells might be due to reduced TH17 and/or ex- TH17 cells in the CNS. In support of this, Ascf/−Lck-Cre CD4+ T cells sorted from the brains of recipient mice were indeed defective in their secretion of key pro-inflammatory cytokines, including those reported to be produced by TH17 and ex-TH17 cells (Il17a, Ifng, Csf2 and Tnf) and, most interestingly, IL-1β (Fig. 3g). Taken together, these findings indicated that ASC-deficient TH17 cells are markedly deficient in their production of key pro-inflammatory cytokines in the CNS.
To exclude the possibility that T cell-intrinsic ASC deficiency impacts CD4+ TH17 migration to the CNS, we adoptively transferred MOG-primed TH17 cells from Ascf/−Lck-Cre and Ascf/+Lck-Cre mice into Rag1−/− recipient mice. We found that TH17 cells from mice with T cell-specific ASC deficiency egressed from the spleen and migrated to the CNS normally during the first wave of CNS infiltration (days 3 and 6 post-transfer) (Fig 3h–i). However, by day 10 after adoptive transfer, CD4+ T cell numbers in the CNS were reduced in mice receiving TH17 cells from Ascf/−Lck-Cre mice, suggesting that TH17 cell survival/expansion in the CNS might be defective in the absence of ASC. In support of this, Ki67 staining in the spinal cord revealed proliferating infiltrated CD4+ T cells after adoptive transfer of wild-type TH17 cells, which was markedly reduced in mice receiving ASC-deficient- TH17 cells (Fig. 3j).
TH17 cells actively secrete mature IL-1β
Given that ASC-deficient TH17 cells failed to expand and secrete pro-inflammatory cytokines – including IL-1β - in the CNS, we explored whether ASC might serve as an adaptor for inflammasome activation in TH17 cells. During in vitro TH17 cell differentiation, pro-IL-1β expression was strongly induced (Fig. 4a–b). ATP is released by cells during stress and cell death, and is abundant in the extracellular space in the CNS, where it also acts as a key excitatory neurotransmitter20–22. We thus speculated that the observed pro-IL-1β in TH17 cells might undergo processing in response to stimulation with extracellular ATP. Indeed, in vitro polarized TH17 cells were capable of processing and secreting the active form of IL-1β upon ATP stimulation (Fig. 4c–g), although the amount of mature IL-1β produced by TH17 cells was less compared to that produced by macrophages (Supplementary. Fig. 4). However, in ASC- and NLRP3-deficient TH17 cells, IL-1β processing was completely abolished (Fig. 4c–d, g). Caspase-1 is the prototypical interleukin-1β converting enzyme in myeloid-lineage cells, and the regulation and function of the canonical caspase-1 inflammasome has been the subject of intense research. However, TH17 cells deficient in both caspase-1 (Casp1–/–) and caspase-11 (Casp4–/–; called ‘Casp11–/–’ here) showed only a moderate reduction in IL-1β production as compared to controls (Fig. 4e,g). Caspase-8 was recently identified as a non-canonical inflammatory caspase in myeloid-lineage cells, and has been shown to be capable of processing pro-IL-1β23–25. This caspase-8-dependent activity was also shown to be important for TH17 cell-mediated host defense24. Additionally, caspase-8 has been revealed to play a critical non-apoptotic role in T cells26–31. We thus speculated that in TH17 cells, caspase-8 might participate in an ASC-dependent non-canonical inflammasome capable of processing pro-IL-1β. In response to ATP stimulation, IL-1β production was significantly reduced in TH17 cells from Caspase-8−/−Ripk3−/− mice compared to those from Caspase-8+/−Ripk3−/− mice, implying a role for caspase-8 in TH17 cells for the ASC-dependent processing of pro-IL-1β (Fig. 4f,g). In support of this, peptide inhibitor of caspase-8 but not caspase-1 specifically blocked IL-1β production in TH17 cells (Fig. 4h). Analysis of caspase-8 activation demonstrated that extracellular ATP indeed activates caspase-8 in a dose-dependent manner, which was substantially reduced in ASC- and NLRP3-deficient TH17 cells (Fig. 4i). Given the finding that ATP stimulation of TH17 cells induced processing of pro-IL-1β in an ASC- and caspase-8-dependent manner, we sought to further define the responsible molecular complex. Stimulation of TH17 cells with ATP induced the recruitment of both caspase-8 and NLRP3 - but not caspase-1 - to ASC, suggesting that an ASC-NLRP3-caspase-8 complex forms in response to ATP stimulation in TH17 cells (Fig. 4j).
Figure 4.
