

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a deadly malignancy that often presents clinically at an advanced stage and that may be confused with chronic pancreatitis (CP). Conversely, CP may be misdiagnosed as PDAC leading to unwarranted pancreas resection. Therefore, early PDAC diagnosis and clear differentiation between PDAC and CP are crucial for improved care. Exosomes are circulating micro-vesicles whose components can serve as cancer biomarkers. We compared exosomal glypican-1 (GPC1) and microRNA levels in normal control subjects and in patients with PDAC and CP. We report that exosomal GPC1 is not diagnostic for PDAC, whereas high exosomal levels of microRNA-10b, (miR-10b), miR-21, miR-30c, and miR-181a and low miR-let7a readily differentiate PDAC from normal control and CP samples. By contrast with GPC1, elevated exosomal miR levels decreased to normal values within 24 h following PDAC resection. All 29 PDAC cases exhibited significantly elevated exosomal miR-10b and miR-30c levels, whereas 8 cases had normal or slightly increased CA 19-9 levels. Thus, our exosomal miR signature is superior to exosomal GPC1 or plasma CA 19-9 levels in establishing a diagnosis of PDAC and differentiating between PDAC and CP.
Keywords: Glypican-1, MicroRNAs, Exosomes, Pancreatic cancer
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a deadly malignancy characterized by an abundant stroma, marked resistance to chemotherapy and radiotherapy, and advanced stage at clinical presentation precluding resection in 80% of patients [1,2]. Consequently, the 5-year survival rate is only 9% [3]. Moreover, PDAC is currently the third leading cause of cancer death in the United States, and is projected to become the second leading cause of cancer death by 2020 [3,4]. There is an urgent need, therefore, to devise strategies for early PDAC detection to allow for the potential of a surgical cure. This has been an elusive goal due to the absence of sufficiently specific and sensitive biomarkers.
Heparan sulfate proteoglycans (HSPGs) are ubiquitous macromolecules that are found on cell surfaces and in the extracellular matrix [5]. They consist of core proteins to which are covalently attached glycosaminoglycans (GAGs) polysaccharide chains that are variable in size and type. GAGs consist of disaccharide repeats, most often of D-glucuronic or L-iduronic acid and either N-acetylglucosamine or N-acetylgalactosamine [5,6]. GAGs exhibit marked diversity due to variations in the disaccharides and sites of sulfation [6]. In addition to heparin and heparan sulfate (HS), GAGs may consist of keratan, chondroitin or dermatan sulfates, and hyaluronan [6,7]. Two families of HSPGs, glypicans and syndecans, carry the majority of the HS on mammalian cells [8–10]. There are six glypican genes encoding glypican proteins, which are attached to the plasma membrane via glycophosphatidyl inositol anchors, and four syndecan genes encoding four transmembrane proteins [8–10].
We previously reported that glypican-1 (GPC1) expression (but not other glypicans) is upregulated in PDAC, and that heparin-binding growth factors require its presence on pancreatic cancer cell (PCC) membranes in order to enhance proliferation and migration [11]. We also reported that high GPC1 levels promote angiogenesis and metastasis in an orthotopic mouse model of PDAC [12], and that in an oncogenic KRAS-driven genetically engineered mouse model of PDAC in which the Ink4a locus has been deleted, the absence of GPC1 is associated with attenuated angiogenesis and tumor growth [13].
HSPGs such as glypicans and syndecans can also act as receptors that internalize exosomes [14]. Conversely, during the internalization process, HSPGs are taken up by exosomes [14]. Recently, exosome-associated GPC1 has been reported as a highly specific and sensitive biomarker for early PDAC detection [15]. Further validation of this observation is required since highly specific anti-GPC1 antibodies are not readily available.
MicroRNAs (miRs) are small, non-coding RNAs that are highly conserved and that modulate developmental, physiological and pathological cellular processes by repressing mRNA translation [16,17]. In addition, miRs exert crucial roles in numerous cancers [18–20]. Due to their marked stability miRs may serve as diagnostic biomarkers in the systemic circulation [21]. Moreover, miRs are present in circulating exosomes where they remain stable under appropriate storage conditions [22,23]. By in situ hybridization using highly specific locked nucleic acid probes, we previously reported that miR-10b and miR-21 are abundant in the cancer cells in PDAC [24,25]. We subsequently showed that miRs are present in the circulation, and that high plasma miR-10b levels are seen in patients with PDAC but not in patients with chronic pancreatitis (CP) or in normal controls [26].
We now describe a method to quantitatively analyze exosomal GPC1 by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Using the peptide QIYGAK that is unique for GPC1 and is not shared with glypican-2, -3, -4, -5, or -6 or with other human proteins, we demonstrate that this novel method is rapid, sensitive, and robust, enabling efficient high-throughput analysis of clinical samples. Using exosomes prepared from the plasma of individuals without pancreatic disease and from patients with PDAC and CP prior to and following pancreas resection, we compared the levels of GPC1, miR-10b, -21, -30c, -106b, -181a, -483, -20, -let7a, and -122a and assessed their utility as potential PDAC biomarkers. GPC1 levels were relatively similar in exosomes derived from normal controls, and patients with PDAC or CP. By contrast, exosomal levels of miR-10b, -20a, -21, -30c, -106b, and -181a were significantly higher in PDAC by comparison with corresponding control and CP levels, and the elevated levels of all six miRs normalized following PDAC resection, whereas the levels of exosomal GPC1 were only slightly lower post-resection.
Materials and methods
Materials
Light peptide QIYGAK (LG021) was purchased from Peptide 2.0 Inc. (Chantilly, VA, USA). Isotopically labeled heavy peptide QIYGAK* (Lysine, 13C6, 15N2) (HG021) used as an internal standard was obtained from New England Peptide Inc. (Gardner, MA, USA). Bovine serum albumin (BSA) was purchased from Sigma–Aldrich (St. Louis, MO, USA). Phosphate Buffered Saline (PBS-1X) was obtained from Lonza (Allendale, NJ). High pressure liquid chromatography HPLC grade water (H2O), acetonitrile (ACN), 0.1% formic acid in ACN, and 0.1% formic acid in water (H2O) were all purchased from Burdick & Jackson (Muskegon, MI, USA).
Patient samples
Individuals with PDAC were treatment-naïve prior to undergoing resection, and the diagnosis of PDAC required cytopathological confirmation. Individuals with CP were classified by the Cambridge criteria [27,28] based on endoscopic retrograde cholangiopancreatography (ERCP). Control subjects had previously undergone normal cross sectional imaging of the pancreas as part of an evaluation of abdominal symptoms that did not reveal the presence of cancer. Prior to subject enrollment, the Indiana University Institutional Review Board approved this study protocol and each subject signed informed consent. Blood (10–20 ml) was collected into EDTA-coated tubes prior to surgery and approximately 24 h following surgery for all patients with PDAC (n = 29) or CP (n = 11), and during a clinic visit in the case of the six control subjects. Specimens were promptly placed on ice or in a refrigerator (4°C), taken to the lab and rapidly processed by centrifugation (1000 × g for 10 min) at 4°C (within 1 h of collection). Plasma supernatants were collected and stored at −8°C until analysis.
