

Abstract
Several phosphorylation signaling pathways have been implicated in the pathogenesis of epilepsy arising from both genetic causes and acquired insults to the brain. Identification of dysfunctional signaling pathways in epilepsy may provide novel targets for antiepileptic therapies. We previously described a deficit in phosphorylation signaling mediated by p38 mitogen-activated protein kinase (p38 MAPK) that occurs in an animal model of temporal lobe epilepsy, and that produces neuronal hyperexcitability measured in vitro. We asked whether in vivo pharmacological manipulation of p38 MAPK activity would influence seizure frequency in chronically epileptic animals. Administration of a p38 MAPK inhibitor, SB203580, markedly worsened spontaneous seizure frequency, consistent with prior in vitro results. However, anisomycin, a non-specific p38 MAPK activator, significantly increased seizure frequency. We hypothesized that this unexpected result was due to activation of a related MAPK, c-Jun N-terminal kinase (JNK). Administration of JNK inhibitor SP600125 significantly decreased seizure frequency in a dose-dependent manner without causing overt behavioral abnormalities. Biochemical analysis showed increased JNK expression and activity in untreated epileptic animals. These results show for the first time that JNK is hyperactivated in an animal model of epilepsy, and that phosphorylation signaling mediated by JNK may represent a novel antiepileptic target.
Keywords: c-Jun N-terminal kinase (JNK), antiepileptic drug, p38 mitogen activated kinase (p38 MAPK), temporal lobe epilepsy, pilocarpine, phosphorylation
Introduction
Much attention has been devoted to the causes underlying the development of epilepsy following an acquired insult to the brain. One leading hypothesis suggests that the development of ion channel dysfunction or “acquired channelopathy” leads to neuronal hyperexcitability and seizures. Dysfunction of numerous species of voltage-gated ion channels, such as the K+ channel Kv4.2 and the Ca2+ channel Cav3.2, and ligand-gated channels such as GABAA receptors and AMPA receptors, has been documented in animal models of acquired epilepsy; all of these promote neuronal hyperexcitability (Jacob et al., 2008; Rakhade et al., 2012; Poolos and Johnston, 2012). In studying the relationship of hyperpolarization-activated, cyclic nucleotide-gated (HCN) channel dysfunction and epilepsy, we and others have found downregulation of HCN channel expression following status epilepticus (SE) provoked by chemoconvulsants that precedes the development of epilepsy (Shah et al., 2004; Jung et al., 2007; Jung et al., 2011; Williams et al., 2015). However, in chronic epilepsy, downregulation of HCN channel voltage-dependent activation also occurs, and additionally promotes neuronal hyperexcitability. This gating defect in HCN channel function depended on loss of activity of an upstream signaling cascade mediated by p38 mitogen activated protein kinase (p38 MAPK), and increased activity of protein phosphatase 2B (Jung et al., 2010). These two biochemical alterations produced a net loss of phosphorylation in hippocampal tissue. When hippocampal pyramidal neurons from epileptic animals were studied in vitro, pharmacological reversal of these signaling abnormalities restored neuronal excitability to normal. We therefore hypothesized that pharmacological manipulation of the p38 MAPK pathway in vivo might influence seizure frequency in chronically epileptic rats.
Phosphorylation signaling pathways like p38 MAPK are ubiquitous regulators of cellular function. Pharmacological modulation of phosphorylation signaling is increasingly recognized as an important therapeutic area (Cohen, 2002; Chico et al., 2009). In epilepsy, the numerous ion channelopathies set into motion by a brain insult likely reflect the diverse action of upstream signaling processes; thus it could be advantageous to identify and pharmacologically manipulate those signaling pathways rather than individual ion channels, their downstream targets. Conventional antiepileptic drug (AED) development—mostly focused on voltage- and ligand-gated ion channels—has made disappointingly little progress over recent decades in treating that one-third of all epilepsy patients who are refractory to medical therapy (Loscher and Schmidt, 2011). Thus, there is a compelling need to understand the root causes of the initiation and maintenance of the epileptic state, and to identify novel therapies.
In this study, we used the pilocarpine animal model of epilepsy to study the effect of pharmacologically manipulating MAPK signaling. We chose to study modulation of kinase rather than phosphatase signaling due to the presumably more diverse downstream effects of manipulating phosphatase activity. We first validated that inhibition of p38 MAPK activity in vivo exacerbated spontaneous seizure frequency in chronically epileptic animals. We then fortuitously discovered that inhibition of a related MAPK, c-Jun N-terminal kinase (JNK), reduced seizure frequency. Biochemical analysis of hippocampal tissue from epileptic animals showed chronic elevation of JNK activity. The finding of JNK hyperactivation in an animal model of epilepsy may mark it as a novel therapeutic target in human refractory epilepsy.
Experimental Procedures
Pilocarpine model of epilepsy
We generated chronically epileptic animals using the pilocarpine protocol as previously described (Jung et al., 2007). The University of Washington Institutional Animal Care and Use Committee approved all animal procedures. In brief, 6 week-old male Sprague Dawley rats underwent induction of SE with pilocarpine hydrochloride (385 mg/kg intraperitoneal [i.p.]) after pretreatment with scolpolamine methylnitrate (1 mg/kg i.p.). After 60 min of convulsive SE, seizures were terminated with repeated doses of diazepam (12 mg/kg i.p.) or phenobarbital (PB; 15 mg/kg i.p.) every 30 – 45 min until cessation of convulsive motor activity. In cohorts of chronically epileptic animals evaluated at 6 weeks post-SE, average baseline seizure frequency ranged from 1.6 to 4.3 convulsive seizures per day. Baseline seizure frequency was lower (0.88 seizures per day) in one cohort of animals that received more frequent post-SE dosing of PB (at 20–30 min intervals) in an attempt to reduce post-SE mortality.
Surgical protocol and video-EEG recording
Five weeks post-SE, animals were implanted with epidural electroencephalogram (EEG) electrodes according to a modification of our previous published protocol (Jung et al., 2007). We implanted two electrodes positioned over the right and left parietal convexities, plus a reference electrode approximately 2 mm anterior and to the right of bregma. We also placed an intracerebroventricular (i.c.v.) cannula (DURECT Corp., Cupertino, CA) into the right lateral ventricle (at position AP= −1.2; ML= 2.2). Epidural electrodes, cannula, and a cable pedestal were secured with dental acrylic. A subcutaneous pocket was then created over the dorsal thorax, and an osmotic pump (Alzet 2ML2 or 2ML4; DURECT Corp.) containing vehicle solution was inserted and connected via polyethylene tubing to the cannula. Following closure of the incision, animals were given buprenorphine (0.10 mg/kg; Bedford Laboratories, Bedford, OH) to minimize discomfort.
Animals underwent baseline video-EEG recordings after a one-week post-surgery recovery period. Their headsets were attached to a wire cable, and they were placed in individual acrylic cages. EEG signals were amplified with a Grass Model 8–10 D amplifier (Grass Tech., West Warwick, RI) and acquired using a PowerLab 8/30 module equipped with Chart Pro software (AD Instruments, Colorado Springs, CO). Video recording utilized a ZR500 digital camcorder (Canon, Lake Success, NY), which interfaced with the Chart software, allowing for synchronization of the video and EEG traces. During baseline and drug treatment phases, animals underwent continuous 24/7 video-EEG recording for an average of 10 d during each phase. Seizures were classified by visual inspection of the video-EEG records according to the Racine scale, with event severity ranging from class 1 (C1) to class 5 (C5) (Racine, 1972). Although all classes of seizure were recorded, only C3-5 seizures (which were the predominant classes) were used for measurement of seizure frequency. Seizure duration was measured as the time between electrographic onset and offset. After baseline video-EEG recording, animals underwent a brief surgical procedure to replace the vehicle osmotic pump with drug-containing solution. After a three-day recovery period, a repeat video-EEG recording session occurred during drug delivery.
Fig. 1A shows our experimental protocol, in which monitoring during baseline and drug treatment occurred during weeks 6–9 post-SE. An important advantage of this protocol is that within-subject comparisons of baseline and drug treatment phases normalize for the inherent variability in seizure frequency among pilocarpine-treated animals. Thus each animal served as its own control. Also, i.c.v. delivery of drugs via osmotic pump achieves steady-state drug levels without diurnal variation; avoids issues with drug blood-brain barrier permeability; avoids the animal stress that would occur with repeated drug injection or gavage; and minimizes drug amounts used.
Fig. 1.
