

Sec1/Munc18 proteins are essential for fusion but of unknown function. The yeast vacuole SM protein is a subunit of the HOPS tethering complex. HOPS catalyzes the interdependent association among the vacuole SNAREs at a membrane surface, and the associated SNAREs can be disassembled by the physiological system Sec17/Sec18/ATP.
Abstract
Rab GTPases, their effectors, SNAREs of the R, Qa, Qb, and Qc families, and SM SNARE-binding proteins catalyze intracellular membrane fusion. At the vacuole/lysosome, they are integrated by the homotypic fusion and vacuole protein sorting (HOPS) complex. Two HOPS subunits bind vacuolar Rabs for tethering, another binds the Qc SNARE, and a fourth HOPS subunit, an SM protein, has conserved grooves that bind R- and Qa-SNARE domains. Spontaneous quaternary SNARE complex assembly is very slow. We report an assay of SNARE complex assembly that does not rely on fusion and for which tethering does not coenrich the four SNAREs. HOPS is required in this assay for rapid SNARE complex assembly. Optimal assembly needs HOPS, lipid membranes to which the R- or Qa-SNARE and Ypt7:GTP are integrally bound, and each of the other three SNAREs. Each SNARE assembles into this complex relying on the others, suggesting four-SNARE complex assembly rather than direct binding of each to HOPS. SNAREs can be disassociated by Sec 17/Sec 18/ATP, completing a catalyzed cycle of SNARE assembly and disassembly.
INTRODUCTION
Regulated intracellular membrane fusion supports each step of exocytic and endocytic vesicular traffic and is essential for cell growth, hormone secretion, and neurotransmission (Wickner and Schekman, 2008). The fundamental mechanism of fusion is conserved throughout eukaryotes, from yeast to plants and humans. Rab/Ypt family GTPases are the master switches of trafficking, active when in complex with GTP (Grosshans et al., 2006). They bind effector proteins, including large multisubunit tethering complexes that tether membranes in stable proximity (Yu and Hughson, 2010). When membranes are tethered, their soluble N-ethylmaleimide–sensitive factor attachment protein receptors (SNAREs) bind to each other in-trans, that is, with SNAREs anchored in each apposed tethered membrane. SNARE complex assembly is believed to require proteins of the Sec 1/Munc18 (SM) family (Rizo and Südhof, 2012), although it is not known whether SM proteins act through tethering, catalysis of trans-SNARE complex assembly, lipid rearrangements, or blockage to SNARE complex disassembly by the Sec 18 (NSF)/Sec 17 (α-SNAP) chaperones. SNARE proteins are defined by their heptad-repeat SNARE domains (Fasshauer et al., 1998), which form α-helices that can assemble with each other in a rod-shaped four–helical coiled coil termed a four-SNARE bundle or SNARE complex. Whereas most of the amino acyl residues of each SNARE domain that face the interior of the SNARE bundle are apolar, the center of the bundle, termed the 0-layer, has inward-facing arginyl (R-) and glutaminyl (Q-) residues. Conserved families of SNAREs are termed R-, Qa-, Qb-, or Qc-SNAREs, and four-SNARE bundles have the composition RQaQbQc (Fasshauer et al., 1998). SNAREs often consist of an N-terminal domain, a SNARE domain, and a C-terminal transmembrane anchor domain. SNARE complexes assemble (zipper) from the N-terminus to their C-terminus of their SNARE domains, drawing the tethered membranes into yet closer apposition. Lipids then rearrange through nonbilayer intermediates, fusing the two membrane bilayers and two noncytoplasmic aqueous compartments while continuously retaining the separation of these compartments from the cytoplasm. Fusion converts trans-SNARE complexes anchored in two apposed bilayers into cis-SNARE complexes, which are anchored in the single fused membrane. The ATP-driven chaperone Sec 18/NSF, aided by its cochaperone, Sec 17/α-SNAP, liberates SNAREs from cis-complexes to permit subsequent rounds of trans-complex assembly (Mayer et al., 1996). We are only beginning to appreciate how these proteins function together and how fusion is regulated.
We study membrane fusion with the vacuole (lysosome) of Saccharomyces cerevisiae. Vacuoles, which are large and readily visualized in the light microscope, undergo constant fission and fusion in the cell. Genetic blockage of fusion allows unimpeded fission, a striking phenotype that allowed early determination of the vacuole-specific membrane fusion factors (Wada et al., 1992). On development of a colorimetric assay for the fusion of purified vacuoles (Haas et al., 1994), it was shown that each of the genetically identified fusion factors is directly required for the fusion event rather than only having an indirect role, for example, for biosynthetic trafficking of other fusion proteins to the organelle. Vacuole tethering requires the Rab-family GTPase Ypt7 (Haas et al., 1995; Stroupe et al., 2009) and a heterohexameric tethering complex (Stroupe et al., 2006; Hickey and Wickner, 2010) termed the homotypic fusion and vacuole protein sorting (HOPS) complex. Two HOPS subunits have direct affinity for Ypt7 (Brett et al., 2008; Bröcker et al., 2012), explaining how HOPS alone can efficiently tether two vacuoles together. Other HOPS subunits have direct affinity for SNAREs. The Qc-SNARE of the vacuole has an N-terminal PX domain with affinities for phosphatidylinositol 3-phosphate (PtdIns3P; Cheever et al., 2001) and HOPS (Stroupe et al., 2006) through the HOPS Vps16 subunit (Krämer and Ungermann, 2011). The R-SNARE Nyv1, the Qa-SNARE Vam3, the Qb-SNARE Vti1, the Qc-SNARE Vam7 (henceforth termed R-, Qa-, Qb-, and Qc-SNAREs, or simply R, Qa, Qb, and Qc), the Rab Ypt7, HOPS, and several lipids that are required for fusion become highly enriched in an interdependent manner in a fusion-competent microdomain on tethered membranes before fusion (Wang et al., 2002, 2003; Fratti et al., 2004).
The functional and physical relationships among these proteins, and the function of SM proteins in particular, have remained obscure despite extensive study (Rizo and Südhof, 2012). Vps33, the SM subunit of HOPS, has conserved grooves that bind the α-helical SNARE domains of R- and Qa-SNAREs in parallel and in register with respect to the arginyl and glutaminyl residues at the center of their SNARE domains, the 0-layer (Baker et al., 2015). This might represent an early step in the catalysis of trans-SNARE complex assembly. This idea was supported by functional studies in which SNARE complex assembly was inferred from assays of the consequent fusion. When R-SNARE proteoliposomes were incubated with proteoliposomes in which the three Q-SNAREs were preassembled, HOPS was needed only for its tethering function because HOPS with mutations in its SM subunit that blocked SNARE domain binding supported this fusion, and the nonspecific membrane clustering agent polyethylene glycol could substitute for HOPS. When the Q-SNAREs were not preassembled on the starting proteoliposomes and the fusion reaction depended on separate addition of the Qc-SNARE, these HOPS mutants still supported vesicle clustering but no longer supported fusion, and polyethylene glycol no longer sufficed. It was therefore suggested (Baker et al., 2015) that HOPS may function beyond tethering when maximal SNARE complex assembly is required. To test this model, further direct assays of the amounts of newly assembled SNARE complex that form in the presence or absence of HOPS are required to establish whether HOPS suffices to mediate such assembly.