We analyzed GPC1 levels in exosomes from the plasma of six normal control subjects who did not have pancreatic disease, as well as from pre-operative and post-operative exosomes from 3 patients with CP and from 3 patients with PDAC. To prepare exosomes, plasma samples were thawed on ice and centrifuged at 10,000 × g for 30 min at 4 °C. Supernatants were filtered through a 0.22 mm filter using a syringe and the filtrate (250 μL) was centrifuged twice at 110,000 × g for 2 h (4 °C) with an intervening PBS wash [29]. Exosomes were suspended in 100 μL of 1 M Tris (pH 7.4) for analysis. An aliquot of the exosome-rich solution was diluted 1:10 in PBS for diameter and concentration quantitation, utilizing the qNano system and the NP100 nanopore, both from Izon Science (Cambridge, MA), under optimized principles of tunable resistive pulse sensing (RSP) as provided by Izon Science [30,31].
The diameter of the vast majority of normal control exosomes ranged from 60 to 100 nm (Supplemental Fig. S1), whereas the diameter of CP (Supplemental Fig. S2) and PDAC exosomes ranged from 70 to 120 nm (Supplemental Fig. S3). Importantly, the smallest exosomes were at the lowest limit of detection by RSP. Exosome purity was confirmed by standard electron microscopy (EM) (Supplemental Fig. S4). The diameter of the majority of exosomes ranged from 54 to 64 nm, which is consistent with imaging by this method due to surface adherence, and changes caused by adherence, fixation and dessication [29,30]. For immunoblotting, exosomes were suspended in exosome lysis buffer [15] and Western blotting was performed [32] with an anti-CD63 antibody (Santa Cruz). As expected, exosomes expressed the exosomal marker CD63 (Supplemental Fig. S5).
Preparation of standards and quality control samples for LC-MS/MS
A stock standard solution of LG021 (“light”, non-labeled analyte) was prepared at a concentration of 30 mM using the appropriate weight for lot purity correction and 50% acetonitrile (ACN) in water with 0.1% formic acid. Duplicate solutions (Set 1 and Set 2) were compared before they were used for preparing intermediate standard solutions. The intermediate standard solutions of LG021 were prepared from Set 1 of the serial dilution stock standard solutions at concentrations of 3.0, 2.4, 1.2, 0.96, 0.48, 0.24, 0.12, and 0.06 μM for the preparation of calibration curves. Intermediate quality control (QC) standard solutions of LG021 were prepared from Set 2 of the serial dilution stock standard solutions at concentrations of 1.5, 0.6, 0.3, and 0.06 μM for the preparation of quality controls, using HPLC grade water with 0.1% formic acid by dilution from the stock standard solutions. The calibration standard samples of LG021 were prepared by spiking the corresponding intermediate calibration standard solutions of 3.0, 2.4, 1.2, 0.96, 0.48, 0.24, 0.12, and 0.06 μM, respectively, into digested BSA to provide concentrations of 0.50, 0.40, 0.02, 0.16, 0.08, 0.04, 0.02, and 0.01 μM, respectively. The QC standard samples of LG021 were prepared at concentrations of 0.25, 0.10, 0.05, and 0.01 μM, by spiking digested BSA with the intermediate QC standard solutions.
In addition to the above, a stock internal standard solution of HG021 (“heavy”, stable-isotope labeled analyte) was prepared at a concentration of 30 mM using the appropriate weight for lot purity correction and 50% ACN in water with 0.1% formic acid. Working internal standard solutions were prepared from the stock internal standard solution at a concentration of 0.75 μM.
Sample preparation and chromatographic conditions for LC-MS/MS
Samples were prepared as follows: 50 μL of each sample (1% BSA and clinical patient samples) were dried in a Speed-vac for 1 h and reconstituted with 50 μL of 8 M urea. Dithiothreitol (50 μL of 10 mM stock solution) was added to reduce disulfide bonds. The mixture was then incubated at 60 °C for 60 min prior to adding 50 μL of 55 mM iodoacetamide in 100 mM NH4HCO3. The mixture was then incubated in the dark for 60 min (room temperature), followed by the addition of trypsin (50 μL of 0.1 μg/μL stock solution) to digest the proteins and an 18 h incubation at 37 °C.
After the tryptic digestion, 60 μL of each patient sample were spiked with 10 μL of 0.75 μM internal standard; 50 μL of each calibration standard and QC sample were spiked with 10 μL of each intermediate calibration standard solution and 10 μL of 0.75 μM internal standard. Each sample was mixed, cleaned up with C18 Silica Macro Spincolumns (Fisher Scientific, Waltham, MA, Cat. #: NC9840653), filtered with Ultrafree-MC centrifugal filter units with microporous membrane (Millipore, Billerica, MA, Cat. #: UFC30GVNB), transferred to a vial, and prepared for mass spec analysis.
A Dionex Ultimate 3000 UHPLC system (Sunnyvale, CA) consisting of a dual pump, autosampler, column oven, and a UV detector was used for peptide separation. A SCIEX 4000 QTRAP triple quadrupole mass spectrometer (Toronto, Canada) with a TurboIonSpray probe was used in positive ion mode for multiple reaction monitoring (MRM). SCIEX Analyst 1.5 was used for data collection and SCIEX MultiQuant v3.0.1 was used for peak integration and peptide concentration calculation.
HPLC separation was performed on a Waters XBridge BEH C8 XP Column (2.1 × 100 mm, 2.6 μm) (Cat. #: 186006043, Milford, MA) at 35 °C. The mobile phase A was 0.1% formic acid in water and the mobile phase B was 0.1% formic acid in ACN. The gradient was 0.0–1.6 min, 5% B; 1.6–1.61 min, gradient to 16% B; 1.6–4.0 min, gradient to 25% B; 4.0–4.7 min, gradient to 80% B; 4.7–5.3 min, 80% B; and 5.4–7.0 min, 5% B. The flow rate was set at 0.3 ml/min and the injection volume was 10 μL.
MS/MS detection and validation procedure
Scheduled MRM (sMRM) transitions monitored for each analyte (precursor ion/ product ion in m/z) were: LG021, 340.2/275.2 at retention time (RT) 3.30 min; HG021, 344.2/283.2 at RT 3.30 min. Ionization source and shared compound parameters were as follows: CUR, 40; CAD, 7; IS, 5500; TEM, 500; GS1, 50; and GS2, 50. DP, EP, CE, and CXP were 57/10/19.5/14, and 57/7/20/14 for LG021 and HG021, respectively.
Validations were performed according to the Guidance for Industry: Bioanalytical Methods Validation issued (May 2001) by the U.S. Department of Health and Human Services, the Food and Drug Administration, and the Centers for Drug Evaluation and Research and for Veterinary Medicine (http://www.fda.gov/downloads/Drugs/Guidance/ucm070107.pdf). Calibration curves and linearity, intra-assay and inter-assay precision and accuracy, selectivity, matrix effect, spike-recovery, and stability were performed as described in our previous publication [33].