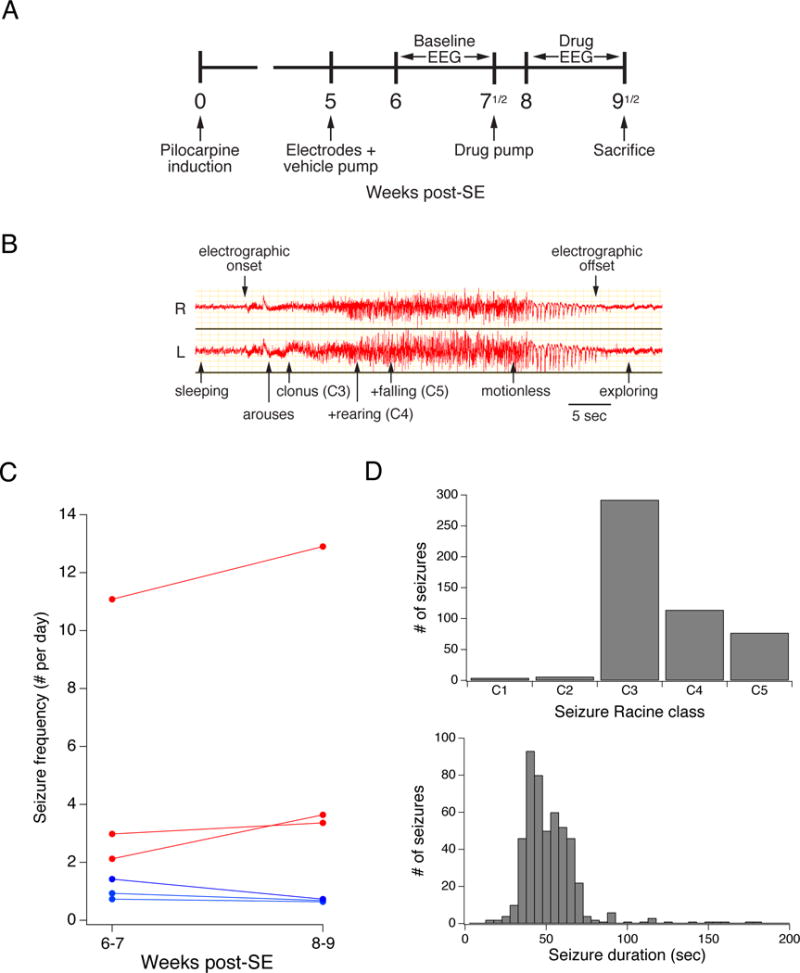
Experimental protocol and characteristics of epilepsy animal model. A. Experimental protocol. Adult animals underwent pilocarpine induction and then were implanted with epidural EEG electrodes and an i.c.v. cannula connected to a vehicle pump at week 5 post-SE. A 10-day continuous baseline video-EEG monitoring period began at week 6, followed by a change to a drug-containing pump, and repeat 10-day monitoring. Following this monitoring period, the animals were sacrificed. B. EEG tracing during a typical Racine class 5 seizure. Shown are right (R) and left (L) epidural cortical electrode channels, with behavioral changes and Racine classifications noted during the course of the electrographic seizure. The typical convulsive seizure in this model is readily apparent from the onset of a high amplitude, high frequency electrographic discharge that gradually increases in amplitude followed by a waning phase. C. Seizure frequency in animals treated throughout weeks 6–9 with a saline pump. Traces show average C3–5 seizure frequency during the two 14-day periods for six animals. Animals with increased seizure frequency are depicted in red; those with decreased seizure frequency in blue. There was variability in the baseline seizure frequency, but little change in seizure frequency in individual animals between weeks 6–7 and 8–9. D. Distribution of Racine classifications and seizure durations for all seizures recorded as shown in Fig. 1C. C3–5 seizures with major motor manifestations (C3–4) or convulsions (C5) were the predominant seizure types. The median seizure duration was 50 sec.
Drug delivery
All drugs were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. SB203580 was dissolved in sterile 150 mM NaCl solution plus 1% DMSO to yield 0.5 mM solution. This solution was loaded into Alzet 2ML2 pumps that deliver at 5 μl/hr to achieve a predicted steady-state cerebrospinal fluid (CSF) concentration of 20 μM (with DMSO concentration of 0.03%), based on rat CSF volume of 400 μl and turnover rate of 2.2 μl/min (3.2 ml/d; Davson et al., 1987). We determined in ex vivo trials that drug-containing fluid would transit the tubing connecting the osmotic pump to the i.c.v. cannula within 24 hours, and thus estimated the drug should be at steady-state CSF concentration well within the 72 hours between pump changes and repeat video-EEG monitoring. Anisomycin was dissolved in saline plus 1% DMSO to yield 1.1 mM solution, and was predicted to yield a steady-state CSF concentration of 40 μM. SP600125 was dissolved in saline plus 1% DMSO to yield either 0.9 or 1.8 mM solution, and was predicted to yield a steady-state CSF concentration of 20 or 40 μM, respectively. Vehicle solution consisted of saline and was delivered by either 2ML2 or 2ML4 pumps (2.5 μl/hr delivery rate). Lamotrigine isethionate (GlaxoSmithKline, Research Triangle Park, NC) was dissolved in saline at a concentration to deliver 20 mg/kg/d, loaded into 2ML4 pumps, and implanted subcutaneously over the dorsal thorax to deliver drug subcutaneously.
Open field behavioral testing
The animal was placed in a 1 m2 square enclosure composed of 25 cm tall black Plexiglas walls with a non-reflective, rubberized floor. The center was defined as the central 50 × 50 cm zone and each corner was defined as a 25 × 25 cm zone. Behavior was monitored by video under dim light illumination for 10 minutes. Position in the field was measured by motion detection software (ANY-Maze, Stoelting Co., Wood Dale, IL), and anxiety-like behavior was quantified by the amount of time spent in the center of the field compared to the corners, with increased anxiety correlating with decreased time in the center of the field. Additional measures of motor activity, such as locomotion speed and time spent mobile were also extracted.
Biochemistry
Animals were sacrificed with ketamine/xylazine anesthesia (87/13 mg/kg i.p.), followed by decapitation and harvest of bilateral CA1 hippocampal tissue. In each experiment, we pooled 6–8 hippocampal slices from a single animal. The CA1 regions were microdissected on dry ice and homogenized in solution containing (in mM): 4 para-nitrophenylphosphate, 1 sodium orthovanadate, 1 phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor mixture (1 tab per 10 ml; Roche, Indianapolis, IN). Homogenates were centrifuged at 15,000× g for 40 min at 4 °C. The pellet was then resuspended with sonication in homogenization solution then run in Laëmmli buffer (with final concentration of 4.5% sodium dodecyl sulfate [SDS]) on a 10% acrylamide gel, transferred to a nitrocellulose membrane, and incubated with the appropriate primary antibody, followed by incubation in secondary antibody, and visualized by enhanced chemiluminescence and film exposure. Three different protein loading amounts were used in each condition so to verify that signal detection was in the linear range, as described previously (Jung et al., 2010). The membrane was then reprobed using anti-β-tubulin III antibody as a marker for neuronal protein content (1:20,000; Sigma-Aldrich) so to verify that protein loading was correct in each experiment. Test and control conditions were always analyzed within the same gel, with results reported as a percentage of control values. All antibodies were used at 1:1000 titers and were purchased from Cell Signaling Technology (Danvers, MA). Antibodies for phospho-JNK recognized phosphorylation at Thr183/Tyr185; and for phospho-c-Jun at Ser63.
Statistics
Seizure data was analyzed by calculating within-subject ratios of seizure frequency under control and drug conditions. This data was then log-transformed, and two-tailed t-tests calculated, followed by back-transformation of the mean and 95% confidence intervals (CIs). This method, used in analyses of both human and animal antiepileptic drug studies, provides a robust means of assessing drug effects in within-subject comparisons: calculation of seizure frequency ratios normalizes for the variability in baseline seizure frequency among subjects, while log-transformation produces normally-distributed data that can be analyzed with parametric statistics (Eastman et al., 2010; Poolos et al., 2012). For comparison purposes, we also report group average data for control and drug-treated animals. The remaining statistics are reported as means ± SEM, with significance determined by two-tailed t-tests unless otherwise mentioned.
Results
Seizure characteristics in the pilocarpine model
To investigate the effects of p38 MAPK signaling modulation on seizure frequency, we used the pilocarpine model (Turski et al., 1983). This model has several benefits for the study of AED efficacy. First, animals display complex partial and secondarily generalized convulsive seizures that are prolonged and behaviorally obvious to an observer. Seizures predominately begin in the hippocampus then spread to extratemporal areas, as occurs in human temporal lobe epilepsy (Toyoda et al., 2013). These are accompanied by electrographic discharges that when recorded from epidural electrodes are easily detected. An EEG recording of a typical Racine class 5 (C5) convulsive seizure is shown in Fig. 1B. Here, an electrographic discharge recorded from right and left parietal cortical epidural electrodes had its onset several seconds before the first behavioral change, in this case arousal from sleep. The discharge gradually gained in amplitude, accompanied by forelimb clonus (C3), followed by forelimb clonus with rearing (C4), and loss of posture with generalized convulsion (C5). As the electrographic discharge waned, the animal became motionless, followed shortly by exploring behavior. Because of these easily identified electrographic and behavioral changes, in this study we were able to rapidly review seizure occurrences in 24 hr/d video-EEG monitoring over prolonged 7–10 d epochs. The obvious nature of the behavioral and electrographic changes with C3-5 seizures meant that their detection was not appreciably subject to observer interpretation, and in our experience inter-observer reliability was very high. In total, our data for this study comprised 20,867 animal-hours (869 animal-days) of video-EEG recordings.