We report new assays of the interactions of SNAREs, HOPS, and membranes in the absence of fusion. They provide direct physical evidence of the central role of HOPS in catalyzing SNARE complex assembly. This SNARE complex can be disassembled by Sec 18/Sec 17/ATP.
RESULTS
The R-, Qa-, and Qb-SNAREs of the yeast vacuole (Figure 1A) have N-terminal domains, SNARE domains, and C-terminal apolar transmembrane (TM) anchors. The Qc SNARE Vam7 lacks an apolar membrane anchor but binds to the membrane by the affinities of its N-terminal PX domain for PtdIns3P and HOPS (Stroupe et al., 2006), as well as through its association with the other SNAREs. To assay the physical associations among vacuole fusion proteins, we purified recombinant “soluble” SNAREs (termed sSNAREs), which lacked their apolar membrane anchors (Figure 1A), and also purified Vam7-t.m. (transmembrane), a previously studied (Xu and Wickner, 2012), integrally anchored synthetic derivative of the Qc-SNARE. For certain studies, we used Vam7 with a glutathione S-transferase (GST) tag at its N-terminus. Proteoliposomes, prepared with one full-length, integrally-anchored SNARE where indicated and with or without Ypt7:GTP, were incubated with sSNAREs and HOPS (Figure 1B). Although these proteoliposomes can cluster, there is no fusion because there is at most one integrally bound SNARE. Because clustering per se would enhance only the local concentration of the one membrane-anchored SNARE, the tethering function alone could not promote assembly among the four SNAREs. After incubation, density medium was added, and the proteoliposomes were isolated by flotation through overlaid solutions of lower density (Figure 1B). Isolated proteoliposomes were then assayed for bound proteins by immunoblot. In other experiments, and after the initial incubation of proteoliposomes, HOPS, and sSNAREs, isolation by flotation was replaced by the addition of detergent and pull down with either antibody to the Qa-SNARE or (when GST–Qc-SNARE was present in place of wild-type Qc-SNARE) with glutathione beads. These three assays allow the measurement of new SNARE complex formation in the absence of either tethering-induced colocalization of the different SNAREs or of fusion. All experiments used affinity-purified HOPS that had been further purified by sucrose gradient velocity sedimentation. In this gradient purification, the coincident peak of protein concentration, HOPS subunits, and fusion-promoting activity was resolved from inactive aggregated HOPS and incompletely assembled subunits (Supplemental Figure S1). This pure HOPS sedimented somewhat more slowly than thyroglobulin, as expected from their molecular masses and from the rather elongate multilobed HOPS structure (Bröcker et al., 2012).
FIGURE 1:
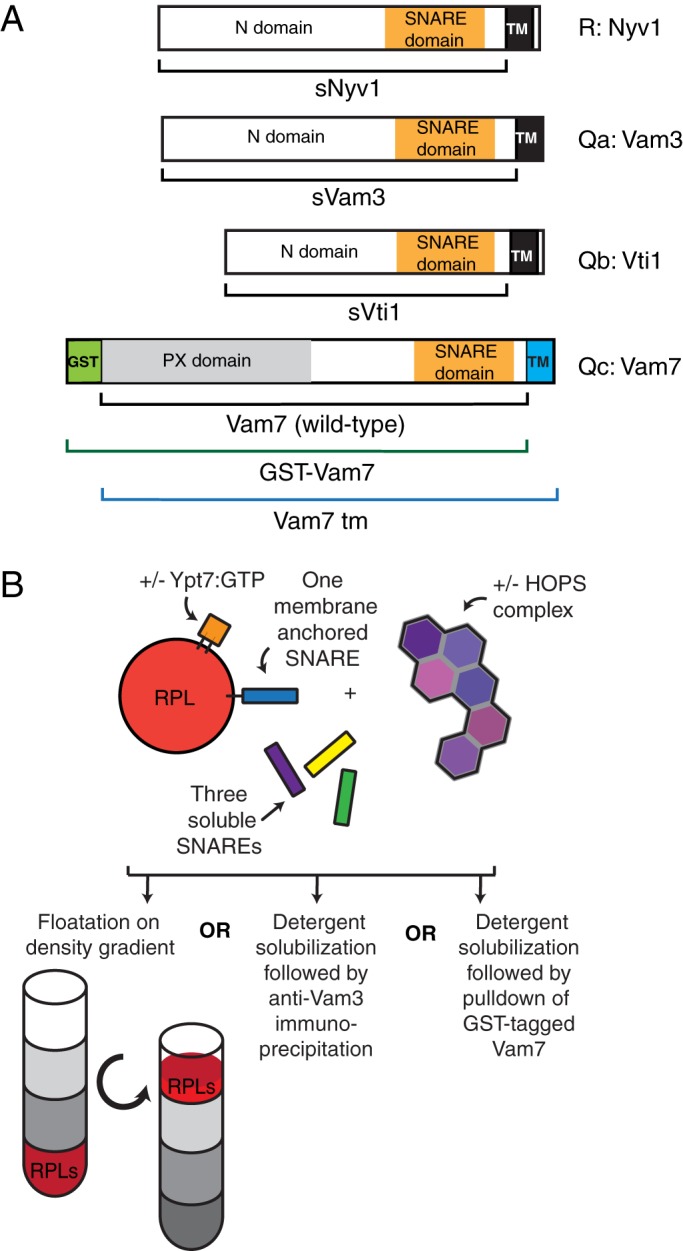
Assays of four-SNARE complex assembly. (A) The R-SNARE Nyv1 and the Qa- and Qb-SNAREs Vam3 and Vti1 were isolated without their transmembrane domains as sSNAREs. For some experiments, the naturally soluble Qc-SNARE Vam7 (wild type) was joined at its C-terminus to a transmembrane domain from Vti1 (Vam7tm; Xu and Wickner, 2012) or was prepared with a noncleavable GST tag (Fratti et al., 2007). (B) Proteoliposomes prepared with or without the Rab Ypt7 and either with or without a single integrally bound SNARE were incubated with soluble SNAREs and HOPS. Reactions were analyzed for complex assembly by any of three methods: reisolation by flotation through density medium or, after detergent solubilization, immunoprecipitation by Qa pull down or glutathione-bead pull down of the GST-tagged Qc-SNARE Vam7.
Flotation assays of assembly
As a baseline control, liposomes that either had no proteins or bore Ypt7:GTP alone were incubated with each of the four sSNAREs in the presence or absence of HOPS either separately (without other SNAREs) or with the other three soluble SNAREs. Liposomes were then isolated by flotation. Only low levels of sR, sQa, or sQb were recovered with the proteoliposomes under any of these conditions (Figure 2, top three graphs). The Qc-SNARE shows a reproducible direct affinity for PtdIns3P (Cheever et al., 2001), which is present in all proteoliposomes, for HOPS (Stroupe et al., 2006), and for Ypt7 (Stroupe et al., 2009), with enhanced binding in the presence of both HOPS and Ypt7 (Figure 2, bottom). Even in the presence of Ypt7 and HOPS, there is no enhancement of the liposomal association of any of the sSNARES when premixed with the other three sSNAREs before incubation (Figure 2, lane 1 vs. lane 5).