Reverse-transcriptase quantitative PCR
Total exosomal RNA was isolated using Trizol-LS (Life Technologies, Carlsbad, CA, USA) and Direct-zol RNA MiniPrep kit (Zymo Research). cDNA was generated using 10 ng of RNA per reaction for miR-10b, -20a, -21, -30c, -106b, -122, -181a, -483, -let7a, and -425-5p RT primers and a miR reverse transcription kit (Life Technologies), as previously reported [26]. Reverse transcriptase quantitative PCR (RT-qPCR) was performed in duplicate for each miR using the corresponding miR Taqman® probes and TaqMan Fast Advanced Master Mix. Expression was normalized to miR-425-5p, and the ΔCt values for miRNAs in 6 control samples were averaged and subtracted from the ΔCt values of each sample, yielding a ΔΔCt value. The levels of each miRNA were calculated using the 2−ΔΔCt method [34].
CA 19-9 assay
CA 19-9 is the only FDA-approved biomarker for PDAC management. To compare the utility of exosomal miR levels with CA19-9 levels in establishing the diagnosis of PDAC, plasma CA19-9 was measured in duplicate by ELISA using CA19-9 AccuBind ELISA Microwells (Monobind Inc, Lake Forest, CA) according to the manufacturer’s instructions.
Statistics
Single-factor ANOVA was performed for RT-qPCR and LC-MS/MS data analysis, followed by Bonferroni adjustment. When indicated, paired t-tests were performed to compare pre- and post-surgery values. In addition, predictive performance was assessed using the Receiver Operating Characteristic (ROC) and the area under the curve (AUC). Excellent accuracy was defined as AUC>0.90.
Results
MS/MS detection
The molecular weight of the two standards, LG021 (MW: 678.4) and HG021 (MW: 686.4), has a difference of 8 Da. Their precursor ion (2+, 340.2 vs. 344.2) mass has a difference of 4 Da. Their unique product ion (1+, 275.2 vs. 283.2) has a difference of 8 Da. Thus, we were able to target these pairs of precursor and product ions without interference from each other. Representative chromatograms of the LC-MS/MS analysis using the chosen transitions from a patient sample are shown in Fig. 1.
Fig. 1.
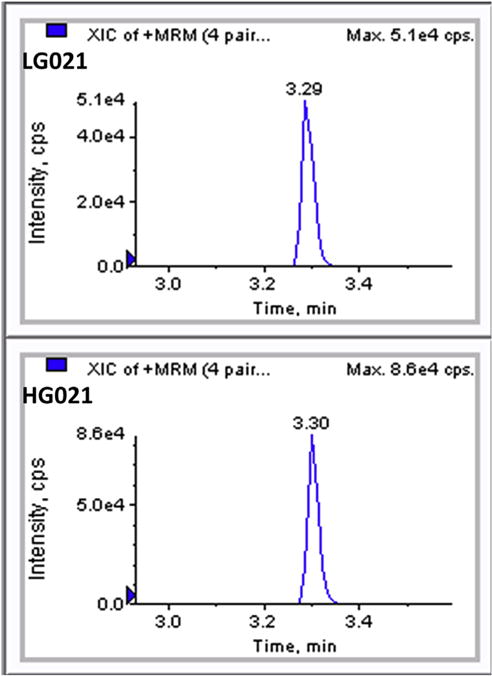
Representative chromatograms of the current LC-MS/MS analysis using the chosen transitions from a patient sample spiked with internal standards. Peak intensity and elution times (min) are shown for authentic analyte (LG021) and internal standard (HG021).
Calibration curve, linearity, precision and accuracy
The calibration curves from three consecutive batches using BSA as blank matrix showed an overall accuracy of 96.0–107.0% with relative standard deviation (RSD) of no more than 6.4% (Table 1). The linear range of LG021 was 10.0 nM–500.0 nM. The lower limit of quantification was 10.0 nM. The intra- and inter-assay precision and accuracy from three consecutive batches using BSA as a blank matrix are shown in Table 2. The intra-assay accuracy is 93.7–111.5% with RSD of 0.7–10.1%. The inter-assay accuracy is 96.0–104.4% with RSD of 2.0–11.0%.
Table 1.
Precision and accuracy of calibration standards of GPC1 in 1% BSA from three validation batches.
| Analysis group | Theoretical concentration (nM)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 10.0 | 20.0 | 40.0 | 80.0 | 160.0 | 200.0 | 400.0 | 500.0 | |
|
| ||||||||
| Measured concentration (nM) | ||||||||
| 001 | 11.0 | 20.0 | 36.0 | 80.0 | 150.0 | 196.0 | 400.0 | 506.0 |
| 002 | 11.0 | 20.0 | n/a | 76.0 | 157.0 | 199.0 | 407.0 | 501.0 |
| 003 | 11.0 | 19.0 | n/a | 76.0 | 170.0 | 199.0 | 396.0 | 499.0 |
| n | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| Mean | 11.0 | 19.7 | 36.0 | 77.3 | 159.0 | 198.0 | 401.0 | 502.0 |
| RSD (%) | 0.0 | 2.9 | n/a | 3.0 | 6.4 | 0.9 | 1.4 | 0.7 |
| Accuracy (%) | 110.0 | 98.3 | 90.0 | 96.7 | 99.4 | 99.0 | 100.3 | 100.4 |
Table 2.
Precision and accuracy of quality control samples for GPC1 in 1% BSA from three validation batches.
| Analysis group | Statistics | Theoretical concentration (nM)
|
|||
|---|---|---|---|---|---|
| 10.0 | 50.0 | 100.0 | 250.0 | ||
| n | 6 | 6 | 6 | 6 | |
| 001 | Intra-assay mean | 11.0 | 49.0 | 98.0 | 246.0 |
| RSD (%) | 10.1 | 2.3 | 2.5 | 1.4 | |
| Accuracy (%) | 107.2 | 97.6 | 98.4 | 98.4 | |
| n | 6 | 6 | 6 | 6 | |
| 002 | Intra-assay mean | 11.00 | 48.00 | 96.00 | 247.00 |
| RSD (%) | 10.1 | 0.7 | 1.8 | 1.5 | |
| Accuracy (%) | 111.5 | 96.3 | 96.0 | 99.0 | |
| n | 6 | 6 | 6 | 6 | |
| 003 | Intra-assay mean | 9.0 | 48.0 | 94.0 | 247.0 |
| RSD (%) | 3.8 | 1.8 | 1.3 | 2.2 | |
| Accuracy (%) | 94.9 | 95.3 | 93.7 | 98.7 | |
| n | 18 | 18 | 18 | 18 | |
| Overall | Inter-assay mean | 10.0 | 48.0 | 96.0 | 247.0 |
| RSD (%) | 11.0 | 2.0 | 2.7 | 1.6 | |
| Accuracy (%) | 104.4 | 96.4 | 96.0 | 98.6 | |
Selectivity and matrix effect
Selectivity of lower QCs was determined using BSA as a blank matrix, yielding an accuracy of 100.0% with a relative standard deviation of 0.0% (Supplemental Table 1). Matrix effect of lower QCs was determined using BSA as blank matrix, exhibiting −12.5% for LG021 and −12.4% for HG021.