To validate that seizure frequency is stable in our animal model during the period of evaluation, weeks 6–9 post-SE, we implanted epileptic animals with saline-filled osmotic pumps and measured spontaneous seizures over two 14 day periods, weeks 6–7, and weeks 8–9 post-SE. As seen in Fig. 1C, baseline average seizure frequency in weeks 6–7 clustered between 1 and 3 seizures per 24-hour day in the majority of animals, but with one animal exhibiting a baseline of 11 seizures per day. Of the six animals, three showed modest decreases in seizure frequency in weeks 8–9, while three showed modest increases. The average change in seizure frequency in weeks 8–9 compared to weeks 6–7 for all animals was a 4.8% decrease, which was not statistically significant. This data demonstrates that while there was significant variability in baseline seizure frequency, there was little change in within-animal comparisons of seizure frequency during the evaluation period. We also quantified the Racine class of the spontaneous seizures observed, which demonstrated a predominance of C3-5 seizures, with a median duration of 50 sec (range 15 – 180 sec; Fig. 1D), and only rare C1-2 seizures. Because C1-2 seizures were behaviorally subtle and infrequent, we chose to quantify only C3-5 seizures for this study.
p38 MAPK inhibition worsens spontaneous seizure frequency
We first asked whether pharmacologic modulation of p38 MAPK activity would influence seizure frequency. Our previous work had shown that p38 MAPK activity in reduced in chronic epilepsy. For our initial experiments we chose to further inhibit p38 MAPK pharmacologically owing to the availability of a widely used specific inhibitor, SB203580 (SB; Martin and Arthur, 2012). We hypothesized that p38 MAPK inhibition should increase spontaneous seizure frequency. We delivered SB at 12 μg/d i.c.v. to yield an estimated steady-state CSF concentration of 20 μM.
Inhibition of p38 MAPK with SB markedly increased seizure frequency. As shown in Fig. 2A, eight out of nine animals experienced an increase in C3-5 seizure frequency. The average group baseline seizure frequency under control conditions was 1.6 seizures/d; in the presence of SB, seizure frequency was 3.3 seizures/d. Calculated as within-subject ratios of seizure frequency under drug and control conditions followed by log transformation of the ratio data (as described in Methods), SB treatment increased seizure frequency by 164% (95% CIs [118%, 229%]; n=9; p<.01). The mean Racine class for the group under control conditions was 4.3 (± 0.09, n=124) while during SB treatment was 4.2 (± 0.07, n=245); the within-subject increase in mean Racine class with SB treatment was 0.07 (± 0.25, n=9, p>.05), and was not statistically significant. Similarly, mean seizure duration for the cohort under control conditions was 53 sec (± 2.3 sec, n=108), while in SB was 43 sec (± 1.3 sec, n=220). However, within-subject comparisons showed a mean 5.9 sec (± 2.3 sec, n=9, p>.05) decrease in seizure duration with SB treatment that was not significant. A histogram of seizure durations in the two groups is shown in Fig. 2B.
Fig. 2.
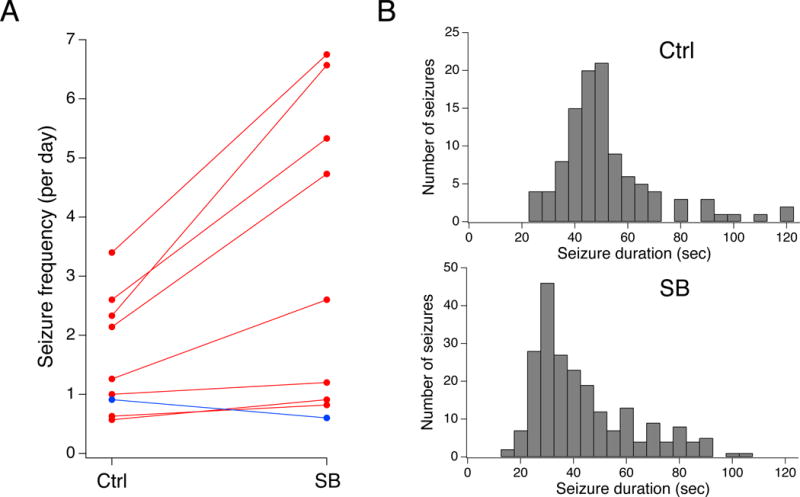
Inhibition of p38 MAPK increases seizure frequency. A. Plot of seizure frequencies for individual animals under control and drug-treated conditions. Inhibition of p38 MAPK with SB increased spontaneous seizure frequency in eight of nine animals. B. Histograms of aggregate seizure durations for the SB-treated and control groups are shown.
These results obtained with in vivo p38 MAPK inhibition validated our initial hypothesis based on prior in vitro work that showed p38 MAPK inhibition significantly increases hippocampal pyramidal neuron excitability, in part via downregulation of HCN channel gating (Poolos et al., 2006; Jung et al., 2010). We then asked whether activation of p38 MAPK might conversely decrease seizure frequency. However, there are no known selective pharmacologic activators of p38 MAPK. Anisomycin (ANI) is a non-specific activator of both p38 MAPK and the related stress-activated kinase JNK (Rosser et al., 2004). ANI also acts to inhibit protein synthesis at 10-fold higher concentrations than those that produce activation of stress kinases (Iordanov et al., 1997). Since there were no published reports on the effects of ANI on seizures in chronically epileptic animals, we proceeded with an experiment to deliver ANI in vivo to our pilocarpine-induced animals.
Anisomycin unexpectedly exacerbates spontaneous seizure frequency
Contrary to our prediction, anisomycin administration at 19 μg/d i.c.v. to yield an estimated steady-state CSF concentration of 40 μM, produced a significant increase in C3-5 seizure frequency (Fig. 3A). All four ANI-treated animals showed an increase in seizure frequency; one animal with a high baseline seizure rate entered into SE within two days of change to an ANI-containing pump, ultimately resulting in near-continuous seizures before euthanization. The other three animals showed more moderate increases in seizure frequency. On average, the ANI cohort had 3.5 seizures/d at baseline, and 12.4 seizures/d with ANI treatment. The mean seizure frequency ratio of treated to control was 256% ([135%, 485%], n=4, p<.05). Seizure Racine class for the group under control conditions was 4.2 (± 0.06, n=111), while during ANI administration was 3.5 (± 0.08, n=1820); in within-subject comparisons, the mean decrease in Racine class was not significant (0.17 ± 0.28, n=4, p>.05). Seizure duration for the cohort under control conditions was 44 sec (± 0.69 sec, n=108), and following ANI 57 sec (± 2.5 sec, n=134). This apparent increase was driven mostly by a single subject in SE; in within-subject comparisons the mean increase in seizure duration was 24 sec (± 13 sec, n=4, p>.05), and was not significant; Fig. 3B). Because of the consistent effect of ANI in exacerbating seizure frequency, and the occurrence of provoked SE, we terminated this experiment after exposure of just four animals.
Fig. 3.
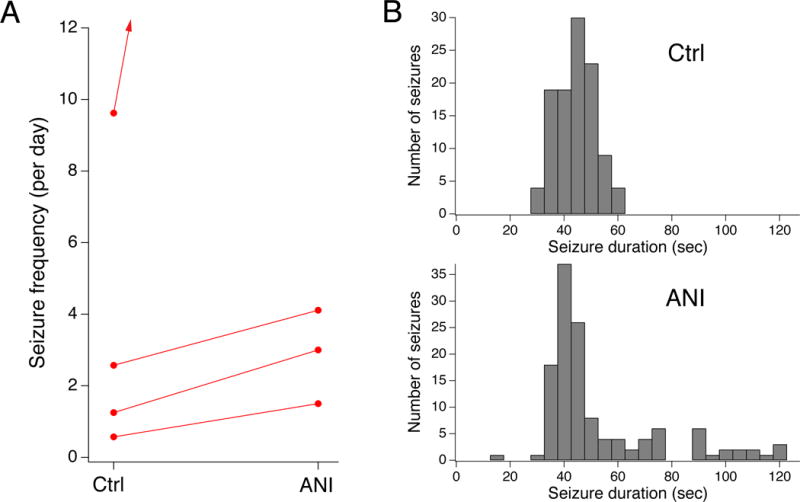
Nonspecific activation of p38 MAPK with anisomycin increases seizure frequency. A. Plot of seizure frequencies for individual animals under control and drug-treated conditions. Non-specific activation of p38 MAPK with ANI increased spontaneous convulsive seizure frequency in all four treated animals, in one case precipitating SE. B. Histograms of seizure duration are shown under control and ANI treatment.