FIGURE 2:
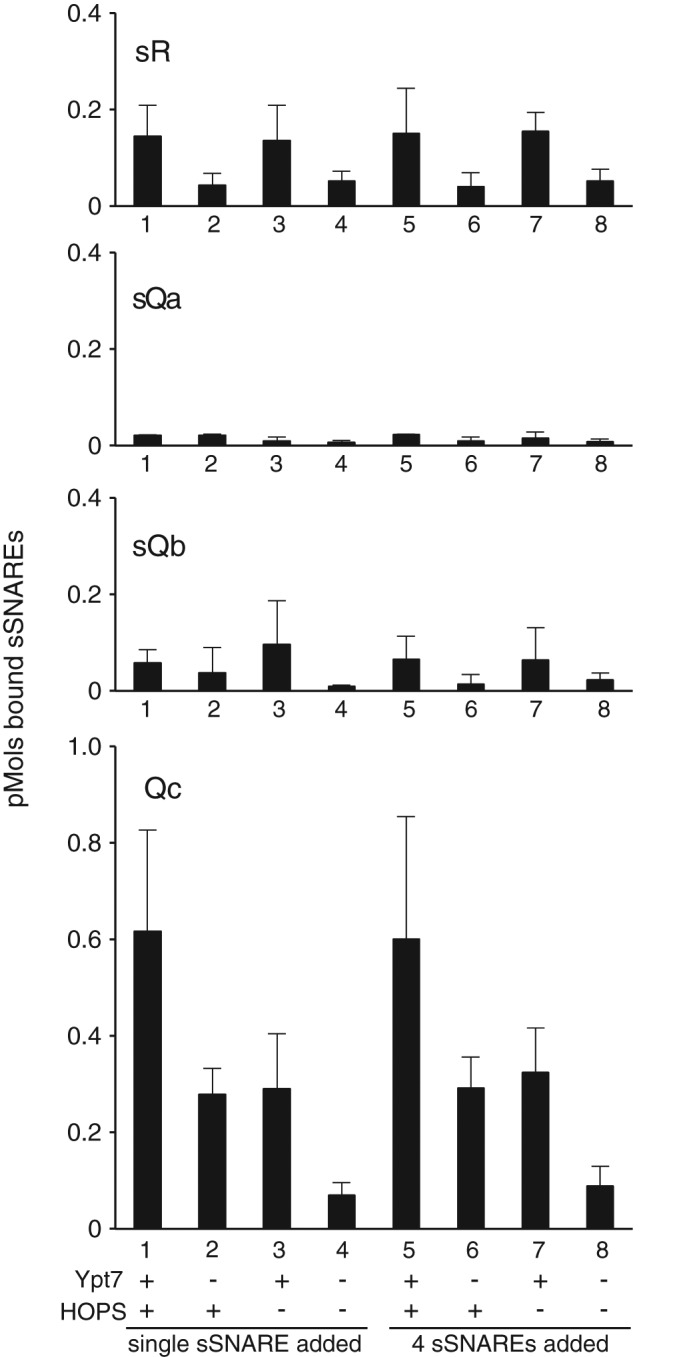
Soluble SNAREs other than the Qc-SNARE have low affinity for membranes. Liposomes were prepared as described in Materials and Methods with or without Ypt7 and without any integral SNAREs. Liposomes were incubated as described in Materials and Methods with single sSNAREs or all four sSNAREs with or without HOPS and then assayed for liposome-bound sSNAREs via flotation through density medium. The portion of the reaction subjected to flotation analysis contained 8 pmol of sR, sQb, and Qc, 2 pmol of sQa, and 0.5 pmol of HOPS. Data were analyzed by immunoblot as described in Materials and Methods and represent the average and SD of three independent experiments.
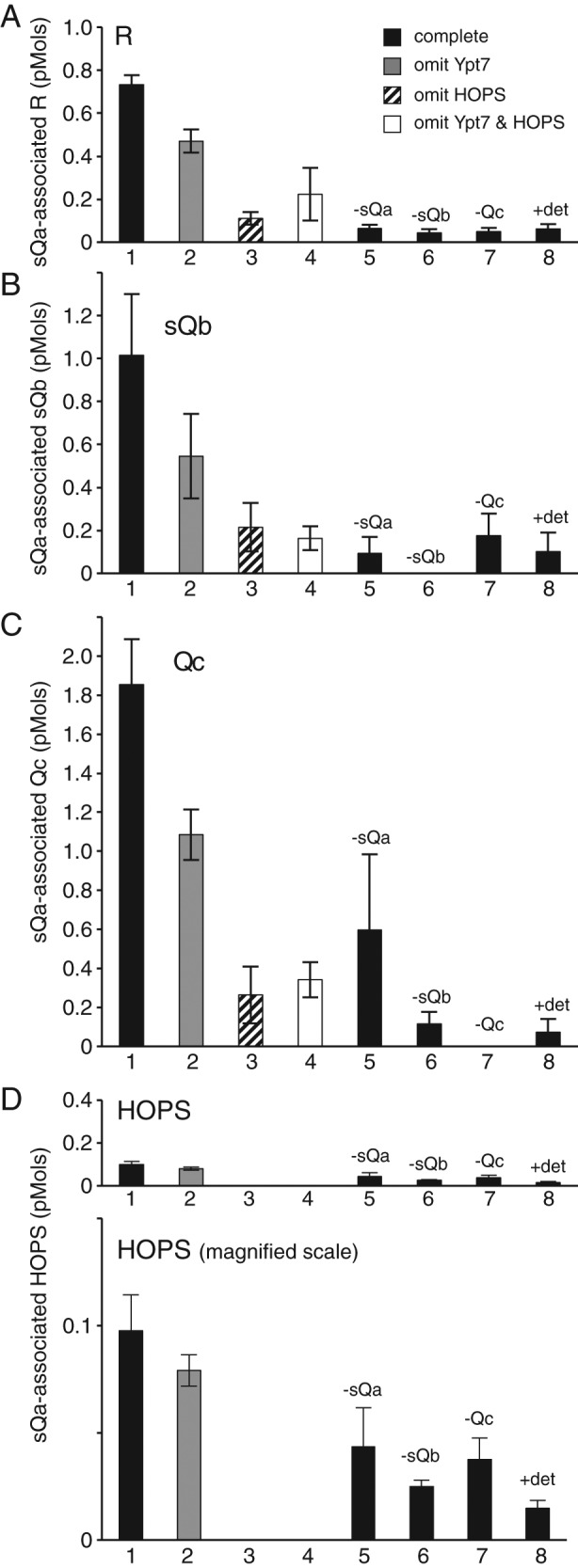
Proteoliposomes were also prepared with Ypt7:GTP and a single integrally bound SNARE (Figure 3). These were incubated with HOPS and the three remaining sSNAREs. After flotation, the proteoliposomes were assayed by immunoblot for bound proteins. HOPS binding to the proteoliposomes was enhanced severalfold by Ypt7:GTP (Figure 3E, lane 1 vs. lane 2) but did not require an integrally-bound SNARE (Figure 3E, lane 1 vs. lane 29) and was unaffected by the absence of individual soluble SNAREs (Figure 3E, lane 1 vs. lanes 5–7).