Spike-recovery and stability
Overall recoveries from low, mid, and high QCs using BSA as a blank matrix were 73.5% for LG021 and 64.8% for HG021, respectively (Table 3). The results indicate that a consistent recovery has been achieved at all three QC levels of the analyte and its internal standard. The analyte was stable in BSA under different conditions (Table 4). The accuracy and precision of 72-hr processed sample reinjection and three freeze/thaw cycles satisfy the ±15% criteria. However, the examination of 48-hr room temperature storage stability shows the analyte was not stable at room temperature for 48 h.
Table 3.
Recoveries at low, mid and high QC levels for GPC1 and the IS in 1% BSA.
| QC levels | Statistics | GPC1 | IS |
|---|---|---|---|
| Low | RSD (%)a | 21.3/5.6 | 20.7/6.2 |
| Recovery (%) | 50.8 | 49.3 | |
| Mid | RSD (%)a | 24.0/40.4 | 23.5/33.3 |
| Recovery (%) | 99.3 | 79.0 | |
| High | RSD (%)a | 31.3/43.3 | 28.4/41.0 |
| Recovery (%) | 70.4 | 65.9 | |
| Overall | Recovery (%) | 73.5 | 64.8 |
RSD: RSD of six replicates of extracted QC samples/RSD of three replicates of recovery samples.
Table 4.
Stability of GPC1 at low, mid, and high QC levels in 1% BSA under different conditions.
| QC levels | Statistics | GPC1 |
|---|---|---|
| 72 h 4 °C processed-sample reinjection | ||
| n | 6 | |
| Low | RSD (%) | 0.0 |
| Accuracy (%) | 100.0 | |
| n | 6 | |
| Mid | RSD (%) | 4.0 |
| Accuracy (%) | 101.7 | |
| n | 6 | |
| High | RSD (%) | 3.3 |
| Accuracy (%) | 98.7 | |
| Three freeze/thaw matrix stability | ||
| n | 6 | |
| Low | RSD (%) | 9.8 |
| Accuracy (%) | 83.3 | |
| n | 6 | |
| Mid | RSD (%) | 0.0 |
| Accuracy (%) | 90.0 | |
| n | 6 | |
| High | RSD (%) | 3.6 |
| Accuracy (%) | 94.0 | |
| 48 h room temperature matrix stability | ||
| n | 6 | |
| Low | RSD (%) | 14.4 |
| Accuracy (%) | 56.7 | |
| n | 6 | |
| Mid | RSD (%) | 8.4 |
| Accuracy (%) | 48.3 | |
| n | 6 | |
| High | RSD (%) | 9.8 |
| Accuracy (%) | 50.0 | |
Clinical samples
The mean ± SE level of GPC1 protein in the 6 control samples was 56.8 ± 13.0 nM (Table 5). By contrast, prior to cancer resection, the corresponding GPC1 levels in the 3 cancer samples and the 3 CP samples were 88.7 ± 15.2 nM and 44.3 ± 9.3 nM, respectively. Approximately 24 h following surgery mean ± SE GPC1 levels decreased to 73.3 ± 21.2 nM and 33.3 ± 3.2 nM in the PDAC and CP samples, respectively (Table 5). As determined by one-way ANOVA, GPC1 levels were not significantly different between normal, PDAC and CP samples, pre-resection, or between PDAC and CP post-resection (Table 5). There was considerable overlap in exosomal GPC1 levels between normal, CP, and PDAC groups (Fig. 2). Moreover, the AUC was 0.75 when comparing PDAC and control samples (Supplemental Fig. S6), with p = 0.09 and p = 0.13 by paired t-tests, respectively, vs normal control. Thus, the predictive value of GPC1 levels for PDAC was fair at best, and clearly below our cutoff for excellence (defined as AUC>0.90).
Table 5.
GPC1 levels in human exosomes.
| Analysis group | GPC1 Levels (nM, mean ± SE) |
|---|---|
| Normal controls | 56.8 ± 13.0 |
| PDAC | 88.7 ± 15.2 |
| Pre-surgery | |
| PDAC | 73.3 ± 21.2 |
| Post-surgery | |
| CP | 44.3 ± 9.3 |
| Pre-surgery | |
| CP | 33.3 ± 3.2 |
| Post-surgery |
Exosomes were obtained from 6 normal controls, 3 PDAC cases pre- and post-surgery, and 3 CP cases pre- and post-surgery.
Fig. 2.
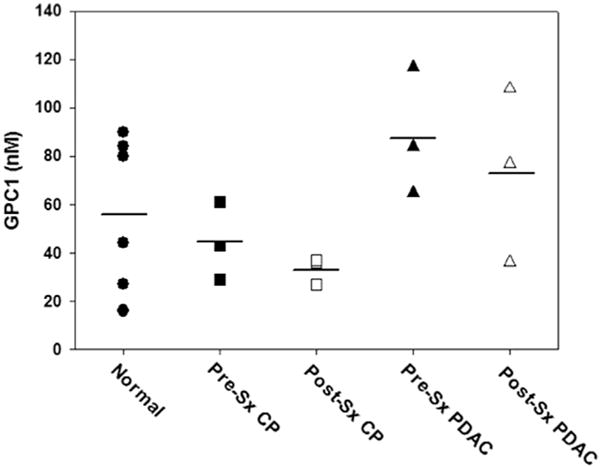
Dot plot of exosomal glypican-1 levels in normal controls, pancreatic cancer, and chronic pancreatitis. Glypican-1 (GPC1) levels were measured by LC-MS/MS using exosomes isolated from6 normal controls (circles), 3 patientswith pancreatic cancer (PDAC; squares), and 3 patients with chronic pancreatitis (CP; triangles). Pre-surgery (Pre-Sx; solid symbols) and post-Sx (open symbols) sampleswere assayed separately. Horizontal lines: mean values. There were no significant differences between the three groups, or between the pre- and post-surgery samples, either by ANOVA or paired t-tests.
RT-qPCR was next used to determine the levels of specific miRs in exosomes from the above samples that included the 6 normal controls, 26 additional PDAC patients (total = 29) and 8 additional CP patients (total = 11). The levels of miR-10b, -20a, -21, -30c, -106b, -181a, -483, -20, -let7a, and -122 were assayed in exosomes and plasma, since these miRs have either been implicated in PDAC or in modulation of cancer cell proliferation and migration [24–26,35–41]. Exosomal miR-10b, -20a, -21, -30c, -106b, and -181a were present at high levels in PDAC and were low in the CP and normal samples, whereas exosomal miR-let7a (which is also known as let7a or let-7a) and miR-122 were lower in PDAC samples by comparison with either normal or CP samples, while miR-483 levels were similar in all three groups (Table 6 and Fig. 3). Moreover, post-resection, the elevated levels of exosomal miR-10b, -20a, -21, -30c, -106b decreased to normal values, but there was only a partial decrease in miR-181a levels (Table 6). By contrast, the levels of exosomal miR-let7a and miR-122 were lower in PDAC than in normal or CP samples (Fig. 3). Moreover, miR-let7a normalized post-resection whereas miR-122 levels increased slightly but significantly (p < 0.01) above normal levels (Table 6). Importantly, based on the ROC curves (Fig. 4), the AUC for exosomal miR-10b, -21, -30c, -181a, and -let7a revealed that these miRs had 100% sensitivity and specificity with respect to their accuracy in distinguishing PDAC from normal controls; only miR-106b and -483 failed to have an excellent AUC (Table 7). Similar trends for elevated miR levels in PDAC plasma by comparison with normal and CP plasma were observed for all 6 miRs (Tables 6 and 7). However, only miR-10b and miR-30c had 100% sensitivity and specificity with respect to their accuracy in distinguishing PDAC plasma from normal control plasma, and only miR-483 failed to have an excellent AUC (Table 7). Moreover, in contrast to exosomal miR-106b, which has an AUC of 0.85, plasma miR-106b had an AUC of 0.98, indicating that plasma levels of this miR were more sensitive for differentiating PDAC from normal samples than exosomal miR-106 levels.