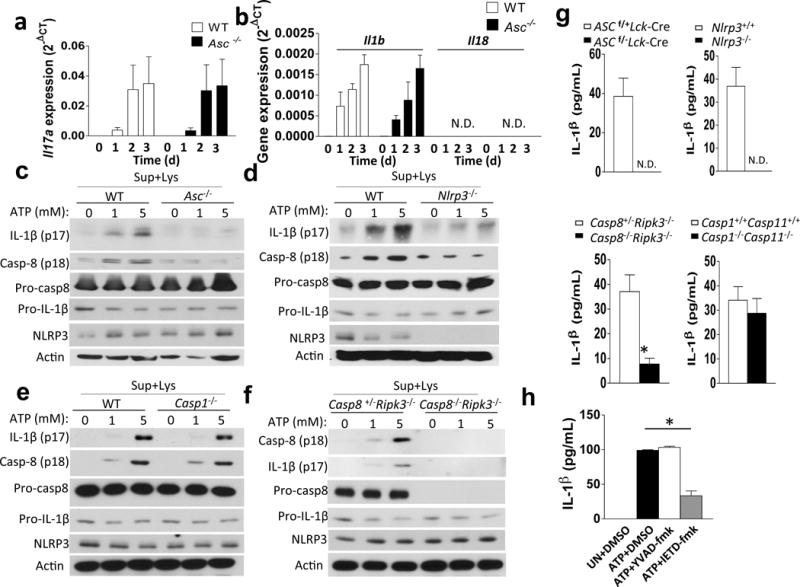
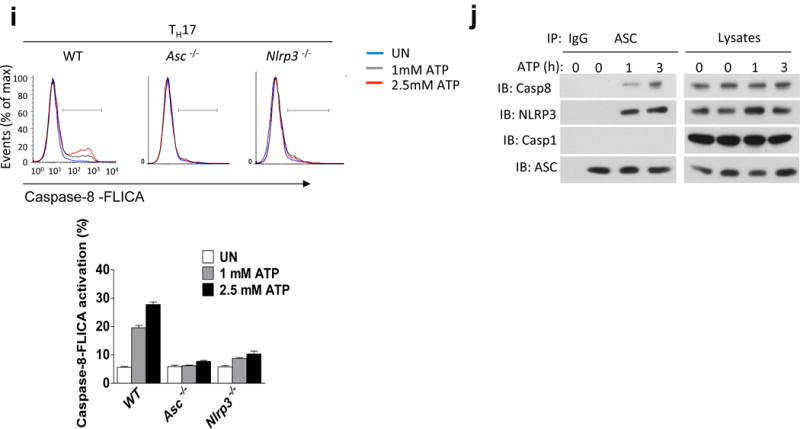
TH17 cells process IL-1β in response to stimulation with extracellular ATP in an ASC/NLRP3/Caspase-8-dependent manner. (a–j) FACS-sorted (CD4+CD44lo) naïve CD4+ T cells were subjected to TH17 polarization. (a, b) TH17 cells were analyzed by real-time PCR analysis of the indicated genes. (c–f) TH17 cells were stimulated with ATP (8 h), and cells were lysed, followed by Western analysis with the indicated antibodies. (g) TH17 cells were stimulated with ATP (8 h), and IL-1β ELISA was performed on cell-free supernatants. (h) TH17 cells were treated with the indicated inhibitors and ATP (8 h), followed by IL-1β ELISA. (i) TH17 cells were stimulated with ATP for 8 h; caspase-8 FLICA (IETD) reagent was added directly to the media for the last hour of incubation. Cells were then harvested and analyzed by flow cytometry (upper). Qutification of Caspase 8 FLICA activation (lower). (j) TH17 cells were stimulated with 1 mM ATP for 0, 1, or 3 h. Cells were then harvested and lysed, followed by immunoprecipitation with anti-ASC antibody and Western blot with indicated antibodies. n=5 mice per group in each experiment. n=2 mice per group in each experiment (i). Error bars represent s.e.m. *P < 0.05 (Unpaired t test for a, b, g, h and j). Data are representative of three independent experiments (a–j).
TH17-derived IL-1β supports cell survival via an autocrine manner
We next sought to examine the mechanism by which TH17-polarizing conditions induce pro-IL-1β expression in T cells. Since all T helper cell populations also require TCR stimulation for their initial activation and differentiation from the naïve state, we first tested whether IL-1β expression was shared by these other T cell lineages. Interestingly, we found that Th0, TH1, and TH17 cells all showed up-regulation of Il1b and Nalp3 mRNA expression (Fig. 5a–b), a process that has been shown in myeloid cells to be strictly regulated, and which is required for optimal inflammasome activation32. Additionally, Il18 mRNA levels were undetectable in TH0 and TH17 cells, with very low expression in TH1 cells (Fig. 5a). ASC deficiency had no impact on expression of Il1b, Il18 and Nalp3 mRNA in TH0, TH1 and TH17 cells (Fig. 5a). ASC, which was located primarily in the nucleus of resting naïve CD4+ T cells, translocated to the perinuclear area in response to TCR stimulation, reflecting what we and others have observed in murine macrophages (Fig. 5c)33,34. Further stimulation of these cells with ATP resulted in the formation of large perinuclear ASC specks, which are a well-described marker of inflammasome formation (Fig. 5c). We further found that all three tested T helper lineages robustly activated caspase-8 and produced mature IL-1β following ATP treatment, which was abolished in the corresponding NLRP3- and ASC-deficient T cells (Fig. 5d,e).
Figure 5.
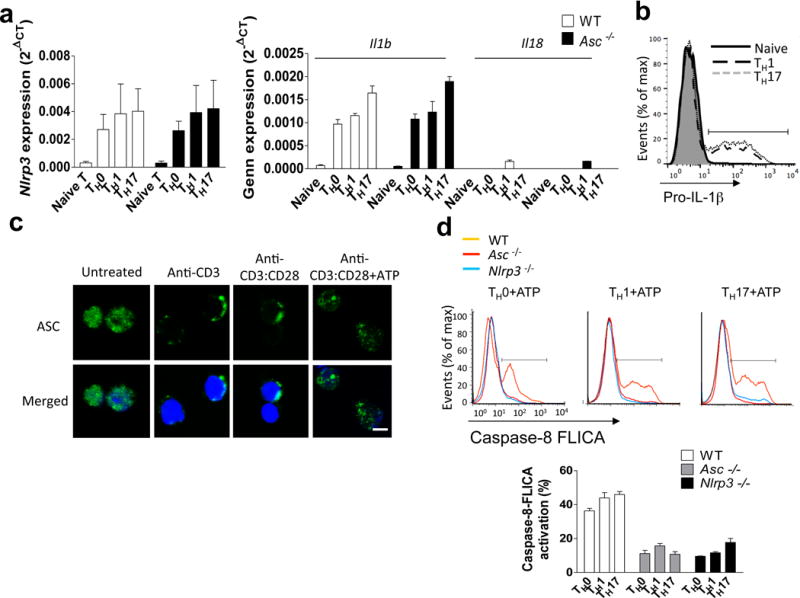
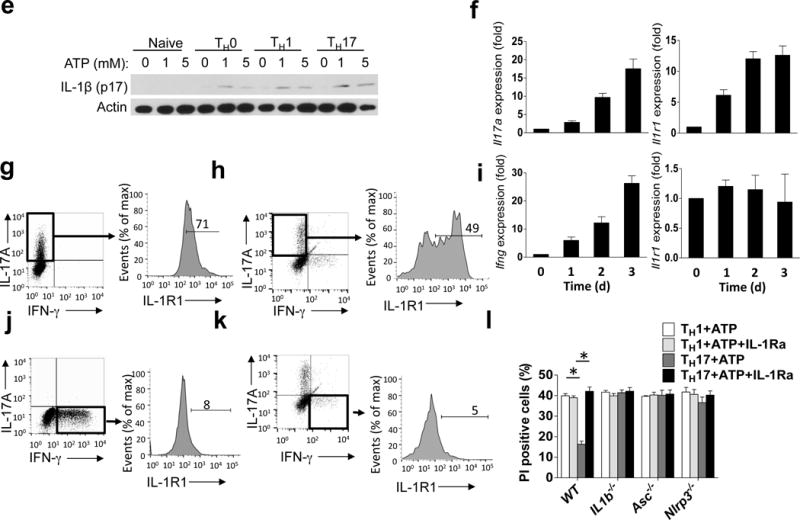
TH17 cells express IL-1R and pro-IL-1β. (a) mRNA expression for the indicated genes in naïve CD4+ T cells, TH0, TH1 and TH17 cells. (b) Naïve CD4+ T cells, TH1 and TH17 cells were stained for pro-IL-1β and analyzed by flow cytometry. (c) Naive CD4+ T cells were stimulated with the antibodies, followed by staining with anti-ASC and confocal microscopic analysis. Scale bar, 5μm. (d) TH0, TH1 and TH17 cells were treated with 5 mM ATP for 8 h; caspase-8 FLICA (IETD) was added, followed by flow cytometry. (e) Naive CD4+ T cells, TH0, TH1 and TH17 cells were treated with ATP for 8 h. Cell lysates were immunoblotted with the antibodies. (f, i) TH1 and TH17 cells were analyzed by real-time PCR on days 0–3 of differentiation. (g, j) TH17 (g) and TH1 (j) cells were stained with the indicated antibodies and analyzed by flow cytometry. (h, k) Cells of draining lymph nodes from MOG-immunized mice were analyzed by flow cytometric analysis with the indicated antibodies. (l) TH1 and TH17 cells were pre-treated for 15 minutes with IL-1ra (1 μg/ml), then stimulated with 5 mM ATP for 8 h. Cells were stained with propidium iodide and analyzed by flow cytometry. Error bars represent s.e.m. *P < 0.05 (Unpaired t test for a, d, f, i, l). Data are representative of three independent experiments (a–i). n=5 mice per group in each experiment (b, g–h, j–l). n=2 mice per group in each experiment (a, d, f and i).