FIGURE 3:

SNARE complex formation depends on HOPS and the R- or Qa–integral SNARE. Liposomes were prepared as described with or without Ypt7 and with one or no integral SNAREs, incubated with HOPS and the other soluble SNAREs, isolated by flotation, and assayed by immunoblot for bound (A) sR, (B) sQa, (C) sQb, (D) Qc, and (E) HOPS as measured by its Vps16 subunit. The complete binding reaction (black bars), containing RPLs with Ypt7 and one integrally bound SNARE plus the three remaining sSNAREs and HOPS, is compared with reactions without Ypt7 (gray bars), without HOPS (striped bars), without Ypt7 or HOPS (white bars), or with singly omitted sSNAREs as indicated. Integrally bound SNARE inputs were ∼5 pmol each, soluble protein inputs were 8 pmol of sR, sQb, and Qc, 2 pmol of sQa, and 0.5 pmol of HOPS. Data were analyzed by immunoblot as described in Materials and Methods and represent the average and SD of three independent experiments.
In contrast to the findings when there were no integrally bound SNAREs (Figure 2), floated proteoliposomes with full-length, wild-type R-SNARE and Ypt7:GTP (Figure 3, column I, lanes 1–7) exhibited considerable binding of sQa-, sQb-, and Qc-SNAREs after incubation with HOPS (Figure 3, B–D, lane 1). The proteoliposome association of the three sQ-SNAREs was specific in several crucial regards. Without HOPS, binding of each sQ-SNARE was strongly reduced (Figure 3, B–D, lane 1 vs. lanes 3 and 4). Even in the presence of HOPS and Ypt7:GTP, the single omission of any one of the three sQ-SNAREs reduced proteoliposomal association of the sQa- or sQb-SNARE to background levels (Figure 3, B and C, lane 1 vs. lanes 5–7); the specific membrane association of the Qc-SNARE was obscured by high background binding (Figure 3D, lane 29). Membrane association was also lost when sR-SNARE replaced membrane-anchored R-SNARE in the reactions (Figure 3B and C, lane 29; also see Figure 2). Because HOPS is required for the proteoliposomal association of each of the sQ-SNAREs and they each depend on the others for this association, the simplest model is that HOPS is catalyzing the assembly of the four-SNARE complex.
When full-length R-SNARE was replaced by full-length Qa on the proteoliposomes and sR was present along with sQb and Qc (Figure 3, column II, lanes 8–14), each of the sSNAREs bound to the proteoliposomes (Figure 3, A, C, and D, lane 8). This complex assembly was also sensitive to omission of HOPS (lane 8 vs. lanes 10 and 11) or of individual sSNAREs (lane 8 vs. lanes 12–14). When the Qb-SNARE or the Qc-SNARE with an artificial transmembrane domain were integrally bound to the proteoliposomes and incubations bore HOPS and the other three sSNAREs (Figure 3, columns III and IV, lanes 15–28), there was little binding of the other sSNAREs above background levels. Optimal assembly thus required HOPS, Ypt7:GTP, integrally bound R- or Qa-SNARE, and the remaining three soluble SNAREs.
Pull-down assays of SNARE complex assembly
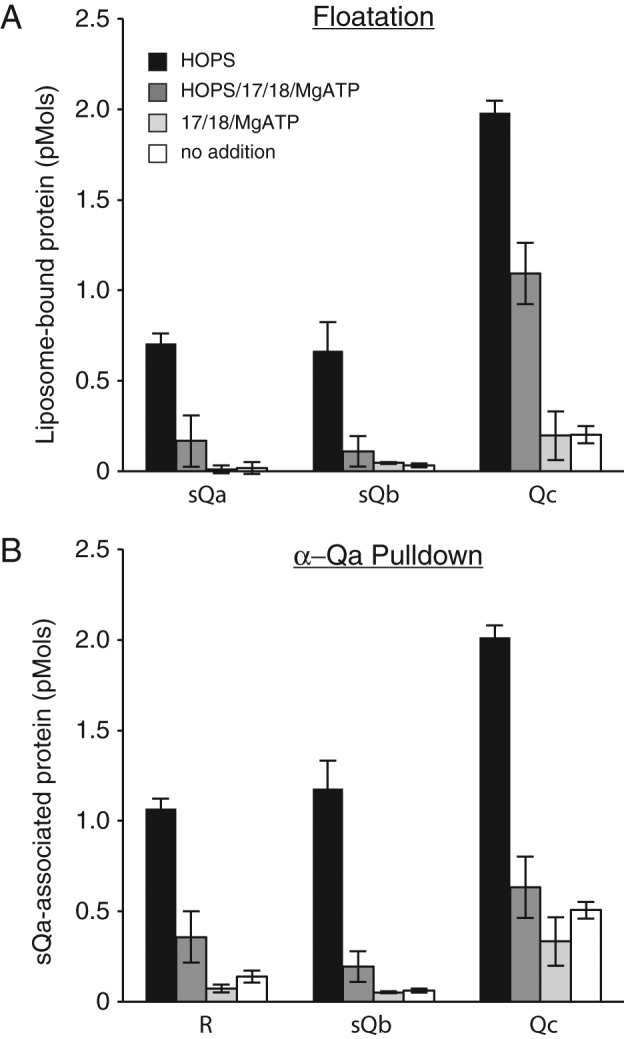
We also performed two independent pull-down assays of HOPS-mediated SNARE complex assembly. Proteoliposomes with Ypt7:GTP and full-length R-SNARE were incubated with HOPS and the three soluble Q-SNAREs and then dissolved in CHAPS without prior flotation. The Qa-SNARE was immunoprecipitated with affinity-purified antibody, and the Qa-associated SNAREs and HOPS were assayed by immunoblot. Each of the other three SNAREs became associated with the Qa-SNARE (Figure 4, A–C, lane 1) in a manner that was reduced by the omission of either HOPS (lanes 3 and 4) or any one of the soluble Q-SNAREs (lanes 5–7) or by addition of the 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) detergent from the start of the assembly incubation (lane 8). A substantially lower molar level of HOPS was recovered in association with Qa (Figure 4D, lane 1) than the other SNAREs (Figure 4, A–C, lane 1). Approximately half of this HOPS was bound nonspecifically and half through association with the Qa-SNARE or SNARE complex, as the pull down of HOPS was partially decreased by omission of individual sSNAREs (Figure 4D, lane 1 vs. lanes 5–7).
FIGURE 4:
Antibody pull down of Qa indicates that SNARE complex formation requires HOPS, all four SNAREs, and an intact membrane. Liposomes were prepared with Ypt7 and the integral R-SNARE Nyv1. After a 30-min incubation with HOPS and the soluble Q-SNAREs, membranes were dissolved with detergent, and the proteins associated with Qa were isolated by α-Qa pull down and analyzed by immunoblot, as described in Materials and Methods. Protein inputs were 5 pmol of integrally bound R, 8 pmol each of sQa, sQb, and Qc, and 0.5 pmol of HOPS. (A–D) Eluted samples were assayed by immunoblot for R, sQb, Qc, and HOPS via its Vps16 subunit, respectively. Data represent the averages and SDs of three independent experiments.