Table 6.
miRNA levels in plasma and plasma-derived exosomes.
| Diagnosis | Pre/Post | miR-lOb
|
miR-21
|
miR-30c
|
miR-106b
|
miR-20a
|
miR-181a
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P | Exo | P | Exo | P | Exo | P | Exo | P | Exo | P | Exo | ||
| Normal (N = 6) | Pre | 0.92 ± 0.21 | 1.13 ± 0.25 | 1.08 ± 0.23 | 1.04 ± 0.18 | 0.85 ± 0.23 | 0.74 ± 0.03 | 0.73 ± 0.19 | 1.09 ±0.24 | 1.25 ± 0.30 | 1.01 ± 0.15 | 1.00 ± 0.07 | 0.90 ± 0.12 |
| CP (N = 11) | Pre | 0.76 ±0.14 | 1.04 ± 0.10 | 1.01 ± 0.11 | 0.94 ±0.16 | 0.83 ± 0.18 | 0.74 ±0.11 | 1.20 ± 0.21 | 0.93 ±0.17 | 1.63 ± 0.29 | 0.95 ±0.12 | 0.80 ± 0.18 | 1.49 ± 0.27 |
| Post | 0.66 ± 0.08 | 0.95 ± 0.10 | 1.20 ± 0.21 | 0.88 ± 0.12 | 0.72 ± 0.17 | 0.78 ± 0.13 | 0.89 ± 0.11 | 0.81 ±0.12 | 1.93 ± 0.31 | 0.72 ±0.15 | 0.75 ± 0.18 | 1.65 ± 0.29 | |
| PDAC (N = 29) | Pre | 12.11 ± 1.38** | 34.76 + 6.24**† | 5.34 ± 0.66** | 7.93 ± 1.52** | 9.77 ± 1.48** | 31.71 + 7.04**† | 5.21 ± 0.62** | 4.03 ± 0.74* | 6.00 ± 0.55** | 2.59 + 0.18**† | 3.06 ± 0.34* | 8.45 + 0.88*1 |
| Post | 1.87 ±0.19## | 3.37 ± 0.47†## | 2.12 ± 0.33## | 1.61 ± 0.15## | 2.12 ± 0.27** | 2.60 ± 0.31## | 1.56 ± 0.21## | 1.28 ±0.09## | 2.00 ± 0.21## | 1.28 ± 0.10†## | 2.65 ± 0.32 | 5.65 ± 0.60†#+ | |
| Diagnosis | Pre/Post | miR-483
|
miR-let7a
|
miR-122
|
|||
| P | Exo | P | Exo | P | Exo | ||
| Normal (N = 6) | Pre | 1.27 ± 0.14 | 1.21 ± 0.15 | 1.01 ± 0.04 | 0.97 ± 0.06 | 1.08 ± 0.19 | 1.00 ± 0.08 |
| CP (N = 11) | Pre | 1.33 ± 0.20 | 1.40 ± 0.43 | 1.66 ± 0.25 | 1.60 ± 0.26 | 1.21 ± 0.25 | 1.47 ± 0.34 |
| Post | 1.45 ± 0.25 | 1.25 ± 0.26 | 1.46 ± 0.15 | 1.60 ± 0.18† | 1.51 ± 0.38 | 1.21 ± 0.11 | |
| PDAC (N = 29) | Pre | 2.23 ± 0.32 | 1.77 ± 0.31 | 0.56 ± 0.04** | 0.40 ± 0.03**† | 0.52 ± 0.04** | 0.39 ± 0.04**† |
| Post | 2.07 ± 0.23 | 2.27 ± 0.34 | 1.48 ± 0.09## | 1.70 ± 0.12## | 3.93 ± 0.57** | 1.77 ± 0.10†##+ | |
Data are based on assays using plasma (P) or exosomes (Exo) from 6 normal controls, 11 chronic pancreatitis (CP) patients, and 29 patients with pancreatic ductal adenocarcinoma (PDAC). Data were analyzed by ANOVA followed by Bonferroni adjustment.
p < 0.01, and
p < 0.001 by comparison with corresponding values in normal or CP plasma or exosomes;
p < 0.02 by comparison with corresponding values in plasma;
p < 0.05 and
p < 0.01 by comparison with corresponding pre-resection values;
p < 0.01 and by comparison with corresponding values in normal controls.
Fig. 3.
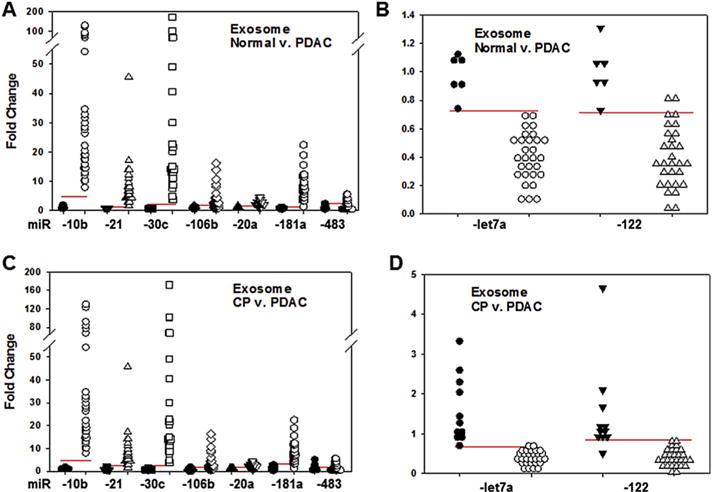
Dot plots of exosomal miR levels. The levels of exosomal miRs were assayed in normal controls, pancreatic cancer (PDAC), and chronic pancreatitis (CP) samples. Using RT-qPCR, miR-10b, -21, -30c, -106b, -20a, -181a, -483, -let7a, and -122 were assessed. (A and B) Individual normal control values were expressed as fold change from the mean value for controls (closed symbols). These values clustered separately from PDAC values (open symbols) for miRs which were overexpressed (A) or underexpressed (B) compared to respective mean normal values. (C and D) Similarly, CP values (closed symbols) clustered separately from PDAC values (open symbols). Horizontal lines represent cutoff values generated by ROC statistics.
Fig. 4.