Given these data indicating that inflammasome-mediated IL-1β production is not restricted to a particular CD4+ T cell lineage, which was in contrast to our observation that T cell-intrinsic ASC is specifically required for TH17-mediated neuroinflammation, we sought to examine the functional TH17 cell-specific role of the ASC-dependent inflammasome. We and others previously reported that IL-1R signaling mediates TH17 cell maintenance through promotion of cell survival and proliferation14,15,35. Il1r1 gene expression was specifically induced during in vitro TH17- but not TH1- cell differentiation (Fig. 5f–g; i–j). Upon MOG immunization, IL-1R expression was detected in IL-17+ CD4+ T cells, but not in IFN-γ+ CD4+ T cells (Fig. 5h; 5k). We therefore hypothesized that IL-1β produced by TH17 cells might act on IL-1R+ TH17 cells in an autocrine fashion. To test this hypothesis, we examined TH1 and TH17 cell survival following 8 hours of ATP stimulation. While both TH1 and TH17 cells exhibited caspase-8 activation and IL-1β production, ATP-treated TH17 cells showed significantly increased cell survival compared to ATP-treated TH1 cells, which was abrogated by deficiency of ASC, NLRP3, or IL-1β, or by addition of IL-1 receptor antagonist (IL-1Ra) (Fig. 5l). These results suggest that TH17-derived IL-1β may act in an autocrine fashion to promote survival and proliferation of TH17 cells with high IL-1R expression (Supplementary. Fig. 5).
CD4+ T cell-derived IL-1β is critical for EAE pathogenesis
To further examine the impact of TH17 cell-derived IL-1β during EAE, we transferred wild-type or Il1b−/−Il18−/− CD4+ T cells into Rag1−/− mice, followed by MOG35–55 immunization of the recipients. We found that mice reconstituted with Il1b−/−Il18−/− CD4+ T cells were significantly protected from EAE development compared to controls (Fig. 6a). The mRNA expression of Il17A and Ifng, as well as their target genes, was significantly decreased in the spinal cords of Il1b−/−Il18−/− CD4+ T cell recipients (Fig. 6b). Inflammatory mononuclear cell infiltration in the CNS, including CD4+ T cells, was also dramatically reduced in Il1b−/−Il18−/− CD4+ T cell recipient mice (Fig. 6C). Histopathological analysis of lumbar spinal cords from Il1b−/−Il18−/− CD4+ T cell recipients showed reduced infiltrating immune cell accumulation and marked protection from demyelinating lesion development (Fig. 6d). Since pro-IL-18 was only slightly induced in TH1 and not detected in TH17 cells (Fig. 5a), these results suggested that CD4+ T cell-derived IL-1β is probably critical for EAE pathogenesis. We indeed found that mice reconstituted with Il1b−/− CD4+ T cells were almost completely protected from EAE development (Fig. 6e–g), mirroring the results in Il1b−/−Il18−/− CD4+ T cell recipients. Taken together, these results confirm that CD4+ T cell-derived IL-1β is critical for EAE pathogenesis. As an alternative approach to Rag1−/− recipient mice, we generated mixed chimeras using Cd3e−/− bone marrow together with either wild-type or Il1b−/− bone marrow to assess the function of T cell-derived IL-1β. Lethal irradiated WT mice were reconstituted with WT+Cd3e−/− bone marrow or Il1b−/−+Cd3e−/− bone marrow. Six weeks after reconstitution, mice were immunized with MOG35–55 peptide. The recipient mice received WT+Cd3e−/− BM developed EAE, whereas the recipient mice received Il1b−/−+Cd3e−/− BM were protected from EAE (Supplementary. Fig. 6). These results independently validated the critical role of T cell-derived IL-1β in EAE pathogenesis.
Figure 6.