JNK inhibition produces an antiepileptic action
The exacerbation of seizure frequency by ANI was unexpected since p38 MAPK inhibition by SB203580 had resulted in significant worsening of seizure frequency, and ANI is a potent p38 MAPK activator. However, ANI also activates related stress-activated kinase JNK (as well as inhibits protein synthesis). There had been no previous reports that modulation of JNK activity affects spontaneous seizure frequency, or that JNK is activated in chronic epilepsy. However, JNK knockout mice have reduced intensity of seizures acutely provoked by kainic acid (Brecht et al., 2005), thus we hypothesized that pharmacologic inhibition of JNK might have an antiepileptic effect. We administered the selective JNK inhibitor SP600125 to chronically epileptic rats (Martin and Arthur, 2012). To determine the maximal JNK inhibition that might be achieved with a concentration of SP that could be reasonably delivered via osmotic pump, we applied SP at 40 μM to hippocampal slices from naive animals incubated in vitro for one hour at 32° C. (Of note, 10 μM SP applied in vitro to cultured cells produced approximately 50% inhibition of JNK activity; Vogel et al., 2009). We measured phosphorylation of the canonical target of JNK, c-Jun, using a phosphospecific antibody that recognizes phosphorylation at Ser63 by all JNK isoforms. SP applied at 40 μM to hippocampal slices reduced the ratio of phosphorylated c-Jun to total c-Jun to only 55% of control (±8.1%, n=3, p<.05; data not shown). Thus we concluded that SP delivered to hippocampal tissue at the maximal concentration accommodated by our osmotic pumps produces only partial inhibition of JNK activity, possibly due to poor tissue penetration of the drug. Nonetheless, we proceeded to test the efficacy of partial JNK inhibition with SP at two dosage levels.
JNK inhibition produced an antiepileptic effect. SP delivered at 14 μg/d (predicted to yield 20 μM CSF concentration) reduced the group seizure frequency to 0.59 seizures per day (± 0.09, n=7). Six of seven treated animals demonstrated decreases in seizure frequency, as shown in Fig. 4A. In the generation of this cohort of chronically epileptic animals, more aggressive phenobarbital treatment was used in an attempt to improve mortality, but unexpectedly the resulting average baseline seizure frequency was 0.88 per day (± 0.09, n=7), less than in the other cohorts. However, the mean seizure frequency ratio of treated to control was significantly reduced to 66% ([45%, 96%], n=7, p<.05). JNK inhibition also produced a small decrease in seizure duration: in the control group mean duration was 43 sec (± 0.97 sec, n=64) while the group treated with SP 14 μg/d had a mean duration of 41 sec (± 1.0 sec, n=38). Within-subject comparisons showed a small but significant mean decrease of 2.1 sec (± 1.0 sec, n=7, p<.05); Fig. 4B. Racine class was not significantly different with a mean of 4.1 (± 0.09, n=64) in the control group and 4.3 (± 0.11, n=38) in the SP-treated group, and a within-subjects increase of 0.26 (± 0.27, n=7, p>.05).
Fig. 4.
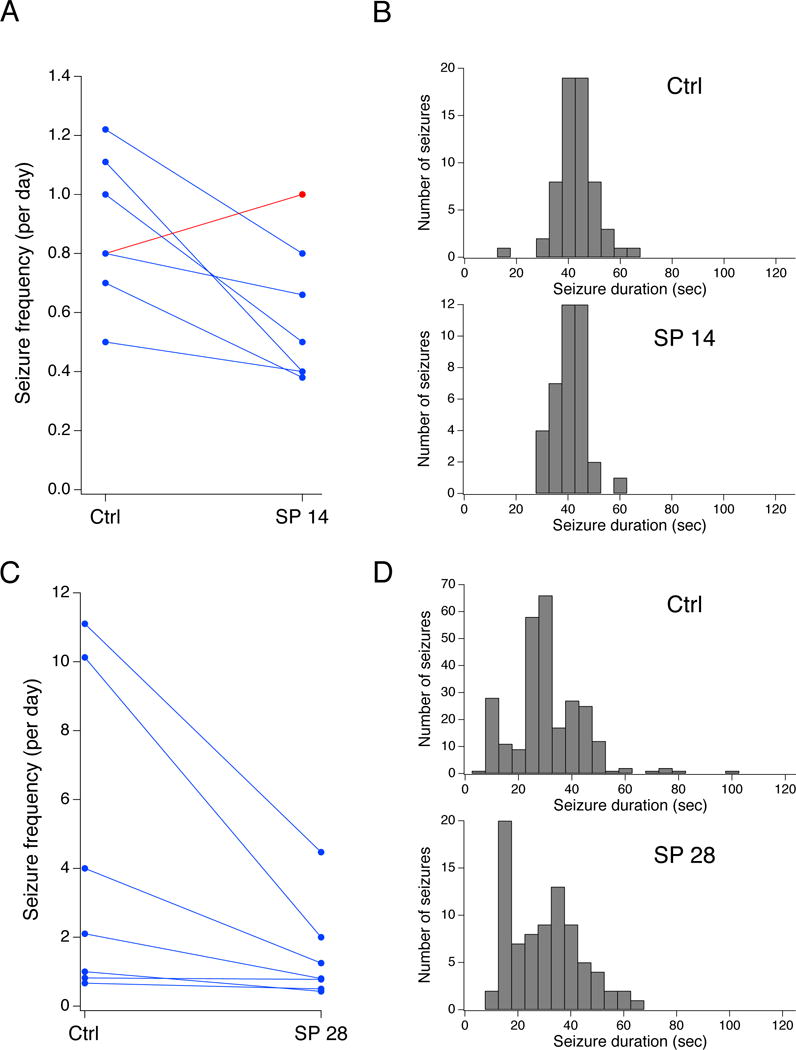
JNK inhibition reduces seizure frequency. A. Plot of seizure frequencies for individual animals under control and drug-treated conditions. Inhibition of JNK with SP delivered at 14 μg/d (SP 14) decreased spontaneous convulsive seizure frequency in six of seven animals. B. Histograms of seizure duration are shown under control and lower-dose JNK inhibition conditions. C. Inhibition of JNK with SP delivered at 28 μg/d (SP 28) decreased spontaneous convulsive seizure frequency in all seven animals from a separate experimental series. D. Histograms of seizure duration are shown under control and higher-dose JNK inhibition conditions.
In order to determine whether a positive dose-response relationship existed for JNK inhibition and seizure frequency, we delivered SP at 28 μg/d i.c.v. (predicted to achieve steady-state 40 μM CSF concentration) to a second cohort of epileptic animals. SP at 28 μg/d produced an increased antiepileptic effect compared to SP at 14 μg/d. As shown in Fig. 4C, all seven animals exposed to SP had decreased convulsive seizure frequency. Average baseline seizure frequency of this cohort was 4.3 seizures per day (± 1.7, n=7), while after SP treatment the group seizure frequency was 1.5 seizures per day (± 0.51). The mean seizure frequency ratio of treated to control was 43% ([27%, 71%], n=7, p<.01). JNK inhibition in this cohort produced a similar small change in seizure duration (control: 31 ± 0.82 sec, n=262; SP 28 μg/d: 30 ± 1.0 sec, n=82) that in within-subjects comparisons was not statistically significant (mean decrease 2.0 ± 3.8 sec, n=7, p>.05). These results demonstrated that JNK inhibition by SP produced a dose-dependent reduction in seizure frequency, even with only partial inhibition of JNK activity.
As a comparison for the antiepileptic effect of JNK inhibition, we treated a cohort of chronically epileptic animals with the AED lamotrigine (LTG). LTG is a commonly used antiepileptic drug, with a well-defined human therapeutic concentration range of 2–20 μg/ml (Shorvon, 2000). LTG isethionate was delivered subcutaneously via osmotic pump instead of i.c.v. because of its well-characterized efficacy when systemically administered. We first treated a pilot series of animals with LTG 20 mg/kg/d and measured serum LTG levels at three time points post-implantation of drug-filled osmotic pumps. At 3 d post-implant, the mean LTG level was 8.8 ± 1.5 μg/ml; at 7 d it was 10.6 ± 0.6; and at 10 d it was 10.4 ± 1.1 (all n=3). Seven days post-pump removal, LTG levels were undetectable (all <0.5). Thus, within three days of LTG pump implantation, serum levels corresponding to the middle of the human therapeutic range were stably attained. We then administered LTG 20 mg/kg/d to epileptic animals. The average baseline convulsive seizure frequency in this cohort was 4.0 (± 1.1, n=11) seizures/d. Average seizure frequency during LTG treatment was 1.2 seizures/d (± 0.43, n=11). The ratio of seizure frequency in within-animal comparisons of LTG to control was 26% ([7.3%, 94%], n=11, p<.05). This provides a point of comparison with the efficacy of a standard AED, and demonstrates that partial JNK inhibition has a similar antiepileptic effect as LTG dosed to achieve mid-therapeutic levels. A graphical comparison of the results from all five treatment groups is shown in Fig. 5.