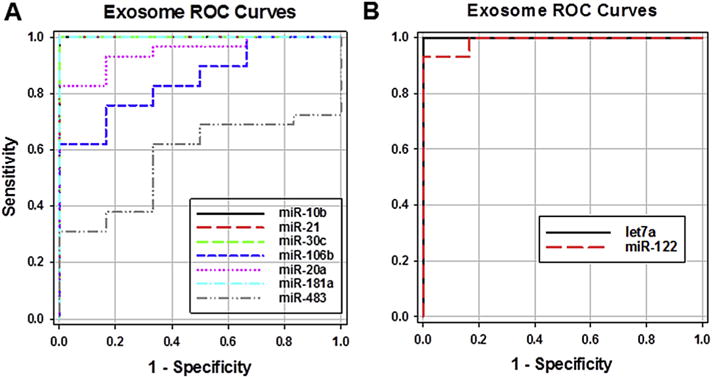
Receiver operating characteristic (ROC). The levels of the indicated miRs in pre-surgery exosomes from 29 PDAC patients and, separately, from 6 normal controls, were used to generate ROC curves for overexpressed (A) and underexpressed (B) miRs.
Table 7.
ROC curves performance characteristics normal v. PDAC.
| miRNA | Sensitivity | Specificity | Cutoff | AUC | p-value |
|---|---|---|---|---|---|
| Exosomes | |||||
| miR-10b | 100% | 100% | >4.94 | 1.00 | p < 0.001 |
| miR-21 | 100% | 100% | >1.38 | 1.00 | p < 0.001 |
| miR-30c | 100% | 100% | >2.14 | 1.00 | p < 0.001 |
| miR-106b | 62% | 100% | >1.84 | 0.85 | p = 0.007 |
| miR-20a | 83% | 100% | >1.73 | 0.95 | p < 0.001 |
| miR-181a | 100% | 100% | >1.32 | 1.00 | p < 0.001 |
| miR-483 | 31% | 100% | >2.57 | 0.57 | p = 0.599 |
| miR-let7a | 100% | 100% | <0.72 | 1.00 | p < 0.001 |
| miR-122 | 93% | 100% | <0.71 | 0.99 | p < 0.001 |
| Plasma | |||||
| miR-10b | 100% | 100% | >3.05 | 1.00 | p < 0.001 |
| miR-21 | 86% | 100% | >1.68 | 0.95 | p < 0.001 |
| miR-30c | 100% | 100% | >1.40 | 1.00 | p < 0.001 |
| miR-106b | 97% | 100% | >1.14 | 0.98 | p < 0.001 |
| miR-20a | 93% | 100% | >2.46 | 0.99 | p < 0.001 |
| miR-181a | 97% | 100% | >1.25 | 0.97 | p < 0.001 |
| miR-483 | 66% | 67% | >1.49 | 0.67 | p = 0.20 |
| miR-let7a | 93% | 100% | <0.86 | 0.99 | p < 0.001 |
| miR-122 | 100% | 67% | <0.94 | 0.89 | p = 0.003 |
Receiver operating characteristic (ROC) curves were generated with the SigmaPlot 13.0 ROC Curves macro tool, based on the levels of indicated miRs in exosomes or plasma.
Pre-operatively, PDAC, exosomes were especially numerous in the 85–115 nm range (Supplemental Fig. S3A), whereas post-resection their diameter distribution changed to that observed in CP and control samples (Supplemental Fig. S3B), suggesting that the high level of mid-range exosomes most likely derived from the cancer. However, the vast majority of PDAC patients had stage IIB disease (Supplemental Table 2). Therefore, it was not possible to assess whether there was a correlation between exosomal miR levels and tumor stage. Because CA 19-9 is an FDA-approved PDAC biomarker, its levels were assayed in all 29 patients (Supplemental Table 2) yielding a sensitivity of 86% with an AUC of 0.92 (Supplemental Fig. 7). There were variable decreases in CA 19-9 levels post-resection, perhaps due to varying clearance rates for this biomarker among patients. Importantly, 8 patients had normal or slightly elevated CA 19-9 levels (Supplemental Table 2). Based on the calculated cutoff values (Table 7) all 8 of these patients had high levels of exosomal and plasma miR-10b and -30c, and low levels of exosomal let7a (Supplemental Tables 3–4). Moreover, exosomal miR-21 and miR-181a levels were elevated and miR-122 levels were low in 7 of these 8 patients, underscoring the potential clinical utility of exosomal miR assays.
Discussion
PDAC is most often insidious in onset and its clinical presentation is preceded for a variable time period by non-specific symptoms. Consequently, the diagnosis of PDAC is frequently delayed and requires the use of CT scans, MRIs, ERCP and endoscopic ultrasonography with tissue sampling, each of which is a costly test and has potential risk. Moreover, the levels of circulating biomarkers in the blood, most notably CA19-9, have limited sensitivity and specificity. Thus, ~14% of the populations may not express CA19-9 and ~25% of PDAC patients do not exhibit a rise in CA19-9 [42]. Conversely, CA19-9 levels may be increased in patients with CP, and fine needle aspirations of pancreatic lesions may sometimes be difficult to interpret due to the presence of CP-like histological changes in PDAC. These clinical circumstances could cause confusion regarding the specific diagnosis, an especially vexing dilemma since the risk of PDAC is increased ~10 to 16-fold in patients with CP [43]. The combination of non-specific symptomatology, absence of specific and sensitive screening test, and occasional inconclusive diagnostic tests means that PDAC diagnosis is most often established at an advanced stage when the cancer has either metastasized or has invaded major organs, precluding resection [2]. Moreover, PDAC is treatment recalcitrant due to intrinsic chemo-resistance of the cancer cells and the intense desmoplasia that interferes with efficient drug delivery [2]. Thus, there is an urgent need to develop new biomarkers for PDAC that are both sensitive and specific, and that would allow for an earlier diagnosis.
Exosomes are membranous extracellular vesicles that are shed by many cell types and that range in size from 40 to 120 nM [29,30]. Exosomes carry RNAs, miRNAs, DNA, lipids, and proteins, and their internalization into cells results in the uptake of HSPGs, including GPC1, by the exosomes [14]. Given the overexpression of GPC1 in PDAC [11] and the report that exosomal GPC1 may serve as a biomarker for early PDAC detection [15], we used LC-MS/MS to establish a highly sensitive and reproducible assay for GPC1, allowing for a quantitative analysis of GPC1 levels. We determined that GPC1 protein levels in exosomes from PDAC patients tend to be slightly higher than in exosomes from individual without pancreatic disease and from CP patients, but these differences are not statistically different. Although the number of subjects in our study in each of these categories is small, the abundance of GPC1 protein in normal and CP-derived exosomes, and the persistent presence of GPC1 protein in exosomes following PDAC resection make it unlikely that this assay, which measures the levels of the core protein of GPC1, could serve as a test for early PDAC diagnosis or for differentiating between PDAC and CP.
GPC1 is decorated with three glycosaminoglycans (GAGs) side chains, and it is conceivable that glycanation of these side chains is qualitatively different in PDAC by comparison with CP. Such differences may arise, for example, as a result of the addition of chondroitin sulfate instead of heparan sulfate (HS) to the GAGs, or as a result of altered expression of sulfatases and sulfotransferases, leading to HS modifications [44]. Indeed, cancer cells can exhibit aberrant glycan structure and glycosylation, which can lead to the generation of cancer-specific epitopes. It is therefore conceivable that antibodies could be generated that recognize cancer-specific GAGs on GPC1, which could be used to establish a PDAC-specific test using exosomes.