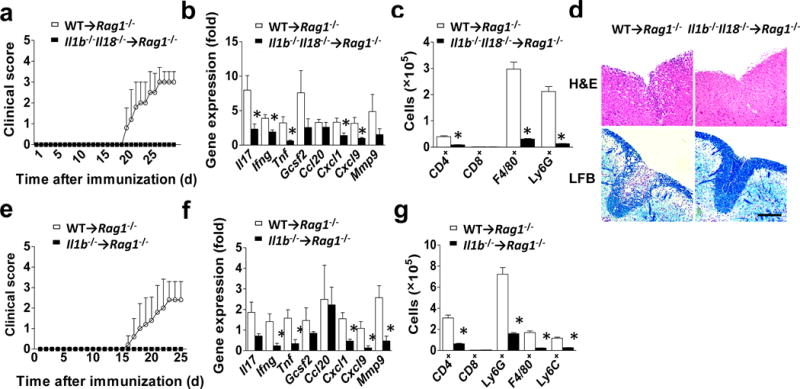
IL-1β produced by CD4+ T cells in vivo. (a–e) Rag1−/− mice (n=5 mice per group) were reconstituted with 2×106 sorted CD4+ T cells from wild-type or Il1b−/−Il18−/− mice, followed by immunization with MOG35–55 peptide. (a) Mean clinical score. (b) Inflammatory gene expression in the lumbar spinal cord was assessed at the peak of disease. (c) Absolute numbers of CNS-infiltrating cells were determined at the peak of disease. Brains and spinal cords were harvested and mononuclear infiltrating cells were stained with the indicated antibodies, followed by flow cytometric analysis. (d) H&E staining (upper panels) and Luxol fast blue staining (lower panels) of lumbar spinal cords from Rag1−/− recipient mice harvested at the peak of disease. Scale bar, 200μm. (e–g) Rag1−/− mice were reconstituted with 2×106 sorted CD4+ T cells from wild-type or Il1b−/− mice, followed by immunization with MOG35–55 peptide. (e) Mean clinical score. (f) Inflammatory gene expression in the lumbar spinal cord was assessed at the peak of disease. (g) Absolute numbers of CNS-infiltrating cells were determined at the peak of disease. Brains and spinal cords were harvested and mononuclear infiltrating cells were stained with the indicated antibodies, followed by flow cytometric analysis. Error bars represent s.e.m. *P < 0.05 (Unpaired t test for b, c, f and g). Error bars represent s.d. *P < 0.05 (Two-way ANOVA for a and e). Data are representative of three independent experiments (a–g). n=5 mice per group in each experiment.
Given the potential role of IL-1β in promoting chemokine signals17, we completed a timecourse analysis to examine the trafficking of donor Il1b−/−Il18−/− CD4+ T cells to the CNS following MOG immunization. The results indicated that T cell-intrinsic IL-1β is dispensible for the first wave of T cell infiltration to the brain and spinal cord, as we observed no defect in CD4+ T cell egression from the spleen or migration to the CNS over the first 3–6 days post-immunization (Fig. 7a). However, by day 10 after adoptive transfer, CD4+ T cell numbers in the CNS were dramatically reduced in mice receiving CD4+ T cells from Il1b−/−Il18−/− mice (Fig. 7a), suggesting CD4+ T cell-derived IL-1β might play an important role in the survival/proliferation of CD4+ T cells in the CNS during EAE. In support of this, Ki-67 immunofluorescent staining of spinal cord tissue revealed a significant reduction of proliferating CD4+ T cells in the absence of T cell-derived IL-1β (Fig. 7b). Flow cytometry indicated that 79% of CD4+ T cells with high IL-1R in the CNS of mice receiving wild-type CD4+ T cells were Ki67+ proliferating cells (IL-1RhiKi67+CD4+ T cells), whereas only 28% of CD4+ T cells with low IL-1R were Ki67+ cells. These IL-1RhiKi67+CD4+ T cells were diminished in mice receiving CD4+ cells from Il1b−/−Il18−/− mice (Fig. 7c). While previous studies reported that T cell-intrinsic IL-1R signaling is required for TH17 cell survival/proliferation, the study here indicated that T cell-derived IL-1β is necessary to maintain the proliferation of CD4+ T cells in the CNS. In addition, between days 3, 6, and 10 of EAE development, mice that received wild-type CD4+ T cells showed a marked induction in IFN-γ and GM-CSF gene expression in the spinal cord, while IL-17A and IL-1R gene expression increased proportionally (Fig. 7d). This progressive cytokine induction in the CNS during the course of EAE was abolished in mice receiving CD4+ cells from Il1b−/−Il18−/− mice, indicating the critical role of T cell-derived IL-1β in driving production of pro-inflammatory cytokines to promote CNS inflammation (Fig. 7d). The effect of IL-1- and IL-18-deficiency on T cell percentages in the CNS seems much less marked than for ASC-deficiency, suggesting that ASC might also be involved in non-inflammasome functions.
Figure 7.
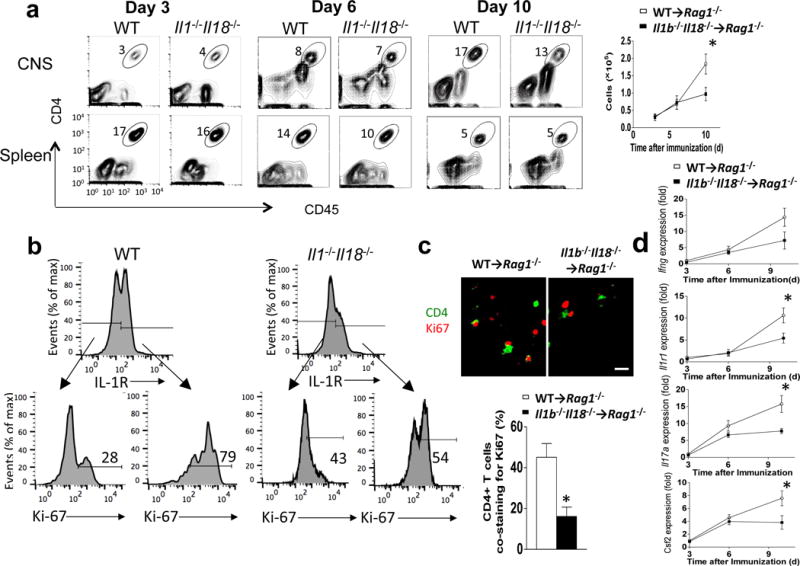
T cell-intrinsic inflammasome activation is required for TH17 cell survival/proliferation in the CNS. (a–d) Rag1−/− mice were reconstituted with 2×106 wild-type or Il1b−/−Il18−/− CD4+ T cells. Rag1−/− mice were actively immunized with MOG35–55 peptide. (a) Flow cytometric analysis of CD4+ T cells from CNS or spleen harvested on the indicated day (post-transfer) from Rag1−/− mice (n=5 mice per group) that received MOG-reactive TH17 cells from wild-type and Il1b−/−Il18−/− mice (left panels). Quantification of total CNS-infiltrating CD4+ cells on indicated day (right panel). (b) CNS-infiltrating CD4+ T cells were isolated on day 10 after the last immunization, and were stained with anti-IL-1R and anti-Ki-67 antibodies, followed by flow cytometry. (c) Anti-CD4 and anti-Ki-67 immunostaining was performed on spinal cords on day 10 after immunization (upper panels). The percentage of CD4+ cells co-staining with Ki67 was calculated for each experimental group (lower panel). Scale bar, 10μm. (d) Gene expression analysis of lumbar spinal cords obtained from Rag1−/− recipient mice on the indicated day post-immunization. Error bars represent s.e.m. *P < 0.05 (Unpaired t test for a, c and d). Data are representative of three independent experiments (a–d). n = 5 mice per group in each experiment.