Fig. 5.
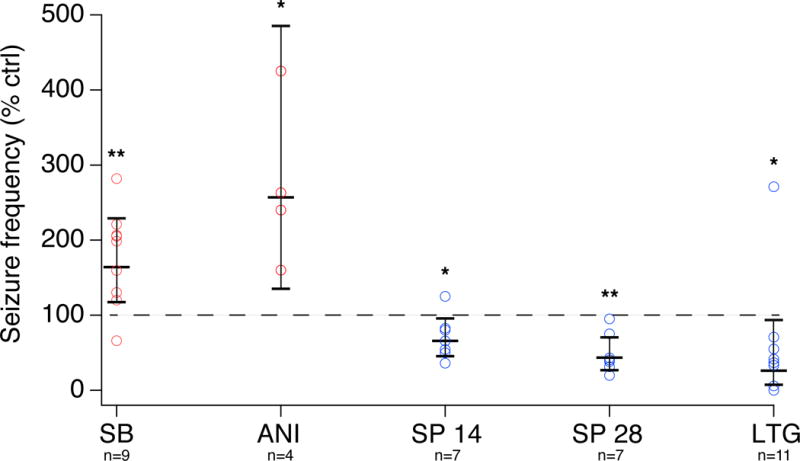
Summary of treatment results. Shown is the ratio of seizure frequency as a percentage of baseline frequency for each animal (circles), with treatments that on average were proconvulsive depicted in red and those that were antiepileptic in blue. Mean and 95% CIs for each group are shown, along with statistical significance level of the change in seizure frequency (* = p<.05; ** = p<.01).
JNK inhibition does not produce behavioral abnormalities
We then asked whether JNK inhibition at the higher SP dose was associated with toxicity manifested by behavioral abnormalities. To test for abnormal locomotion and also for any effects on anxiety-like behavior, we performed open-field testing on epileptic animals treated with SP 28 μg/d; untreated, age-matched epileptic animals; and naive, age-matched controls. Prior studies have shown that increased anxiety-like behavior is a comorbidity of epilepsy induced in mice with pilocarpine or kainic acid (Muller et al., 2009; Liu et al., 2013), while decreased anxiety-like behavior is seen in pilocarpine-induced rats (Inostroza et al., 2012; Cardoso et al., 2009; Detour et al., 2005). As shown in Fig. 6A, SP treatment did not alter mean locomotion speed or the amount of time spent mobile, showing that JNK inhibition did not produce gross motor impairment or lethargy. The untreated epileptic group showed decreased anxiety-like behavior compared to naive controls, as manifested by increased amounts of time spent exploring in the center area of the field (epileptic: 77.5 ±14.3 sec, n=11; naive: 34.0 ± 6.5 sec, n=9, p<.05 by one-way ANOVA with Tukey post-hoc test; Fig. 6B). There were no significant differences in anxiety-like behavior between SP treated and untreated epileptic animals. The decrease in anxiety-like behavior in our epileptic animals is consistent with several prior studies in rats, while mice show increases in anxiety-like behavior; thus it appears that behavioral alterations in post-SE epilepsy models animals have species-dependent differences. Regardless, these results also show that JNK inhibition did not overtly affect behavior in our animals.
Fig. 6.
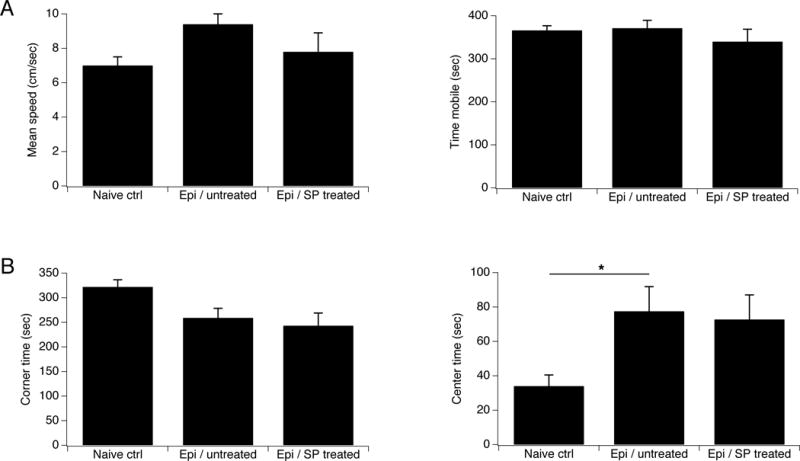
JNK inhibition does not affect behavior. Results of open-field behavioral testing are shown for naive controls, chronically epileptic animals that were untreated, and epileptic animals treated with SP at 28 μg/d. A. Mean speed of locomotion and time spent mobile was similar in all groups. B. Untreated epileptic animals showed decreased anxiety-like behavior as shown by increased time spent in the center of the field, compared to naive controls. There were no differences in these measures between treated and untreated epileptic animals.
Elevated JNK signaling in chronic epilepsy
Since JNK inhibition produced an antiepileptic effect, we asked whether our chronically epileptic animals showed evidence of increased JNK expression or activation. We harvested hippocampal tissue from a cohort of untreated, chronically epileptic animals at 6 weeks post-SE, and used pan-specific antibodies to measure total and phosphorylated JNK protein expression. JNK isoforms tend to segregate electrophoretically in two main bands: isoforms JNK1 and JNK3 run in a 46 kDa band (which sometimes shows an additional weak band at a slightly higher mobility), and JNK2 in a 54 kDa band (Brecht et al., 2005). We quantified the aggregate expression of all three isoforms together. As a positive control for detection of JNK activation, we first measured JNK expression in hippocampal tissue harvested from naive animals and exposed in vitro to 20 μM anisomycin for 60 min, which produces substantial activation of JNK signaling. Phosphorylated JNK (pJNK) levels in ANI-treated tissue were 162% (± 8.0%, n=5, p<.01; Fig. 7A) of control, with a no significant change in total JNK expression (117 ± 6.8%, n=5, p>.05), yielding an increase in pJNK / total JNK ratio of 139% (± 5.4%, n=5, p<.01). This validated our ability to detect JNK activation by a known positive modulator.
Fig. 7.
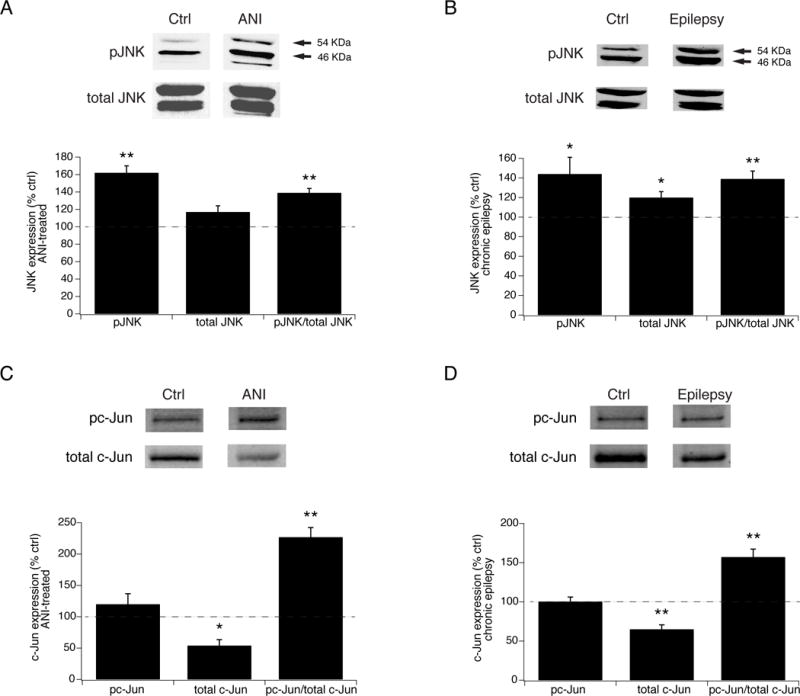
JNK signaling is hyperactivated in chronic epilepsy. A. Summary data showing increased JNK activation in hippocampal tissue from naive animals after in vitro treatment with ANI compared to untreated tissue. Shown are elevated phospho-JNK (pJNK) and phospho- to total JNK levels in ANI-exposed tissue, while total JNK levels remained unchanged. Above are representative blots against pJNK and total JNK in ANI-exposed and naive controls. Arrows denote 54 and 46 kDa bands. B. In hippocampal tissue from chronically epileptic animals, pJNK levels are increased compared to those from naive, non-epileptic controls, along with modestly activated total JNK levels, and an increased ratio of phospho- to total JNK expression. C. In vitro ANI treatment of hippocampal tissue from naive animals produced a significant increase in the fraction of phosphorylated c-Jun, while producing a decrease in the level of total c-Jun expression compared to untreated tissue. D. Phosphorylated c-Jun was increased in tissue from chronically epileptic animals compared to that in naive, nonepileptic animals, and similarly to ANI-exposed tissue, demonstrated a decrease in total c-Jun levels.