Numerous miRs are overexpressed in PDAC, including miR-10b, -20a, -21, -30c, -106b, -196a, -196b, -203, -155, -205, -210, -221, -222, -223, -486, -744, and 17-5p [24–26,35–41]. Some of these miRs are present at high levels in the plasma and serum of patients with PDAC [26,41]. By contrast, certain miRs, such as let7a and miR-122, are tumor suppressor miRs that may be underexpressed in cancer [19]. In the present study we demonstrated that high exosomal levels of miR-10b, -21, -30c, and -181a and low let7a readily differentiate PDAC from normal control and CP samples. By contrast to GPC1, the levels of the elevated exosomal miRs declined to normal values within 24 h following PDAC resection, suggesting that these miR-rich exosomes originated in the cancers. This conclusion is supported by our finding that at 24 h PDAC post-resection, there was a concomitant normalization in the excessive number of exosomes whose diameter ranged from 85 to 105 nm that was only observed in the PDAC samples, and by reports that exosomes clear rapidly from the circulation [45,46]. Importantly, all 29 PDAC cases exhibited high levels of exosomal miR-10b and miR-30c, and with the exception of one plasma sample for miR-30c, the corresponding plasma samples were also elevated. Taken together, our findings indicate that an exosomal miR signature consisting of elevated levels of miR-10b, -21, -30c, -181a, and low let7a is diagnostic for PDAC when used in conjunction with an appropriate clinical evaluation, and yields a clear distinction between PDAC and CP.
Both miR-10b and miR-21 have important roles in PDAC [24,25], and miR-10b has been shown to downregulate TIP30 and thereby to enhance epidermal growth factor receptor (EGFR) signaling [47,48]. EGFR and heparin-binding epidermal growth factor-like growth factor (HB-EGF) are crucial for KRAS-driven oncogenesis in PDAC [49–51], and these pathways are enhanced by miR-10b. Therefore, the abundance of plasma and exosomal miR-10b and miR-21 documented in the present study and miR-10b’s ability to promote EGFR-mediated proliferation, invasion, and EGFR-transforming growth factor–beta interactions that facilitate epithelial to mesenchymal transition [48] raise the possibility that exosomal miR-10b and miR-21 combine to exert a crucial role in PDAC pathobiology. Such deleterious actions may be further enhanced by exosomal GPC1, given the important role of GPC1 in PCCs, where it facilitates the ability of HBGFs to enhance proliferation and invasion [11–13].
Due to the limited number of PDAC cases in the present study it is not possible to establish a training and validation cohort. Future studies with a larger number of PDAC and control samples are necessary to assess the true sensitivity and specificity of this exosomal miR signature. Nonetheless, our findings raise the possibility that our exosomal miR signature could be expanded to include other miRs that are elevated in PDAC-derived exosomes as well as miRs that are preferentially elevated in plasma over exosomes, as in the case of miR-106b. Such a combined exosome and plasma signature could lead to the development of sensitive and specific biomarkers for early PDAC diagnosis and for monitoring PDAC recurrence following resection.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health grant R01 CA-075059 to M. K. The authors thank Dr. Susan Perkins for helpful discussions regarding the statistical methods used in the present study, and Ms. Alicia Winters for outstanding collection of biospecimens. We thank the Indiana University Simon Cancer Center Tissue Bank for providing some of the human plasma samples for this study, and the Indiana University Electron Microscopy Center for performing transmission electron microscopy imaging.
Abbreviations
- ACN
Acetonitrile
- ANOVA
Analysis of variance
- BSA
Bovine serum albumin
- CP
Chronic pancreatitis
- ERCP
Endoscopic retrograde cholangiopancreatography
- GAG
Glycosaminoglycan
- GPC1
Glypican-1
- HPLC
High performance liquid chromatography
- HSPG
Heparan sulfate proteoglycans
- IS
Internal standard
- LC
Liquid chromatography
- miR
MicroRNA
- MRM
Multiple reaction monitoring
- MS/MS
Tandem mass spectrometry
- PBS
Phosphate buffered saline
- PDAC
Pancreatic ductal adenocarcinoma
- QC
Quality control
- RSD
Relative standard deviation
- RSP
Resistive pulse sensing
- RT
Retention time
- sMRM
Scheduled multiple reaction monitoring
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.canlet.2017.02.019.
Footnotes
Conflicts of interest
None.
References
- 1.Ellis L, Benedetti J, Rothenberg M, Willett C, Tempero M, Lowy A, et al. Consensus report of the national cancer institute clinical trials planning meeting on pancreas cancer treatment. J Clin Oncol. 2009;27:5660–5669. doi: 10.1200/JCO.2009.21.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 4.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 5.Häcker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- 6.Rodgers KD, San Antonio JD, Jacenko O. Heparan sulfate proteoglycans: a GAGgle of skeletal-hematopoietic regulators. Dev Dyn. 2008;237:2622–2642. doi: 10.1002/dvdy.21593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamanna WC, Kalus I, Padva M, Baldwin RJ, Merry CL, Dierks T. The heparanome–the enigma of encoding and decoding heparin sulfate sulfation. J Biotechnol. 2007;129:290–307. doi: 10.1016/j.jbiotec.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 8.Mythreye K, Blobe GC. Proteoglycan signaling co-receptors: roles in cell adhesion, migration and invasion. Cell Signal. 2009;21:1548–1558. doi: 10.1016/j.cellsig.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu W, Litwack ED, Stanley MJ, Langford JK, Lander AD, Sanderson RD. Heparan sulfate proteoglycans as adhesive and anti-invasive molecules. Syndecans and glypican have distinct functions. J Biol Chem. 1998;273:22825–22832. doi: 10.1074/jbc.273.35.22825. [DOI] [PubMed] [Google Scholar]
- 10.Iozzo RV, Sanderson RD. Proteoglycans in cancer biology, tumour micro-environment and angiogenesis. J Cell Mol Med. 2011;15:1013–1031. doi: 10.1111/j.1582-4934.2010.01236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleeff J, Ishiwata T, Kumbasar A, Friess H, Büchler MW, Lander AD, et al. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J Clin Invest. 1998;102:1662–1673. doi: 10.1172/JCI4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aikawa T, Whipple CA, Lopez ME, Gunn J, Young A, Lander AD, et al. Glypican-1 modulates the angiogenic and metastatic potential of human and mouse cancer cells. J Clin Invest. 2008;118:89–99. doi: 10.1172/JCI32412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whipple CA, Young AL, Korc M. A KrasG12D-driven genetic mouse model of pancreatic cancer requires glypican-1 for efficient proliferation and angio-genesis. Oncogene. 2012;31:2535–2544. doi: 10.1038/onc.2011.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christianson HC, Svensson KJ, van Kuppevelt TH, Li JP, Belting M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc Natl Acad Sci U S A. 2013;110:17380–17385. doi: 10.1073/pnas.1304266110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177–182. doi: 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ambros V. MicroRNAs: tiny regulators with great potential. Cell. 2001;107:823–826. doi: 10.1016/s0092-8674(01)00616-x. [DOI] [PubMed] [Google Scholar]