DISCUSSION
We used T cell-specific genetic deletion of the inflammasome adaptor molecule ASC and found that ablation of ASC in T cells conferred robust protection from EAE development following immunization of mice with MOG35–55, while having no impact on the activation and expansion of MOG35–55-reactive TH1 and TH17 cells in the secondary lymphoid organs. Adoptive transfer of TH1- and TH17-polarized MOG35–55-reactive cells showed that the protective effect of T cell ASC deficiency was specific to TH17-, but not TH1-mediated neuroinflammation. This study demonstrates the critical role of T cell-intrinsic inflammasome in TH17-mediated EAE pathogenesis.
TH17-polarized ASC-deficient T cells isolated from the brains of recipient mice were markedly defective in their ability to secrete key pro-inflammatory cytokines, including IL-17A, IFN-γ, GM-CSF, TNF, and IL-1β. Intriguingly, a recent report demonstrated that human CD4+ T cells also process IL-1β in an ASC/NLRP3-dependent manner in response to HIV infection36. In addition, findings from the murine EAE model have suggested that IL-1β stimulation actually induces the secretion of IL-17A, IFN-γ, GM-CSF, and TNF from TH17-polarized brain-infiltrating cells19. We further found that TH17 cells differentiated in vitro highly express both IL-1R and pro-IL-1β, and that this pro-IL-1β can be processed and secreted in response to stimulation with extracellular ATP in a manner dependent on both ASC and NLRP3. Seeking to identify the caspase responsible for ATP-mediated IL-1β production by TH17 cells in vitro, we found that while caspase-1/11 deficiency had minimal effect on IL-1β processing, IL-1β maturation and secretion was abrogated by genetic deletion of caspase-8. This data is surprising, since caspase-1 is expressed in T cells. The fact that pro-caspase-1 was not recruited to ASC upon ATP stimulation suggests the possibility that caspase-1 might be suppressed or sequestered from ASC by an unknown mechanism in murine CD4+ T cells. Future studies are required to elucidate the molecular mechanism for the selective recruitment of caspase-8 to ASC in this context.
An abundance of previously published reports support the notion that IL-1β is critical for the expansion and survival of MOG-reactive T cells in the secondary lymphoid organs12,14–16,37,38. However, following immunization of Ascf/−LckCre mice with MOG, we did not observe any defect in antigen-dependent TH17 or TH1 cell polarization in the spleen, suggesting that IL-1β production by T cells is dispensable for MOG-induced priming in the periphery. It is therefore likely that IL-1β produced by macrophages and dendritic cells in the peripheral environment is sufficient to support initial TH17 cell priming and expansion. However, we found that while ASC-deficient TH17 cells are normal in their priming, egress from the spleen, and first-wave infiltration of the CNS following adoptive transfer, they fail to expand and to induce recruitment of second-wave effector cell populations in the CNS. Furthermore, Il1b−/−Il18−/− CD4+ T cells also show marked defects in proliferation and effector function in the CNS and Rag−/− mice reconstituted with Il1b−/− CD4+ T cells were protected from EAE development. Taken together, given the specific IL-1R expression in TH17 cells, these results suggest that ASC-dependent production of IL-1β by T cells in the CNS might be necessary for effective TH17 cell proliferation and effector cell recruitment.
Of particular note is the finding that both IL-17A- and IFN-γ-producing CD4+ T cell populations were reduced in the absence of T cell-intrinsic ASC. This finding mirrors a recent report that elegantly demonstrated the plasticity of TH17 cells during EAE pathogenesis19. In that study, the authors used an IL-17 promoter-driven TH17 lineage-tracing reporter system to show that following MOG immunization, classical TH1 IFN-γ-producing cells were present at a ~1:1 ratio with TH17-lineage IL-17A-producing cells in the draining lymph nodes. However, after arrival in the CNS, classical TH1 lineage cells rapidly disappeared by day 15 post-immunization. On the other hand, some TH17 cells were converted to IL-17- IFN-γ+ cells, so called ex- TH17 cells, which actually became the dominant IFN-γ-producing cell population in the spinal cord. The IFN-γ-secreting ex- TH17 cells continue to express IL-1R, while classical TH1 cells do not. As we observed dramatic defects in both IFN-γ and IL-17A production (and the other inflammatory cytokines) by ASC- and IL-1-deficient CD4+ T cells in the brain, implicating the critical role of T cell-derived IL-1β and consequent IL-1 signaling in TH17 and ex- TH17 cells during EAE. TH1 cells, which lack IL-1R expression, fail to receive the important survival signal provided by T cell-derived IL-1β in the CNS, possibly putting them at a relative disadvantage to TH17 and ex- TH17 cells.