We then assayed JNK activation in epilepsy. Chronically epileptic animals showed elevated levels of activated pJNK at 144% of those in naive, nonepileptic controls (±17%, n=7, p<.05), with a mean phospho- to total JNK ratio of 139 ± 7.7% (n=7, p<.01). There was a modest increase in total JNK protein expression (120 ± 5.6% of control, n=7, p<.05, Fig. 7B) compared to naive controls as well. These results demonstrate that JNK signaling is hyperactivated in hippocampal tissue from chronically epileptic animals, with the degree of JNK hyperactivation seen in epileptic tissue a significant fraction of that seen with stimulation of JNK activity by ANI in naive tissue.
We next determined whether activated JNK produced downstream phosphorylation of its canonical target, c-Jun. We first validated that activation of JNK by ANI in naive tissue in vitro produced c-Jun phosphorylation. As shown in Fig. 7C, ANI produced a marked elevation of the fraction of phosphorylated c-Jun (pc-Jun / total c-Jun) to 227% of control (± 15.3%, n=4, p<.01), demonstrating that JNK activation had functional downstream effects on the phosphorylation of c-Jun, as expected. Interestingly, total c-Jun protein expression declined to 54% of control (±9.4%, p<.05), leaving the total amount of pc-Jun unchanged by ANI activation (120 ± 16.7% of control, p>.05). The mechanism underlying the acute decline of c-Jun expression is unclear, but this phenomenon has been observed previously following stimulation by ANI (Leppa et al., 1998; Morton et al., 2003). We then measured c-Jun phosphorylation in tissue from chronically epileptic animals (Fig. 7D). Similar to the effect seen with in vitro stimulation by ANI, the fraction of phosphorylated pc-Jun to total c-Jun was elevated at 157% of naive, nonepileptic controls (± 10.1%, n=5, p<.01). Likewise, the expression level of total c-Jun was decreased to 65% of control (± 5.6%, p< .01), leaving total pc-Jun expression unchanged from control (110 ± 5.6%, p>.05). The level of pc-Jun activation in epilepsy was concordant with the level of JNK activation, and demonstrates that activated JNK has functional downstream signaling actions in chronic epilepsy.
Discussion
In this study we translated a prior finding, that loss of p38 MAPK signaling in chronic epilepsy contributes to neuronal hyperexcitability, to an in vivo trial of pharmacologic MAPK modulation in order to test its effect on spontaneous seizure frequency in chronically epileptic animals. We employed the pilocarpine model of temporal lobe epilepsy, with prolonged 10 d periods of video-EEG monitoring during both baseline and treatment phases in within-subject comparisons, as a rigorous test of antiepileptic efficacy against spontaneous seizures. We found that inhibition of brain p38 MAPK activity produced a significant increase in seizure frequency, validating the role of this signaling pathway role in modulating seizure activity. Following an unexpected result from treatment with the non-specific MAPK activator anisomycin, we determined that partial inhibition of JNK activity produced a substantial, dose-dependent antiepileptic effect without causing overt behavioral abnormalities. Finding elevated JNK expression and activity in chronically epileptic animals supported the idea that JNK hyperactivation may be relevant to the pathogenesis of chronic epilepsy. These results represent the novel identification of JNK phosphorylation signaling as a potential antiepileptic target in temporal lobe epilepsy.
JNK inhibition produces an antiepileptic action
We demonstrated a significant reduction of spontaneous seizure frequency with inhibition of JNK activity by SP600125. Using two SP dosages, we observed a positive dose-response relationship, with the lower dose producing a 34% reduction of convulsive seizure frequency, while a two-fold higher dose produced a 57% reduction. In both cases, it is likely that only partial inhibition of JNK signaling was achieved. This may reflect poor tissue penetration of the drug, as even in vitro application of 40 μM SP to brain slices, a concentration that is four times the IC50 seen with application to cultured cells (Vogel et al., 2009), yielded only about 50% inhibition of JNK activity. Nonetheless, even partial JNK inhibition produced a significant antiepileptic effect. We included the lamotrigine data as a comparator for the antiepileptic effect of JNK inhibition. Since lamotrigine is a clinically validated antiepileptic drug active at a known concentration range (which we achieved in our study), the LTG data shows that partial JNK inhibition achieved a comparable magnitude of seizure frequency reduction as LTG dosed in the middle of the human therapeutic range. (However, there is no reason to suspect that LTG’s mechanism of action depends on MAPKs.)
This raises the possibility that with more complete inhibition of JNK activity, a more substantial antiepileptic effect might be achieved. JNK inhibition at the higher dose did not produce behavioral abnormalities in tests of open field locomotion.
Prior studies of the association between JNK and epilepsy have demonstrated acute increases in JNK activation following chemoconvulsant-induced SE or kindling (Mielke et al., 1999; Schauwecker, 2000; Jeon et al., 2000; Cole-Edwards et al., 2006). Knockout of the JNK3 isoform diminished the seizure class acutely provoked by kainate administration (Brecht et al., 2005), while knockdown of JNK-interacting protein (JIP3) decreased JNK activation and kindling induced by chemoconvulsants (Wang et al., 2015). Our study appears to be the first to demonstrate that JNK is hyperactivated in chronic epilepsy, and that its pharmacological inhibition reduces spontaneous seizure frequency. This demonstration in the pilocarpine model serves as a rigorous validation of the effects of MAPK modulation on convulsive seizures; since this model replicates complex partial and secondarily generalized seizures seen in humans with TLE, along with hippocampal neurodegeneration typical of mesial temporal sclerosis, these results may have relevance to human epilepsy. It is increasingly recognized that the use of animal models with clear similarities to human epilepsy may aid in the discovery of novel antiepileptic drugs, a pressing clinical need given the lack of progress over recent decades in the medical treatment of refractory epilepsy (Loscher, 2002; Grabenstatter et al., 2005; Loscher and Schmidt, 2011).
Our evidence for an antiepileptic action aside, the role of JNK activation in promoting neurodegeneration is its most well-established action on neuronal pathophysiology. Impending neuronal death following either kainate administration or kindling is associated with JNK activation (Cole-Edwards et al., 2006; Schauwecker, 2000). Both genetic deletion of JNK isoforms and pharmacological inhibition of JNK activity protect against excitotoxic neuronal loss from kainate or kindling (Chen et al., 2010; Spigolon et al., 2010; Yang et al., 1997). JNK activation has also been widely implicated in neurodegeneration acutely following ischemic insults (Borsello et al., 2003; Repici et al., 2007). Interestingly, chronic JNK hyperactivation has been shown in a variety of neurodegenerative disorders characterized by progressive neuronal loss, including Parkinson’s and Alzheimer’s diseases (Mehan et al., 2011; Ploia et al., 2011; Wang et al., 2012; Zhou et al., 2015), but thus far has not been studied in human epilepsy.
Inhibition of p38 MAPK is proconvulsant
We began this project to investigate whether modulation of MAPK signaling could affect seizure frequency in chronically epileptic animals. We had previously demonstrated that hippocampal p38 MAPK activity was diminished in epileptic animals, and contributed to neuronal hyperexcitability as measured in vitro. Our finding here that p38 MAPK inhibition significantly increases spontaneous seizure frequency validated our initial hypothesis. This adds to a growing literature showing dysregulation of phosphorylation signaling pathways in animal models of acquired epilepsy. The most notable example of this is the mammalian target of rapamycin (mTOR) pathway. Although mTOR is a kinase dysregulated in the genetic disorder tuberous sclerosis (TS), evidence has accumulated that it is also hyperactivated in models of acquired epilepsy (Guo et al., 2013; Wong, 2011). While mTOR inhibition in TS patients shows antiepileptic efficacy (Krueger et al., 2013), there is conflicting evidence as to whether mTOR inhibition has antiepileptic efficacy in animal models of acquired epilepsy (Heng et al., 2013; Huang et al., 2010). It is unlikely however that p38 MAPK will prove a target for AED development, since activation of p38 MAPK might have oncogenic potential (Engelberg, 2004). This study also suggests that p38 MAPK inhibition—a strategy pursued for some years in rheumatologic drug development—might have proconvulsive consequences. Indeed, clinical development of at least one p38 MAPK inhibitor was stopped due to unspecified CNS toxicity in animals, and subsequent inhibitor development has largely focused on agents that do not penetrate the blood-brain barrier (Dominguez et al., 2005; Chico et al., 2009). Despite these concerns, JNK inhibition may be a more promising target in refractory epilepsy since JNK activation has a clear pathologic association with neurodegeneration.