As a further assay of SNARE complex assembly, proteoliposomes with Ypt7:GTP and full-length R-SNARE were incubated with HOPS, sQa-, sQb-, and either GST-tagged Qc- or wild-type Qc-SNARE. Samples were harvested after 5, 30, or 90 min of incubation, dissolved in CHAPS, and assayed with glutathione beads for proteins that had bound to GST–Qc-SNARE (Figure 5). In complete incubation conditions, HOPS and the other three SNAREs bound to GST–Qc-SNARE (Figure 5, lanes 1–3). High levels of SNARE association were not seen when HOPS or sQa- or sQb-SNARE was omitted (Figure 5, lanes 1–3 vs. lanes 7–15) or when detergent was present in the initial incubation (lanes 4–6). At each time point, the HOPS that had been present under complete SNARE complex assembly conditions bound at levels that were higher than under control conditions, but there was substantial background binding. This background HOPS binding may have arisen from the known direct association of HOPS with GST-Qc (Stroupe et al., 2006). Scanning and quantitation of these immunoblots (Supplemental Table S1) showed 5–10 times more picomoles of SNAREs than of HOPS associated with the tagged Qc-SNARE.
FIGURE 5:

Kinetics of HOPS-dependent and HOPS-independent SNARE complex assembly revealed by a GST-pull-down assay. Proteoliposomes with Ypt7 and the R-SNARE Nyv1 were incubated with HOPS and sQa, sQb, and GST-tagged Qc for 90 min (lane 3), 30 min (lane 2), or 5 min (lane 1), solubilized in 1% CHAPS, and mixed with glutathione-magnetic beads as described in Materials and Methods. Where indicated, incubations contained detergent from the start (lanes 4–6), were performed without HOPS (lanes 7–9), without sQa (lanes 10–12), without sQb (lanes 13–15), or with wild-type Qc in place of GST-Qc (lanes 16–18). Protein inputs were 5 pmol of integrally bound R, 8 pmol each of sQa, sQb, and Qc, and 0.5 pmol of HOPS. These data are representative of three independent experiments, which are quantified in Supplemental Table S1.
SNARE complex disassembly
When proteoliposomes bearing R-SNARE and Ypt7:GTP that had been incubated the three sQ-SNAREs and with or without HOPS were further incubated with or without Sec 17, Sec 18, and ATP, the four-SNARE complex whose assembly required HOPS was largely disassembled by Sec 17/Sec 18/ATP, as judged by coflotation (Figure 6A, black vs. dark gray bars) or coimmunoprecipitation (Figure 6B), completing the cycle of SNARE complex assembly and disassembly.
FIGURE 6:
The four-SNARE complex is disassembled by Sec 17/Sec 18/MgATP. A 10× scale nucleotide exchange of Ypt7 and R-SNARE bearing RPLs and the sequential addition of the soluble SNAREs were completed as described for the flotation assay in Materials and Methods. Half of this mixture was incubated with HOPS for 30 min at 27°C, and the other half was incubated with HOPS buffer. Each of these tubes was then further divided in two and received either Rb150 or a mixture of Sec 17/18/MgATP. Final concentrations were 0.5 mM lipid, 1 mM EDTA, 0.1 mM GTP, 2 mM MgCl2 (3 mM MgCl2 for treatments containing Sec 17/18/MgATP), 0.2% BSA, 100 nM sQa, 400 nM sQb, 400 nM Qc, 23 nM HOPS, and 1 mM, 600 nM, and 100 nM ATP, Sec 17, and Sec 18, respectively, where added. Tubes were further incubated for 30 min in a 27°C water bath and then (A) 30 µl of each reaction was transferred to four snap-cap 1.5-ml conical tubes containing 90 µl of 54% Histodenz in isoosmolar Rb150 plus Mg and assayed for binding by flotation as described in Materials and Methods, and (B) 25 µl of each reaction was transferred into four snap-cap conical tubes containing 215 µl of buffer C and assayed for Qa association via α-Qa pull down as described in Materials and Methods. Protein inputs were 5 pmol of integrally bound R, 2 pmol of sQa, 8 pmol each of sQb and Qc, and 0.5 pmol of HOPS. All data represent the average and SD of three independent experiments.
DISCUSSION
SM proteins are as essential for fusion as SNAREs themselves. Despite extensive study (Rizo and Südhof, 2012), the mode of SM protein action has remained obscure. The vacuole/lysosome SM protein, Vps33, is a subunit of the HOPS complex, suggesting that its function may be integrated with other HOPS functions, such as tethering, SNARE binding to other non-SM subunits, as seen for the Qc-SNARE Vam7 (Krämer and Ungermann, 2011), and specific lipid associations (Stroupe et al., 2006). Although the capacity of SM proteins to bind Qa-SNAREs has been well described (Rizo and Südhof, 2012), it has been unclear how this promotes fusion, and recombinant SM proteins have even proven inhibitory in some fusion reconstitutions (Schollmeier et al., 2011).
It was reported (Baker et al., 2015) that Vps33 has two SNARE domain–binding sites that accommodate the Qa- and R-SNARE domain and that Vps33-bound Qa- and R-SNARE domains are helical, have parallel orientations, and are in register for SNARE domain zippering. However, the apolar residues of the R- and Qa-SNARE domains, when bound to Vps33, sit in grooves on Vps33 and would have to exit these grooves to assemble a four-SNARE complex in which apolar residues and 0-layers of these SNAREs face each other instead of the Vps33 (Baker et al., 2015). In addition, the Qc-SNARE is not bound by its SNARE domain to HOPS but instead via the affinity of its PX domain for Vps16 (Stroupe et al., 2006; Krämer and Ungermann, 2011). There has been no detection of direct affinity of the Qb-SNARE for HOPS. Many steps or aspects of trans-SNARE complex assembly thus remain obscure. Mutations in the binding sites of Vps33 for R- and Qa-SNAREs that disrupt binding of their SNARE domain peptides also block HOPS-mediated fusion when the Q-SNAREs are not initially fully assembled as a 3Q-SNARE complex (Baker et al., 2015). However, these SNARE-binding grooves on Vps33 are unnecessary, and HOPS is needed only as a tether when the fusing membranes bear 1R- and assembled 3Q-SNAREs. A role for HOPS in SNARE complex assembly was inferred in these studies by the indirect assay of downstream membrane fusion. HOPS might promote other aspects of fusion, such as lipid reorientation, in addition to any role in SNARE pairing. A physical measurement of SNARE complex formation has been needed to test directly whether HOPS catalyzes SNARE complex assembly.
We now provide direct physical evidence of a specific role of HOPS in bringing together the four vacuolar SNAREs. HOPS clustering of reconstituted proteoliposomes (RPLs) in this assay would not promote spontaneous SNARE complex assembly, as the RPLs bear only one SNARE, and the other SNAREs are soluble. The synergy of assembly among the sSNAREs, that is, that each is needed for optimal association of the other, indicates that the SNARE domains are interacting to form a canonical four-helical bundle rather than indicating four separate binding sites on HOPS that interact allosterically. The assembly of the four-SNARE complex requires an intact bilayer, which may simply reflect that Ypt7, phosphoinositide, and Nyv1 are otherwise diluted among detergent micelles.