- 17.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 19.Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol. 2009;27:5848–5856. doi: 10.1200/JCO.2009.24.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fabbri M, Calore F, Paone A, Galli R, Calin GA. Epigenetic regulation of miRNAs in cancer. Adv Exp Med Biol. 2013;754:137–148. doi: 10.1007/978-1-4419-9967-2_6. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valadi H, Ekström K, Bossios A, Sjostrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 23.Ge Q, Zhou Y, Lu J, Bai Y, Xie X, Lu Z. miRNA in plasma exosome is stable under different storage conditions. Molecules. 2014;19:1568–1575. doi: 10.3390/molecules19021568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sempere LF, Preis M, Yezefski T, Ouyang H, Suriawinata AA, Silahtaroglu A, et al. Fluorescence-based co-registration with protein markers reveals distinct cellular compartments for altered microRNA expression in solid tumors. Clin Cancer Res. 2010;16:4246–4255. doi: 10.1158/1078-0432.CCR-10-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Preis M, Gardner TB, Gordon SR, Pipas JM, Mackenzie TA, Klein EE, et al. MicroRNA-10b expression correlates with response to neoadjuvant therapy and survival in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2011;17:5812–5821. doi: 10.1158/1078-0432.CCR-11-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cote GA, Gore AJ, McElyea SD, Heathers LE, Xu H, Sherman S, et al. A pilot study to develop a diagnostic test for pancreatic ductal adenocarcinoma based on differential expression of select miRNA in plasma and bile. Am J Gastroenterol. 2014;109:1942–1952. doi: 10.1038/ajg.2014.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarner M, Cotton PB. Classification of pancreatitis. Gut. 1984;25:756–759. doi: 10.1136/gut.25.7.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chandrasekhara V, Chathadi KV, Acosta RD, Decker GA, Early DS, Eloubeidi MA, et al. ASGE Standards of Practice Committee, The role of endoscopy in benign pancreatic disease. Gastrointest Endosc. 2015;82:203–214. doi: 10.1016/j.gie.2015.04.022. [DOI] [PubMed] [Google Scholar]
- 29.Théry C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006 doi: 10.1002/0471143030.cb0322s30. Chapter 3: Unit 3.22. [DOI] [PubMed] [Google Scholar]
- 30.van der Pol E, Coumans FA, Grootemaat AE, Gardiner C, Sargent IL, Harrison P, et al. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nano-particle tracking analysis, and resistive pulse sensing. J Thromb Haemost. 2014;12:1182–1192. doi: 10.1111/jth.12602. [DOI] [PubMed] [Google Scholar]
- 31.Coumans FA, van der Pol E, Böing AN, Hajji N, Sturk G, van Leeuwen TG, et al. Reproducing extracellular vesicle size and concentration determination with tunable resistive pulse sensing. J Extracell Vesicles. 2014;3:25922. doi: 10.3402/jev.v3.25922. http://dx.doi.org/10.3402/jev.v3.25922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleeff J, Korc M. Up-regulation of transforming growth factor (TGF)-beta receptors by TGF-beta1 in COLO-357 cells. J Biol Chem. 1998;273:7495–7500. doi: 10.1074/jbc.273.13.7495. [DOI] [PubMed] [Google Scholar]
- 33.Lai X, Kline JA, Wang M. Development, validation, and comparison of four methods to simultaneously quantify l-arginine, citrulline, and ornithine in human plasma using hydrophilic interaction liquid chromatography and electrospray tandem mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci. 2015;1005:47–55. doi: 10.1016/j.jchromb.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 34.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 35.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bloomston M, Frankel WL, Petrocca F, Volinia S, Alder H, Hagan JP, et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297:1901–1908. doi: 10.1001/jama.297.17.1901. [DOI] [PubMed] [Google Scholar]
- 37.Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, Frankel WL, et al. Schmittgen, Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer. 2007;120:1046–1054. doi: 10.1002/ijc.22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szafranska AE, Doleshal M, Edmunds HS, Gordon S, Luttges J, Munding JB, et al. Analysis of microRNAs in pancreatic fine-needle aspirates can classify benign and malignant tissues. Clin Chem. 2008;54:1716–1724. doi: 10.1373/clinchem.2008.109603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.du Rieu MC, Torrisani J, Selves J, Al Saati T, Souque A, Dufresne M, et al. MicroRNA-21 is induced early in pancreatic ductal adenocarcinoma precursor lesions. Clin Chem. 2010;56:603–612. doi: 10.1373/clinchem.2009.137364. [DOI] [PubMed] [Google Scholar]
- 40.Greither T, Grochola LF, Udelnow A, Lautenschlager C, Wurl P, Taubert H. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. Int J Cancer. 2010;126:73–80. doi: 10.1002/ijc.24687. [DOI] [PubMed] [Google Scholar]
- 41.Johansen JS, Calatayud D, Albieri V, Schultz NA, Dehlendorff C, Werner J, et al. The potential diagnostic value of serum microRNA signature in patients with pancreatic cancer. Int J Cancer. 2016;139:2312–2324. doi: 10.1002/ijc.30291. [DOI] [PubMed] [Google Scholar]
- 42.Korc M. Sugar-coated proteins pave the way to improving pancreatic cancer diagnosis. Cell Mol Gastroenterol Hepatol. 2016;2:118–119. doi: 10.1016/j.jcmgh.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, et al. Pancreatitis and the risk of pancreatic cancer: international pancreatitis study group. N Engl J Med. 1993;328:1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 44.Knelson EH, Nee JC, Blobe GC. Heparan sulfate signaling in cancer. Trends Biochem Sci. 2014;39:277–288. doi: 10.1016/j.tibs.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imai T, Takahashi Y, Nishikawa M, Kato K, Morishita M, Yamashita T, et al. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J Extracell Vesicles. 2015;4:26238. doi: 10.3402/jev.v4.26238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiklander OP, Nordin JZ, O’Loughlin A, Gustafsson Y, Corso G, Mager I, et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015;4:26316. doi: 10.3402/jev.v4.26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang C, Li A, Zhang X, Xiao H. A novel TIP30 protein complex regulates EGF receptor signaling and endocytic degradation. J Biol Chem. 2011;286:9373–9381. doi: 10.1074/jbc.M110.207720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ouyang H, Gore J, Deitz S, Korc M. microRNA-10b enhances pancreatic cancer cell invasion by suppressing TIP30 expression and promoting EGF and TGF-β actions. Oncogene. 2013;405:1–11. doi: 10.1038/onc.2013.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Navas C, Hern Andez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–330. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ardito CM, Grüner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22:304–317. doi: 10.1016/j.ccr.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ray KC, Moss ME, Franklin JL, Weaver CJ, Higginbotham J, Song Y, et al. Heparin-binding epidermal growth factor-like growth factor eliminates constraints on activated Kras to promote rapid onset of pancreatic neoplasia. Oncogene. 2014;33:823–831. doi: 10.1038/onc.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.