In summary, this study is the first to report that T cell intrinsic inflammasome activity drives IL-1β production via caspase-8 activation, and is required for the pathogenic role of TH17-mediated CNS inflammation. Our findings lead us to speculate that chronic inflammation and tissue destruction in the CNS during EAE provides a prolonged danger signal, potentially in the form of extracellular ATP, to activate ASC-NLRP3-Caspase-8-dependent IL-1β production in T cells. T cell-derived IL-1β in turn promotes the survival/expansion of IL-1R+ TH17cells; and IL-1β stimulation of TH17 cells also promotes secretion of key pro-inflammatory cytokines, resulting in amplification of the TH17-mediated inflammatory cascade. This novel model of IL-1β- TH17 axis yields possible new therapeutic strategies for inhibition of CNS inflammation.
METHODS
Mice
B6(Cg)-Tg(Lck-Cd1d1)1Aben/J mice41 (C57BL/6 background) were purchased from Jackson Laboratory (stock number 019418). Ascflox/flox mice were generated by targeting the entire open reading frame of the Pycard gene on chromosome 7 for conditional deletion. Targeting vector was generated with a pCR4.0 backbone, and C57BL/6 embryonic stem cells were microinjected and screened by Southern hybridization for presence of the mutant allele. Both the targeting vector and the conditional knockout mice were designed and generated by Caliper Discovery Alliances & Services, which is now part of Taconic. Asc−/−, Nlrp3−/− and Il1b−/− mice were described previously42,43. Il1b−/−Il18−/− mice were described previously44. CD45.1 congenic mice were bought from Jackson Laboratory (002014). Rag1−/− mice were bought from Jackson Laboratory (002216). Casp1−/−Casp11−/− mice were bought from Jackson Laboratory (016621)45. Cd3e−/− mice were bought from Jackson Laboratory (004177). Thy1.1+ mice bought from Jackson Laboratory (000406). Casp8−/− Ripk3−/− and control mice were described previously46. All of the mice used in this study were female. For all experiments, mice were 10–12 weeks of age, with age-matched littermates used as experimental groups, and 5 mice pre group. Mice were housed under specific pathogen–free conditions. Experimental protocols were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic.
Reagents
Anti-ASC (1:1000, N-15-R) was purchased from Santa Cruz Biotechnology. Anti-ASC (1:1000, 2EI-7) was purchased from Milipore. Anti-IL-1β (1:1000, 3ZD) was got from Biological Resources Branch of the NIH. Anti-IL-1β (1:1000, AF-401-NA) was purchased from R&D. Anti-Caspase8 (1:1000 for all three antibodies, 4927, 1G12 and PA1-29159) were purchased from Cell Signaling Technology, Adipogen and ThermoFisher Scientific. Anti-NLRP3 (1:500, H-66) was purchased from SANT CRUZ Biotechnology. Anti-Actin (1:5000, A-2228) was purchased from Sigma. Anti-Ki67 (1:1000, ab15580) was purchased from Abcam. Anti-CD45-APC (1:500, 103112), Anti-CD45.2-PE/cy7 (1:500,104) and Anti-F4/80-FITC (1:200, 123108) were purchased from Biolegend. Anti-CD4-FITC (1:200, L3T4), Anti-Ly6C-PE (1:300, HK1.4), Anti-IFN-γ-FITC (1:200, XMG1.2), Anti-CD3 (1:1000, 145-2C11), Anti-CD28 (1:1000, 37.51), Anti-IL-4-FITC (1:200, BVD6-24G2) and Anti-FOXP3-PE (1:300, FJK-16S) were purchased from eBioscience. Anti-IL-17A-PE (1:300, 559502), Anti-CD8-PE (1:300, 553041) and Anti-Ly6G-PE (1:300, 1A8) were purchased from BD. Anti-IL-1R/CD121a (1:500, clone JAMA-147, catalog #113505) was purchased from Biolegend. Luxol Fast Blue MBS Solution (26681) was purchased from Electron Microscopy Sciences. Caspase-8 Flica (Kit#99) was purchased from ImmunoChemistry. YVAD-fmk (ALX-260-154-R100) and IETD-fmk (550380) were purchased from Enzo and BD. Anti-IL-1β-PE antibody (1:300, IC4013P) was purchased from R&D. IL-1Ra (anakinra) was purchased from AMGEN.
Induction and assessment of EAE
Active EAE was induced and assessed as previously described39. Adoptive transfer (passive) EAE was also induced as previously described40. Briefly, recipient mice were injected with 3.0×107 polarized MOG35−55-specific TH1 or TH17 cells 4 hours after 500 Rads sublethal irradiation. To prepare MOG35−55-specific polarized T cells, donor mice were immunized with MOG35−55 subcutaneously; draining lymph node cells were prepared from donor mice 10 days after immunization. Cells were cultured for 5 days with MOG35−55 at a concentration of 25 μg/ml under either TH1-polarizing conditions (20 ng/ml IL-12, R&D; 2 μg/ml anti-IL-23p19, eBioscience) or TH17-polarizing conditions (20 ng/ml IL-23, R&D). The clinical score was assessed in double-blinded manner. For the sample size, we performed power analysis for the clinical score of EAE, which has an average of 25% coefficient of variance. We determined that with n=15 mice we had 90% power to detect 30% difference between the groups.
In vitro CD4+ T cell differentiation
For TH17 cell differentiation, naive CD4+CD44lo T cells from C57BL/6, Ascf/−Lck-Cre, Nlrp3−/−, Casp1−/−/Casp11−/−, Casp8+/−Ripk3−/− and Casp8−/−Ripk3−/− mice were sorted by flow cytometry and activated with plate-bound 1 mg/ml anti-CD3 and 1 mg/ml anti-CD28 and in the presence of 2 ng/ml TGF-β (Peprotech), 20 ng/ml IL-6 (Peprotech), 5 μg/ml anti-IL-4 (11B11), and 5 μg/ml anti-IFN-γ (XMG 1.2). For TH1 cell differentiation, FACS-sorted naïve (CD4+CD44lo) CD4+ T cells were cultured on an anti-CD3/CD28 coated plate in the presence of 20 ng/ml IL-12 (Peprotech) and 5 μg/ml anti-IL-4 for 3 days. For TH2 cell differentiation, FACS-sorted naïve (CD4+CD44lo) CD4+ T cells were cultured on an anti-CD3/CD28 coated plate in TH2-polarizing (IL-4 (10 ng/ml) and anti-IFN-γ (10 μg/ml)) condition for 3 days. For Treg cell differentiation, FACS-sorted naïve (CD4+CD44lo) CD4+ T cells were cultured on an anti-CD3/CD28 coated plate in the presence of TGFβ (5ng/ml) for 3 days.