Although our studies establish that HOPS catalyzes SNARE complex assembly, further work is needed to understand the associations and stoichiometries of the reaction products. HOPS solublized directly from vacuolar membranes sediments as a 65 S particle (Nakamura et al., 1997 ; Price et al., 2000; Seals et al., 2000) in association with at least the R- and Qa-SNAREs (Price et al., 2000). Although it has been unclear what other proteins in addition to SNAREs, if any, were also associated with HOPS to give this large size, the HOPS subunits in this vacuole-derived complex shifted to lower apparent mass after the particle was incubated with added Sec 18p and ATP (Price et al., 2000; Seals et al., 2000), consistent with disassembly of associated SNAREs. The SNARE complex we reconstituted here with recombinant components is also sensitive to disassembly by Sec 17, Sec 18, and ATP (Figure 6). Alternative analysis of the assembly reaction, through detergent solublization and pull down with bead-bound antibody to the Qa SNARE (Figure 4) or to a GST epitope tag on the Qc SNARE (Figure 5), also showed that there is substoichiometric HOPS associated with SNAREs. Further experiments will be required to determine the spatial and binding relationships among the SNAREs and with HOPS. On vacuoles, SNAREs are bound to HOPS or to Sec 17 but not to both (Collins et al., 2005).
Our working model is that the R-, Qa-, and Qc-SNAREs initially bind to their respective sites on HOPS. It is not known whether there is a preferred order or synergy among these bindings or whether the association of one SNARE to HOPS may trigger the release of another from its distinct HOPS binding site. The Qb-SNARE may not be directly recognized by HOPS but instead engage with the other three SNAREs as they are brought together on the HOPS surface; alternatively, the capacity of the Qa- and Qb-SNAREs to form a stable dimer (Fukuda et al., 2000) may mediate Qb associations with HOPS. Understanding the specific aspects of how HOPS promotes SNARE complex assembly may reveal the conserved mechanism of SM protein function. HOPS may be bound to one or more SNARE complex in-trans and remain bound or bind in-cis after fusion and before Sec 18/Sec 17/ATP-mediated disassembly.
MATERIALS AND METHODS
Protein purification
HOPS was purified as described (Zick and Wickner, 2013) with modifications. After affinity isolation via its GST tag, the tag was removed by overnight incubation with tobacco etch virus (TEV) protease, and 200 µl of HOPS was layered on a 3.8-ml linear gradient (30–10% sucrose in 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]-NaOH, pH 8.0, 0.4 M NaCl, 0.2 M sorbitol, 10% [vol/vol] glycerol, 0.004% Triton X-100, and 1 mM β-mercaptoethanol) and centrifuged (SW60, 3°C, 14 h, 55,000 rpm). Tubes were punctured from the bottom with a shortened 18-gauge steel spinal needle, and three drop fractions were collected. Fractions were analyzed for protein by the Bradford method and for activity by a lumenal compartment mixing assay (Zucchi and Zick, 2011; Supplemental Figure 1). Sec 17 (Schwartz and Merz, 2009), Sec 18 (Haas and Wickner, 1996), and Ypt7 (Zick and Wickner, 2013) were prepared as described. Vacuolar SNAREs were isolated (Mima et al., 2008; Schwartz and Merz, 2009; Zucchi and Zick, 2011), and Vti1 and Nyv1 were exchanged into octylglucoside (Zucchi and Zick, 2011). Vam7tm (Xu and Wickner, 2012) was purified as described. MBP-sVti1 and MBP-sVam3 were isolated as described (Zick and Wickner, 2013) and nutated at 4°C for 2 h with a ¼ molar ratio of TEV protease. GST-Vam7, which lacks a TEV cleavage site, was isolated as described (Fratti et al., 2007). The soluble hexahistidine (his6)-sNyv1 was purified as follows; pParallel his6-TEV-sNyv1 was transformed into Rosetta2(DE3) pLysS cells (Novagen 71403). A single colony was inoculated into 10 ml of LB (Difco LB [Luria–Bertani] Broth; Becton, Dickinson, Sparks, MD) plus 100 µg/ml ampicillin (Amp) and 37 µg/ml chloramphenicol (Cam), grown overnight at 37°C with shaking, and then transferred to 3 l of LB plus 100 µg/ml Amp and 37 µg/ml Cam in a 6-l flask. At OD600 ≈ 0.4, isopropyl-β-d-thiogalactoside was added to 0.5 mM, and cultures were moved to 30°C and shaken for 5 h. Cells were harvested by centrifugation, washed in 100 ml of lysis buffer (20 mM HEPES-NaOH, pH 7.8, 250 mM NaCl, 5% glycerol, 10 mM imidazole), and then resuspended in 30 ml of lysis buffer plus 1 mM phenylmethylsulfonyl fluoride and PIC (0.46 µg/ml leupeptin, 3.5 µg/ml pepstatin A, 2.4 µg/ml Pefabloc-SC). This suspension was frozen dropwise in liquid nitrogen, thawed the next day, and lysed via French press. The lysate was centrifuged (60 Ti, 4°C, 45 min, 55,000 rpm), and the supernatant was passed over a 3-ml nickel–nitriloacetic acid (Qiagen) column equilibrated with lysis buffer. The column was then washed with 20 ml of lysis buffer plus 500 mM NaCl and then 20 ml of lysis buffer. The protein was eluted with lysis buffer plus 250 mM imidazole. The protein peak was pooled and dialyzed against 20 mM HEPES-NaOH, pH 7.8, 125 mM NaCl, and 5% glycerol. The resulting protein was nutated at 4°C for 2 h with a ¼ molar ratio of TEV protease.
Proteoliposome preparation
Lipids in chloroform were brought to room temperature. Using Hamilton syringes, 919 µl of 25 mg/ml 18:2 1,2-dilinoleoyl-sn-glycero-3-phosphocholine in chloroform (68 mol%; Avanti Polar Lipids, Alabaster, AL), 418 µl of 25 mg/ml 18:2 1,2-dilinoleoyl-sn-glycero-3-phospho-l-serine (30 mol%; Avanti Polar Lipids), 413 µl of 1 mg/ml di-C16 phosphatidylinositol 3-phosphate in 1:2:0.8 chloroform:methanol:water (1 mol%; Echelon Biosciences, Salt Lake City, UT), and 115 µl of 5 mg/ml Lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt in chloroform (1 mol%; Invitrogen, Eugene, OR) were added to an 8-ml glass vial containing 756 µl of 0.5 M n-octyl-β-d-glucopyranoside (Affymetrix, Maumee, OH) in methanol. The lipid-detergent mixture was dried with a stream of nitrogen, followed by 3 h in a SpeedVac. The film was fully solubilized via nutation in 2.16 ml of 100 mM HEPES-NaOH, pH 7.4, 750 mM NaCl, 50% glycerol, and 5% MgCl2 plus 3.78 ml of water and distributed in 550-µl aliquots into 2-ml glass vials and stored at −80°C. Lipid-detergent preparations were thawed on ice, and transmembrane-bearing SNAREs were added at a 1:3000 protein-to-lipid molar ratio. The Rab Ypt7p was added at a 1:2000 protein-to-lipid molar ratio, and his6-TEV was added to each at 2 µM. Rb150 plus Mg (20 mM HEPES-NaOH, pH 7.4, 150 mM NaCl, 10% glycerol, 1 mM MgCl2) plus 1% β-octylglucoside was added to a final volume of 1 ml. Each 1-ml preparation, containing 4 mM lipid, ∼35 mM β-octylglucoside, 1.33 µM SNAREs (where added), 2 µM Rab (where added), and 2 µM his6-TEV, was gently mixed by inversion, pipetted into 25,000–molecular weight cutoff dialysis tubing with a flat width of 12 mm (Spectrum Laboratories, Rancho Dominguez, CA), and dialyzed against 250 ml of Rb150 plus Mg and 1 g of Bio-Beads SM-2 (Bio-Rad, Hercules, CA) at 4°C for at least 16 h in the dark. Proteoliposomes (∼1 ml) were harvested into 5-ml tubes on ice and mixed with 1 ml of 70% (wt/vol) Histodenz (Sigma-Aldrich, St. Louis, MO) in Rb150 plus Mg prepared with 2% glycerol to achieve isoosmolarity between the proteolipsomes and the density medium. Samples were mixed by gentle inversion and transferred into 11 × 60 mm Ultra-Clear ultracentrifuge tubes (Beckman Coulter, Brea, CA), overlaid with 25% (wt/vol) isoosmolar Histodenz (prepared by diluting 70% isoosmolar Histodenz with Rb150 plus Mg) to the 3.8-ml mark and then with 600 µl of Rb150 plus Mg, and centrifuged (SW60, 4°C, 1.5 h, 55,000 rpm). Proteoliposomes (400–600 µl) were harvested at the highest interface and assayed for lipid phosphorous (Chen et al., 1956). Samples were diluted with Rb150 plus Mg to 2 mM lipid phosphorus and frozen as aliquots in liquid nitrogen.