Limitations and future avenues for investigation
There are several limitations to the findings of this study, and several areas that may warrant further research. The unexpectedly modest degree of JNK antagonism in vivo with SP600125 suggests that further investigation of drug delivery parameters will be needed to produce more substantial JNK inhibition and to test its potential for a complete antiepileptic effect. An alternative approach might include use of D-JNKI1, a novel peptide that inhibits JNK from binding with its substrates (Antoniou et al., 2011; Martin and Arthur, 2012).
JNK encompasses three isoforms, with JNK3 highly implicated in CNS neurodegeneration. We did not separately quantify the expression of these isoforms in our epileptic animals; however, SP inhibits all JNK isoforms, and no subtype-specific antagonists are currently available, thus we cannot pharmacologically test whether the antiepileptic action of JNK inhibition is isoform-dependent.
Finally, despite the clear involvement of JNK activation in neurodegeneration, it is unclear how JNK activation exerts a proconvulsant effect. Several investigations have pointed to a role for JNK activity in neuronal plasticity under nonepileptic, physiological conditions (Haeusgen et al., 2009; Sherrin et al., 2011), but there is little understanding on how JNK influences the intrinsic and synaptic excitability of neurons. Although p38 MAPK is a strong modulator of HCN channels, which in turn affect neuronal excitability and seizure threshold, JNK does not appear to regulate HCN channels, at least under normal conditions (Poolos et al., 2006). Nor is it clear whether JNK hyperactivation is a feature of other animal models of epilepsy or of human epilepsy. These are avenues for further investigation to better understand whether modulation of JNK activity might have clinical potential in refractory epilepsy syndromes.
Conclusions
In this study we have demonstrated for the first time that in vivo pharmacological inhibition of c-Jun N-terminal kinase activity exerts an antiepileptic action, and that JNK is hyperactivated in chronic epilepsy. We also showed that inhibition of p38 MAPK is proconvulsive, in line with our prior results that showed p38 MAPK inhibition increases neuronal excitability when measured in vitro. These findings support the idea that JNK activity may be contributing to maintenance of the epileptic state, and that JNK may be a target for future development of novel antiepileptic therapies.
Highlights.
Inhibition of c-Jun N-terminal kinase (JNK) produces a dose-dependent antiepileptic effect in an animal model of epilepsy.
JNK is hyperactivated in hippocampal tissue from chronically epileptic animals.
JNK inhibition did not produce behavioral abnormalities.
Inhibition of p38 mitogen-activated protein kinase (p38 MAPK) yielded a proconvulsive effect.
Acknowledgments
We appreciated the technical assistance of Katie Krupin and Vicky Herrera in animal surgery and EEG interpretation. Funding support was from NIH grant NS050229 to NPP. Lamotrigine isethionate was a gift from GlaxoSmithKline, Inc.
Abbreviations
- AED
antiepileptic drug
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ANI
anisomycin
- C1-5
Racine class 1-5
- Cav3.2
voltage-gated calcium channel type 3.2
- CI
confidence interval
- CSF
cerebrospinal fluid
- EEG
electroencephalogram
- GABAA
γ-aminobutyric acid type A
- HCN
hyperpolarization-activated, cyclic nucleotide-gated
- i.c.v
intracerebroventricular
- i.p
intraperitoneal
- JIP3
JNK-interacting protein
- JNK
c-Jun N-terminal kinase
- Kv4.2
voltage-gated potassium channel type 4.2
- LTG
lamotrigine
- mTOR
mammalian target of rapamycin
- p38 MAPK
p38 mitogen-activated protein kinase
- PB
phenobarbital
- pJNK
phosphorylated JNK
- SB
SB203580
- SE
status epilepticus
- SEM
standard error of mean
- Ser
serine
- SP
SP600125
- Thr
threonine
- TLE
temporal lobe epilepsy
- TS
tuberous sclerosis
- Tyr
tyrosine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing financial interests.
References
- Antoniou X, Falconi M, Di Marino D, Borsello T. JNK3 as a therapeutic target for neurodegenerative diseases. J Alzheimers Dis. 2011;24:633–642. doi: 10.3233/JAD-2011-091567. [DOI] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Brecht S, Kirchhof R, Chromik A, Willesen M, Nicolaus T, Raivich G, Wessig J, Waetzig V, Goetz M, Claussen M, Pearse D, Kuan CY, Vaudano E, Behrens A, Wagner E, Flavell RA, Davis RJ, Herdegen T. Specific pathophysiological functions of JNK isoforms in the brain. Eur J Neurosci. 2005;21:363–377. doi: 10.1111/j.1460-9568.2005.03857.x. [DOI] [PubMed] [Google Scholar]
- Cardoso A, Carvalho LS, Lukoyanova EA, Lukoyanov NV. Effects of repeated electroconvulsive shock seizures and pilocarpine-induced status epilepticus on emotional behavior in the rat. Epilepsy Behav. 2009;14:293–299. doi: 10.1016/j.yebeh.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Chen X, Wu J, Hua D, Shu K, Wang JZ, Li L, Lei T. The c-Jun N-terminal kinase inhibitor SP600125 is neuroprotective in amygdala kindled rats. Brain Res. 2010;1357:104–114. doi: 10.1016/j.brainres.2010.07.082. [DOI] [PubMed] [Google Scholar]
- Chico LK, Van Eldik LJ, Watterson DM. Targeting protein kinases in central nervous system disorders. Nat Rev Drug Discov. 2009;8:892–909. doi: 10.1038/nrd2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P. Protein kinases–the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Cole-Edwards KK, Musto AE, Bazan NG. c-Jun N-terminal kinase activation responses induced by hippocampal kindling are mediated by reactive astrocytes. J Neurosci. 2006;26:8295–8304. doi: 10.1523/JNEUROSCI.1986-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davson H, Welch K, Segal MB. The secretion of the cerebrospinal fluid. In: Anonymous, editor. Physiology and Pathophysiology of the Cerebrospinal Fluid. London: Churchill Livingstone; 1987. pp. 189–246. [Google Scholar]
- Detour J, Schroeder H, Desor D, Nehlig A. A 5-month period of epilepsy impairs spatial memory, decreases anxiety, but spares object recognition in the lithium-pilocarpine model in adult rats. Epilepsia. 2005;46:499–508. doi: 10.1111/j.0013-9580.2005.38704.x. [DOI] [PubMed] [Google Scholar]
- Dominguez C, Powers DA, Tamayo N. p38 MAP kinase inhibitors: many are made, but few are chosen. Curr Opin Drug Discov Devel. 2005;8:421–430. [PubMed] [Google Scholar]
- Eastman CL, Verley DR, Fender JS, Temkin NR, D’Ambrosio R. ECoG studies of valproate, carbamazepine and halothane in frontal-lobe epilepsy induced by head injury in the rat. Exp Neurol. 2010;224:369–388. doi: 10.1016/j.expneurol.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelberg D. Stress-activated protein kinases-tumor suppressors or tumor initiators? Semin Cancer Biol. 2004;14:271–282. doi: 10.1016/j.semcancer.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Grabenstatter HL, Ferraro DJ, Williams PA, Chapman PL, Dudek FE. Use of chronic epilepsy models in antiepileptic drug discovery: the effect of topiramate on spontaneous motor seizures in rats with kainate-induced epilepsy. Epilepsia. 2005;46:8–14. doi: 10.1111/j.0013-9580.2005.13404.x. [DOI] [PubMed] [Google Scholar]
- Guo D, Zeng L, Brody DL, Wong M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS One. 2013;8:e64078. doi: 10.1371/journal.pone.0064078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusgen W, Boehm R, Zhao Y, Herdegen T, Waetzig V. Specific activities of individual c-Jun N-terminal kinases in the brain. Neuroscience. 2009;161:951–959. doi: 10.1016/j.neuroscience.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Heng K, Haney MM, Buckmaster PS. High-dose rapamycin blocks mossy fiber sprouting but not seizures in a mouse model of temporal lobe epilepsy. Epilepsia. 2013;54:1535–1541. doi: 10.1111/epi.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zhang H, Yang J, Wu J, McMahon J, Lin Y, Cao Z, Gruenthal M, Huang Y. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. 2010;40:193–199. doi: 10.1016/j.nbd.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inostroza M, Cid E, Menendez de la Prida L, Sandi C. Different emotional disturbances in two experimental models of temporal lobe epilepsy in rats. PLoS One. 2012;7:e38959. doi: 10.1371/journal.pone.0038959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iordanov MS, Pribnow D, Magun JL, Dinh T-H, Pearson JA, Chen SL-Y, Magun BE. Ribotoxic stress response: activation of the stress-activated protine kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol. 1997;17:3373–3381. doi: 10.1128/mcb.17.6.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob TC, Moss SJ, Jurd R. GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9:331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SH, Kim YS, Bae CD, Park JB. Activation of JNK and p38 in rat hippocampus after kainic acid induced seizure. Exp Mol Med. 2000;32:227–230. doi: 10.1038/emm.2000.37. [DOI] [PubMed] [Google Scholar]