Inflammasome activation in CD4+ T cells
FACS-sorted naive CD4+CD44lo T cells were polarized to the indicated T helper lineage in vitro for 3 days. For simulation with ATP, cells were washed and re-suspended in 200 μl low-serum 1640 RPMI media (0.1% of serum) containing the indicated concentration of ATP. For detection of inflammasome activation by immunoblot analysis, we followed the protocol described previously47. Briefly, cells were lysed by adding 10× Cell Lysis Buffer (10% Nonidet P-40, 20mM DTT and 10× protease inhibitors) to the well that contains both the cells and culture media (i.e. without removing the condition medium) to a final concentration of 1% Nonidet P-40, 2mM DTT and protease inhibitors. Cells were then lysed (in the presence of culture medium) on ice for 30 minutes, and spun at 16,000 rpm. Protein concentration of the total lysate was measured. 4× Laemlli buffer was then added to the lysate, samples were boiled and approximately 20 μg of sample was run on 12% SDS-PAGE gel, followed by immunoblot analysis.
Reconstitution of T cells in Rag1−/− mice and active induction of EAE
2×106 FACS-sorted CD4+ T cells from wild-type, Il1b−/−/Il18−/− mice or Il1b−/− were i.v. transferred into Rag1−/− mice. On days 1 and 4 after CD4+ T cell reconstitution, mice were immunized subcutaneously in the abdominal flank with 200 mg MOG35–55 and 400 mg Mycobacteria tuberculosis H37RA (Difco) in 400 μl of an emulsion of equal volumes of water and complete Freund’s adjuvant (Difco). Each mouse also received 0.4 mg of purified Bordetella pertussis toxin in 200 μl PBS i.p. on days 0 and 4.
Generation of chimera mice
To generate Cd3e−/−Il1b−/− and Cd3e−/−Il1b+/− chimera mice, 15×106 bone marrow cells from Cd3e−/− and Il1b−/− or Il1b+/− bone marrow were injected into irradiated wild-type mice (550 Rads×2 times) by i.v. injection (15×106 cells per recipient mouse). Recipient mice were allowed to recover for 6 weeks, followed by active immunization as previously described39.
Histological analysis
All spinal cord tissue sections used here were 5 μm thick. For paraffin-embedded tissue, spinal cords collected from phosphate-buffered saline–perfused mice were fixed in 10% formalin and then dehydrated with 70% alcohol. Sections were stained with either hematoxylin and eosin or Luxol fast blue to evaluate inflammation and demyelination, respectively.
Real-time PCR
Total RNA was extracted from spinal cord with TRIzol (Invitrogen) according to the manufacturer’s instructions. All gene expression results are expressed as arbitrary units relative to expression of Actb. Fold induction of gene expression in spinal cord after EAE induction was determined by dividing the relative abundance of experimental samples by the mean relative abundance of control samples from naive mice. Primer sequence is in supplementary table 1.
ELISA
Thy1.2+ CD4+ T cells FACS-sorted from the brains of wild-type Thy1.1+ recipient mice that received TH17 cells from MOG-immunized Ascf/+Lck-Cre and Ascf/−Lck-Cre mice, were cultured for 24 hours at 37°C on plates coated with anti-CD3 (3 μg/ml; 553057; BD Pharmingen) and anti-CD28 (3 μg/ml; 553294; BD Pharmingen). Supernatants were collected for enzyme-linked immunosorbent assay of cytokines with a kit from BioLegend (for IL-17A) or kits from R&D Systems (for all other cytokines). Il1b levels were assayed by Il1b (MLB00C) ELISA kit (R&D systems) according to the manufacturer’s instructions. Ifng (MIF00), csf2 (DY415-05) and Tnf (MTA00B) ELISA kit were purchased from R&D Systems.
Immunoblot and immunoprecipitation
Cells were lysed by lysis buffer (0.5% Triton X-100, 20 mM Hepes pH 7.4,150 mM NaCl, 12.5 mM β-glycerophosphate, 1.5 mM MgCl2, 10 mM NaF, 2 mM dithiothreitol, 1 mM sodium orthovanadate, 2 mM EGTA, 20 mM aprotinin, 1 mM phenylmethylsulfonyl fluoride). 20 μg of protein lysate per lane was run on a 12% SDS-PAGE gel, followed by immune-blotting with different antibodies. Co-immunoprecipitation experiments were performed as described previously17. In brief, cell extracts were incubated overnight with antibodies and protein A beads at 4 °C After incubation, beads were washed four times with lysis buffer, resolved by SDS-PAGE and analyzed by immunoblotting according to standard procedures.
Statistics
Non-parametric statistics was applied to all data set. The P values of clinical scores were determined by two-way multiple-range ANOVA for multiple comparisons. Other P values were determined by Unpaired t test. P < 0.05 was considered to be significant. Unless otherwise specified, all results are shown as mean and the s.e.m. We performed power analysis for a sample size that gives 80% power for an effect size of 1.5. The control and experimental groups were blinded to the investigators who recorded the clinical score, flow analysis, histology and LFB staining. No samples were excluded, and Randomization was not performed.
Supplementary Material
Acknowledgments
We thank M.D. Wewers and A. Sarkar (Ohio State University College of Medicine) for Il1b–/–Il18–/– mice; Edward S. Mocarski (Emory Vaccine Center) for Casp8–/–Rip3–/– and control mice; Amy G. Hise (Case Western Reserve University) for Asc−/−, Nlrp3−/− and Il1b−/− mice. This investigation was supported by a grant from the National Multiple Sclerosis Society (RG5130A2/1) and National Institutes of Health (5R01NS071996-05, 1RO1AA023722 and MSTP-T32GM007250).
Footnotes
AUTHOR CONTRIBUTIONS STATEMENT
B.N.M. and C.W. did the experiments and analyzed the data; C.J Z., Z. K., M.F.G., J.A.Z., J.Z., C.E.S. and B.L. contributed to the experiments; J.D. and P.P. helped to make the Cd3e−/− and Il1b−/− bone marrow chimera mice; A.A., Y.L., A.S., M.D.W., W.J.K., E.S.M., M.E.R. and A.G.H. provided reagents and participated in discussion; B.M. and G.R.D contributed reagents, helped to design experiments and helped to edit the manuscript; B.N.M., C.W. and X.L. wrote the manuscript; R.M.R. and X.L. conceived the study, oversaw the experiments and analyzed the data.