Flotation assay
Proteoliposomes prepared as described bore either no SNAREs or one integral SNARE either with or without Ypt7. The Ypt7-bearing liposomes were nucleotide exchanged by mixing 7.5 µl of liposomes in Rb150 plus Mg with 3 µl of 10 mM EDTA in Rb150 (20 mM HEPES-NaOH, pH 7.4, 150 mM NaCl, 10% glycerol) and 3 µl of 1 mM GTP in Rb150 and incubating for 10 min at 27°C. The nucleotide exchange was completed by adding 1.5 µl of 30 mM MgCl2 in Rb150, 0.6 µl of 10% defatted bovine serum albumin (BSA; Sigma-Aldrich) in Rb150, and 2.4 µl of Rb150, and the mixture was placed on ice. Liposomes without Ypt7 were treated identically. Soluble SNAREs, or their buffers, were added sequentially (2.4 µl each of 1.25 µM sQa and 5 µM sQb, Qc, and sR) where indicated, followed by 2.4 µl of 290 nM HOPS or its buffer, for a total volume of 30 µl. Reactions were incubated for 30 min at 27°C. We added 54% (wt/vol) Histodenz (90 µl) in isoosmolar Rb150 plus Mg and gently vortexed the reactions. Samples (80 µl) were transferred into 7 × 20 mm polycarbonate tubes (Beckman Coulter) and overlaid with 50 µl of 35% and then 50 µl of 30% Histodenz in isoosmolar Rb150 plus Mg, followed by 50 µl of Rb150 plus Mg. The remaining sample (40 µl) was solubilized with 1 µl of 5% (vol/vol) Thesit for lipid recovery determination. Reactions were centrifuged (TLA100, 4°C, 3 h, 100,000 rpm), and 80 µl was harvested from the top of the tube and solubilized with 2 µl of 5% Thesit. Lipid recovery was determined by measuring rhodamine fluorescence (excitation, 560 nm; emission, 580 nm; CO 570) of 10 µl of the starting and floated samples in a SpectraMax Gemini XPS fluorescence plate reader (Molecular Devices, Sunnyvale, CA). Floated samples, adjusted for percentage of lipid recovered, were analyzed for liposome-bound proteins by SDS–PAGE and immunoblot against a standard curve of the starting samples and quantified using UN-SCAN-IT software (Silk Scientific, Orem, UT).
Pull-down analysis
Proteoliposomes bearing the R-SNARE Nyv1 were prepared with or without Ypt7. For nucleotide exchange, 6.25 µl of 2 mM liposomes in Rb150 plus Mg were mixed with 2.5 µl of 10 mM EDTA in Rb150 and 2.5 µl of 1 mM GTP in Rb150 and incubated for 10 min at 27°C before adding 1.25 µl of 30 mM MgCl2 in Rb150, 3.0 µl of Rb150, and 0.5 µl of 10% defatted BSA in Rb150 and placing on ice. For detergent controls, samples received 1 µl of 25% (wt/vol) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) in Rb150; other samples received 1 µl of Rb150. Soluble SNAREs (2 µl of each at 5 µM) were added sequentially (sQa, sQb, wild-type Qc, or GST-Qc) where indicated, followed by 2 µl of 290 nM HOPS or its buffer, for a total volume of 25 µl. Reactions were incubated at 27°C for 30 min for the αQa pull downs and for 5, 30, or 90 min for the pull downs with glutathione beads and then moved to ice. For the αQa pull downs, 215 µl of ice-cold buffer C was added (20 mM HEPES-NaOH pH 7.4, 150 mM NaCl, 0.2% defatted BSA, 1% [wt/vol] CHAPS) along with 10 µl of 1 µg/µl affinity-purified αQa for a total volume of 250 µl. For the GST pull downs, 225 µl of ice-cold buffer C was added. A portion (200 µl) of each reaction was transferred to 1.5 ml of Eppendorf tubes containing either 10 µl of 10 mg/ml Pierce Protein A Magnetic Beads (Thermo Scientific, Rockford, IL) or 2 µl of settled volume of Pierce Glutathione Magnetic Beads previously washed three times in 1 ml of buffer C. The remaining starting sample (50 µl) was boiled for 5 min in sample buffer (2% SDS, 5% glycerol, 50 mM Tris Cl, pH 6.8, 20 mM dithiothreitol). Samples with beads were nutated end over end for 2 h at room temperature. Unbound samples were discarded, and the beads were washed three times in 1 ml of buffer C. The beads were suspended in 100 µl of sample buffer and boiled for 5 min. Samples were analyzed for bound proteins by SDS–PAGE and immunoblot and quantified using UN-SCAN-IT software.
Supplementary Material
Acknowledgments
We thank Deborah Douville for expert assistance and Charles Barlowe and Gustav Lienhard for discussions. This work was supported by National Institutes of Health Grants R01 GM23377-40 and R35GM118037-01 to W.W., R35-GM119455 to A.N.K., and T32-GM008704 to S.R.
Abbreviations used:
- HOPS
homotypic fusion and vacuole protein sorting complex
- SNARE
soluble N-ethylmaleimide–sensitive factor attachment protein receptor.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.975) on February 1, 2017.