- Jung S, Warner LN, Pitsch J, Becker AJ, Poolos NP. Rapid loss of dendritic HCN channel expression in hippocampal pyramidal neurons following status epilepticus. J Neurosci. 2011;31:14291–14295. doi: 10.1523/JNEUROSCI.1148-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Bullis JB, Lau IH, Jones TD, Warner LN, Poolos NP. Downregulation of dendritic HCN channel gating in epilepsy is mediated by altered phosphorylation signalling. J Neurosci. 2010;30:6678–6688. doi: 10.1523/JNEUROSCI.1290-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Jones TD, Lugo J, Jr, Sheerin AH, Miller JW, D’Ambrosio R, Anderson AE, Poolos NP. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DA, Wilfong AA, Holland-Bouley K, Anderson AE, Agricola K, Tudor C, Mays M, Lopez CM, Kim MO, Franz DN. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74:679–687. doi: 10.1002/ana.23960. [DOI] [PubMed] [Google Scholar]
- Leppa S, Saffrich R, Ansorge W, Bohmann D. Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. EMBO J. 1998;17:4404–4413. doi: 10.1093/emboj/17.15.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Gu B, He XP, Joshi RB, Wackerle HD, Rodriguiz RM, Wetsel WC, McNamara JO. Transient inhibition of TrkB kinase after status epilepticus prevents development of temporal lobe epilepsy. Neuron. 2013;79:31–38. doi: 10.1016/j.neuron.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: Ways out of the current dilemma. Epilepsia. 2011;52:657–678. doi: 10.1111/j.1528-1167.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- Loscher W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res. 2002;50:105–123. doi: 10.1016/s0920-1211(02)00073-6. [DOI] [PubMed] [Google Scholar]
- Martin KJ, Arthur JS. Selective kinase inhibitors as tools for neuroscience research. Neuropharmacology. 2012;63:1227–1237. doi: 10.1016/j.neuropharm.2012.07.024. [DOI] [PubMed] [Google Scholar]
- Mehan S, Meena H, Sharma D, Sankhla R. JNK: a stress-activated protein kinase therapeutic strategies and involvement in Alzheimer’s and various neurodegenerative abnormalities. J Mol Neurosci. 2011;43:376–390. doi: 10.1007/s12031-010-9454-6. [DOI] [PubMed] [Google Scholar]
- Mielke K, Brecht S, Dorst A, Herdegen T. Activity and expression of JNK1, p38 and ERK kinases, c-Jun N-terminal phosphorylation, and c-jun promoter binding in the adult rat brain following kainate-induced seizures. Neuroscience. 1999;91:471–483. doi: 10.1016/s0306-4522(98)00667-8. [DOI] [PubMed] [Google Scholar]
- Morton S, Davis RJ, McLaren A, Cohen P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 2003;22:3876–3886. doi: 10.1093/emboj/cdg388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller CJ, Groticke I, Bankstahl M, Loscher W. Behavioral and cognitive alterations, spontaneous seizures, and neuropathology developing after a pilocarpine-induced status epilepticus in C57BL/6 mice. Exp Neurol. 2009;219:284–297. doi: 10.1016/j.expneurol.2009.05.035. [DOI] [PubMed] [Google Scholar]
- Ploia C, Antoniou X, Sclip A, Grande V, Cardinetti D, Colombo A, Canu N, Benussi L, Ghidoni R, Forloni G, Borsello T. JNK plays a key role in tau hyperphosphorylation in Alzheimer’s disease models. J Alzheimers Dis. 2011;26:315–329. doi: 10.3233/JAD-2011-110320. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Bullis JB, Roth MK. Modulation of h-channels in hippocampal pyramidal neurons by p38 mitogen-activated protein kinase. J Neurosci. 2006;26:7995–8003. doi: 10.1523/JNEUROSCI.2069-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Warner LN, Humphreys SZ, Williams S. Comparative efficacy of combination drug therapy in refractory epilepsy. Neurology. 2012;78:62–68. doi: 10.1212/WNL.0b013e31823ed0dd. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Johnston D. Dendritic ion channelopathy in acquired epilepsy. Epilepsia. 2012;53(Suppl 9):32–40. doi: 10.1111/epi.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroenceph Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rakhade SN, Fitzgerald EF, Klein PM, Zhou C, Sun H, Hunganir RL, Jensen FE. Glutamate receptor 1 phosphorylation at serine 831 and 845 modulates seizure susceptibility and hippocampal hyperexcitability after early life seizures. J Neurosci. 2012;32:17800–17812. doi: 10.1523/JNEUROSCI.6121-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repici M, Centeno C, Tomasi S, Forloni G, Bonny C, Vercelli A, Borsello T. Time-course of c-Jun N-terminal kinase activation after cerebral ischemia and effect of D-JNKI1 on c-Jun and caspase-3 activation. Neuroscience. 2007;150:40–49. doi: 10.1016/j.neuroscience.2007.08.021. [DOI] [PubMed] [Google Scholar]
- Rosser EM, Morton S, Ashton KS, Cohen P, Hulme AN. Synthetic anisomycin analogues activating the JNK/SAPK1 and p38/SAPK2 pathways. Org Biomol Chem. 2004;2:142–149. doi: 10.1039/b311242j. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. Seizure-induced neuronal death is associated with induction of c-Jun N-terminal kinase and is dependent on genetic background. Brain Res. 2000;884:116–128. doi: 10.1016/s0006-8993(00)02888-2. [DOI] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrin T, Blank T, Todorovic C. c-Jun N-terminal kinases in memory and synaptic plasticity. Rev Neurosci. 2011;22:403–410. doi: 10.1515/RNS.2011.032. [DOI] [PubMed] [Google Scholar]
- Shorvon SD. Handbook of Epilepsy Treatment. Oxford: Blackwell Science Ltd; 2000. p. 167. [Google Scholar]
- Spigolon G, Veronesi C, Bonny C, Vercelli A. c-Jun N-terminal kinase signaling pathway in excitotoxic cell death following kainic acid-induced status epilepticus. Eur J Neurosci. 2010;31:1261–1272. doi: 10.1111/j.1460-9568.2010.07158.x. [DOI] [PubMed] [Google Scholar]
- Toyoda I, Bower MR, Leyva F, Buckmaster PS. Early activation of ventral hippocampus and subiculum during spontaneous seizures in a rat model of temporal lobe epilepsy. J Neurosci. 2013;33:11100–11115. doi: 10.1523/JNEUROSCI.0472-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res. 1983;9:315–335. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- Vogel J, Anand VS, Ludwig B, Nawoschik S, Dunlop J, Braithwaite SP. The JNK pathway amplifies and drives subcellular changes in tau phosphorylation. Neuropharmacology. 2009;57:539–550. doi: 10.1016/j.neuropharm.2009.07.021. [DOI] [PubMed] [Google Scholar]
- Wang G, Pan J, Chen SD. Kinases and kinase signaling pathways: potential therapeutic targets in Parkinson’s disease. Prog Neurobiol. 2012;98:207–221. doi: 10.1016/j.pneurobio.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Wang Z, Chen Y, Lu Y, Chen X, Cheng L, Mi X, Xu X, Deng W, Zhang Y, Wang N, Li J, Li Y, Wang X. Effects of JIP3 on epileptic seizures: Evidence from temporal lobe epilepsy patients, kainic-induced acute seizures and pentylenetetrazole-induced kindled seizures. Neuroscience. 2015;300:314–324. doi: 10.1016/j.neuroscience.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Williams AD, Jung S, Poolos NP. Protein kinase C bidirectionally modulates Ih and hyperpolarization-activated cyclic nucleotide-gated (HCN) channel surface expression in hippocampal pyramidal neurons. J Physiol. 2015;593:2779–2792. doi: 10.1113/JP270453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M. Rapamycin for treatment of epilepsy: antiseizure, antiepileptogenic, both, or neither? Epilepsy Curr. 2011;11:66–68. doi: 10.5698/1535-7511-11.2.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Wang M, Du Y, Zhang W, Bai M, Zhang Z, Li Z, Miao J. Inhibition of c-Jun N-terminal kinase activation reverses Alzheimer disease phenotypes in APPswe/PS1dE9 mice. Ann Neurol. 2015;77:637–654. doi: 10.1002/ana.24361. [DOI] [PubMed] [Google Scholar]