COMPETING INTERESTS STATEMENT
The authors declare no competing financial interests.
References
- 1.Frohman EM. Multiple sclerosis. The Medical clinics of North America. 2003;87:867–897. viii–ix. doi: 10.1016/s0025-7125(03)00008-7. [DOI] [PubMed] [Google Scholar]
- 2.Ransohoff RM. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat Neurosci. 2012;15:1074–1077. doi: 10.1038/nn.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lees JR, Golumbek PT, Sim J, Dorsey D, Russell JH. Regional CNS responses to IFN-gamma determine lesion localization patterns during EAE pathogenesis. J Exp Med. 2008;205:2633–2642. doi: 10.1084/jem.20080155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jager A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. 2009;183:7169–7177. doi: 10.4049/jimmunol.0901906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cua DJ, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 8.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 10.Chung Y, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gulen MF, et al. The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity. 2010;32:54–66. doi: 10.1016/j.immuni.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Staschke KA, et al. IRAK4 kinase activity is required for Th17 differentiation and Th17-mediated disease. J Immunol. 2009;183:568–577. doi: 10.4049/jimmunol.0802361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuki T, Nakae S, Sudo K, Horai R, Iwakura Y. Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int Immunol. 2006;18:399–407. doi: 10.1093/intimm/dxh379. [DOI] [PubMed] [Google Scholar]
- 14.Gulen MF, et al. Inactivation of the enzyme GSK3alpha by the kinase IKKi promotes AKT-mTOR signaling pathway that mediates interleukin-1-induced Th17 cell maintenance. Immunity. 2012;37:800–812. doi: 10.1016/j.immuni.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang J, et al. MyD88 is essential to sustain mTOR activation necessary to promote T helper 17 cell proliferation by linking IL-1 and IL-23 signaling. Proc Natl Acad Sci USA. 2013;110:2270–2275. doi: 10.1073/pnas.1206048110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gris D, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol. 2010;185:974–981. doi: 10.4049/jimmunol.0904145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2012;109:10480–10485. doi: 10.1073/pnas.1201836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue M, et al. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal. 2012;5:ra38. doi: 10.1126/scisignal.2002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirota K, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arbeloa J, Perez-Samartin A, Gottlieb M, Matute C. P2X7 receptor blockade prevents ATP excitotoxicity in neurons and reduces brain damage after ischemia. Neuro dis. 2012;45:954–961. doi: 10.1016/j.nbd.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 21.Qu Y, et al. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol. 2011;186:6553–6561. doi: 10.4049/jimmunol.1100478. [DOI] [PubMed] [Google Scholar]
- 22.Gordon GR, et al. Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat Neurosci. 2005;8:1078–1086. doi: 10.1038/nn1498. [DOI] [PubMed] [Google Scholar]
- 23.Antonopoulos C, El Sanadi C, Kaiser WJ, Mocarski ES, Dubyak GR. Proapoptotic chemotherapeutic drugs induce noncanonical processing and release of IL-1beta via caspase-8 in dendritic cells. J Immunol. 2013;191:4789–4803. doi: 10.4049/jimmunol.1300645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gringhuis SI, et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat Immunol. 2012;13:246–254. doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 25.Bossaller L, et al. FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol. 2012;189:5508–5512. doi: 10.4049/jimmunol.1202121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oberst A, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leverrier S, Salvesen GS, Walsh CM. Enzymatically active single chain caspase-8 maintains T-cell survival during clonal expansion. Cell Death Differ. 2011;18:90–98. doi: 10.1038/cdd.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bell BD, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci USA. 2008;105:16677–16682. doi: 10.1073/pnas.0808597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang TB, et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol. 2004;173:2976–2984. doi: 10.4049/jimmunol.173.5.2976. [DOI] [PubMed] [Google Scholar]
- 30.Salmena L, et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003;17:883–895. doi: 10.1101/gad.1063703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chun HJ, et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002;419:395–399. doi: 10.1038/nature01063. [DOI] [PubMed] [Google Scholar]
- 32.Bauernfeind FG, et al. NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin BN, et al. IKKalpha negatively regulates ASC-dependent inflammasome activation. Nat Commun. 2014;5:4977. doi: 10.1038/ncomms5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol. 2009;182:3173–3182. doi: 10.4049/jimmunol.0802367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schenten D, et al. Signaling through the adaptor molecule MyD88 in CD4+ T cells is required to overcome suppression by regulatory T cells. Immunity. 2014;40:78–90. doi: 10.1016/j.immuni.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doitsh G, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaw PJ, et al. Critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4610–4614. doi: 10.4049/jimmunol.1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El-Behi M, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian Y, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 40.Kang Z, et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei DG, et al. Expansion and long-rangedifferentiation of the NKT cell lineage in mice expressing CD1d exclusively on cortical thymocytes. J Exp Med. 2005;202:239–48. doi: 10.1084/jem.20050413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shornick LP, et al. Mice deficient in IL-1beta manifest impaired contact hypersensitivity to trinitrochlorobenzone. J Exp Med. 1996;183:1427–36. doi: 10.1084/jem.183.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomalka J, et al. A Novel Role for the NLRC4 Inflammasome in Mucosal Defenses against the Fungal Pathogen Candida albicans. PLoS Pathog. 2011;7:e1002379. doi: 10.1371/journal.ppat.1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anasuya S, et al. Caspase-1 regulates Escherichia coli sepsis and splenic B cell apoptosis independent of interleukin-1β and interleukin-18. Am J Respir Crit Care Med. 2006;174:1003–1010. doi: 10.1164/rccm.200604-546OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuida K, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–3. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 46.William J Kaiser, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shiho Suzuki, et al. Shigella Type III Secretion Protein MxiI Is Recognized by Naip2 to Induce Nlrc4 Inflammasome Activation Independently of Pkcd. PLoS Pathog. 2014;10:e1003926. doi: 10.1371/journal.ppat.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.