REFERENCES
- Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM. A direct role for the Sec 1/Munc18-family protein Vps33 as a template for SNARE assembly. Science. 2015;349:1111–1114. doi: 10.1126/science.aac7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett CL, Plemel RL, Lobingier BT, Vignali M, Fields S, Merz AJ. Efficient termination of vacuolar Rab GTPase signaling requires coordinated action by a GAP and a protein kinase. J Cell Biol. 2008;182:1141–1151. doi: 10.1083/jcb.200801001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bröcker C, Kuhlee A, Gatsogiannis C, Kleine Balderhaar HJ, Hönscher C, Engelbrecht-Vandré S, Ungermann C, Raunser S. Molecular architecture of the multisubunit homotypic fusion and vacuole protein sorting (HOPS) tethering complex. Proc Natl Acad Sci USA. 2012;109:1991–1996. doi: 10.1073/pnas.1117797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheever ML, Sato TK, deBeer T, Kutateladze TG, Emr SD, Overduin M. Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat Cell Biol. 2001;3:613–618. doi: 10.1038/35083000. [DOI] [PubMed] [Google Scholar]
- Chen PS, Toribara TY, Warner H. Microdetermination of phosphorus. Anal Chem. 1956;28:1756–1758. [Google Scholar]
- Collins KM, Thorngren NL, Fratti RA, Wickner WT. Sec 17 and HOPS, in distinct SNARE complexes, mediate SNARE complex disruption or assembly for fusion. EMBO J. 2005;24:1775–1786. doi: 10.1038/sj.emboj.7600658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins KM, Wickner WT. trans-SNARE complex assembly and yeast vacuole membrane fusion. Proc Natl Acad Sci USA. 2007;104:8755–8760. doi: 10.1073/pnas.0702290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D, Sutton RB, Brunger AT, Jahn R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R- SNAREs. Proc Natl Acad Sci USA. 1998;95:15781–15786. doi: 10.1073/pnas.95.26.15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratti RA, Collins KM, Hickey CM, Wickner W. Stringent 3Q 1R composition of the SNARE 0-Layer can be bypassed for fusion by compensatory SNARE mutation or by lipid bilayer modification. J Biol Chem. 2007;282:14861–14867. doi: 10.1074/jbc.M700971200. [DOI] [PubMed] [Google Scholar]
- Fratti RA, Jun Y, Merz AJ, Margolis N, Wickner W. Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles. J Cell Biol. 2004;167:1087–1098. doi: 10.1083/jcb.200409068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda R, McNew JA, Weber T, Parlati F, Engel T, Nickel W, Rothman JE, Söllner TH. Functional architecture of an intracellular membrane t-SNARE. Nature. 2000;407:198–202. doi: 10.1038/35025084. [DOI] [PubMed] [Google Scholar]
- Grosshans BL, Ortiz D, Novick P. Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci USA. 2006;103:11821–11827. doi: 10.1073/pnas.0601617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A, Conradt B, Wickner W. G-protein ligands inhibit in vitro reactions of vacuole inheritance. J Cell Biol. 1994;126:87–97. doi: 10.1083/jcb.126.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A, Scheglmann D, Lazar T, Gallwitz D, Wickner W. The GTPase Ytp7p of Saccharomyces cerevisiae is required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. EMBO J. 1995;14:5258–5270. doi: 10.1002/j.1460-2075.1995.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A, Wickner W. Homotypic vacuole fusion requires Sec 17p (yeast alpha-SNAP) and Sec 18p (yeast NSF) EMBO J. 1996;15:3296–3305. [PMC free article] [PubMed] [Google Scholar]
- Hickey CM, Wickner W. HOPS initiates vacuole docking by tethering membranes before trans-SNARE complex assembly. Mol Biol Cell. 2010;21:2297–2305. doi: 10.1091/mbc.E10-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer L, Ungermann C. HOPS drives vacuole fusion by binding the vacuolar SNARE complex and the Vam7 PX domain via two distinct sites. Mol Biol Cell. 2011;22:2601–2611. doi: 10.1091/mbc.E11-02-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, Wickner W, Haas A. Sec 18p (NSF)-driven release of Sec 17p (α-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Mima J, Hickey CM, Xu H, Jun Y, Wickner W. Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones. EMBO J. 2008;27:2031–2042. doi: 10.1038/emboj.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Hirata A, Ohsumi Y, Wada Y. Vam2/Vps41 and Vam6/Vps39 are components of a protein complex on the vacuolar membranes and involved in the vacuolar assembly in the yeast Saccharomyces cerevisiae. J Biol Chem. 1997;272:11344–11349. doi: 10.1074/jbc.272.17.11344. [DOI] [PubMed] [Google Scholar]
- Price A, Seals D, Wickner W, Ungermann C. The docking stage of yeast vacuole fusion requires the transfer of proteins from a cis- SNARE complex to a Rab/Ypt protein. J Cell Biol. 2000;148:1231–1238. doi: 10.1083/jcb.148.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Südhof TC. The membrane fusion enigma: SNAREs, Sec 1/Munc18 proteins, and their accomplices—guilty as charged? Annu Rev Cell Dev Biol. 2012;28:279–308. doi: 10.1146/annurev-cellbio-101011-155818. [DOI] [PubMed] [Google Scholar]
- Schollmeier Y, Krause JM, Kreye S, Malsam J, Söllner TH. Resolving the function of distinct Munc18-1/SNARE protein interaction modes in a reconstituted membrane fusion assay. J Biol Chem. 2011;286:30582–30590. doi: 10.1074/jbc.M111.269886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz ML, Merz AJ. Capture and release of partially zipped trans-SNARE complexes on intact organelles. J Cell Biol. 2009;185:535–549. doi: 10.1083/jcb.200811082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DF, Eitzen G, Margolis N, Wickner WT, Price A. A Ypt/Rab effector complex containing the Sec 1 homolog Vps33 is required for homotypic vacuole fusion. Proc Natl Acad Sci USA. 2000;97:9402–9407. doi: 10.1073/pnas.97.17.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroupe C, Collins KM, Fratti RA, Wickner W. Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J. 2006;25:1579–1589. doi: 10.1038/sj.emboj.7601051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroupe C, Hickey CM, Mima J, Burfeind AS, Wickner W. Minimal membrane docking requirements revealed by reconstitution of Rab GTPase-dependent membrane fusion from purified components. Proc Natl Acad Sci USA. 2009;106:17626–17633. doi: 10.1073/pnas.0903801106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Ohsumi Y, Anraku Y. Genes for directing vacuolar morphogenesis in Saccharomyces cerevisiae. I. Isolation and characterization of two classes of vam mutants. J Biol Chem. 1992;267:18665–18670. [PubMed] [Google Scholar]
- Wang L, Merz AJ, Collins KM, Wickner W. Hierarchy of protein assembly at the vertex ring domain for yeast vacuole docking and fusion. J Cell Biol. 2003;160:365–374. doi: 10.1083/jcb.200209095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Seeley ES, Wickner W, Merz AJ. Vacuole fusion at a ring of vertex docking sites leaves membrane fragments within the organelle. Cell. 2002;108:357–369. doi: 10.1016/s0092-8674(02)00632-3. [DOI] [PubMed] [Google Scholar]
- Wickner W, Schekman R. Membrane fusion. Nat Struct Mol Biol. 2008;15:658–664. doi: 10.1038/nsmb.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wickner W. N-terminal domain of vacuolar SNARE Vam7p promotes trans-SNARE complex assembly. Proc Natl Acad Sci USA. 2012;109:17936–17941. doi: 10.1073/pnas.1216201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu IM, Hughson FM. Tethering factors as organizers of intracellular vesicular traffic. Annu Rev Cell Dev Biol. 2010;26:137–156. doi: 10.1146/annurev.cellbio.042308.113327. [DOI] [PubMed] [Google Scholar]
- Zick M, Wickner W. The tethering complex HOPS catalyzes assembly of the soluble SNARE Vam7 into fusogenic trans-SNARE complexes. Mol Biol Cell. 2013;24:3746–3753. doi: 10.1091/mbc.E13-07-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchi PC, Zick M. Membrane fusion catalyzed by a Rab, SNAREs, and SNARE chaperones is accompanied by enhanced permeability to small molecules and by lysis. Mol Biol Cell. 2011;22:4635–4646. doi: 10.1091/mbc.E11-08-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.