

1. Disease characteristics
1.1 Name of the Disease (synonyms)
Cantú syndrome (Hypertrichotic osteochondrodysplasia).
1.2 OMIM# of the Disease
239850.
1.3 Name of the Analysed Genes or DNA/Chromosome Segments
ABCC9 at 12p12.1; KCNJ8 also at 12p12.1.
1.4 OMIM# of the Genes
ABCC9 – *601439.
KCNJ8 – *600935.
1.5 Mutational spectrum
All reported variants to date are heterozygous single-nucleotide variants which are predicted to have an activating effect on the function of ATP-sensitive potassium channels generated by the SUR2 and Kir6.1 proteins encoded by ABCC9 and KCNJ8. At least 15 different variants in ABCC9 and two variants in KCNJ8 have been reported to date1, 2, 3, 4 (list of variants available in the GeneReviews entry for Cantú syndrome;5 see also the LOVD database for ABCC9 at http://databases.lovd.nl/shared/genes/ABCC9). Variants in ABCC9 to date cluster in transmembrane domain 2, with ~80% of reported patients having variants in this domain. Recurrent ABCC9 variants affecting the amino acid residues p.Arg1116 and p.Arg1154 have been identified in approximately half of the individuals in whom pathogenic variants have been found to date. Copy number variants (small or large scale) have not been associated with Cantú syndrome.
1.6 Analytical methods
Sequencing of all coding exons and intron-exon boundaries of ABCC9 by Sanger sequencing, as part of a targeted gene panel for Mendelian syndromes, or by exome or whole genome sequencing. Sequencing of KCNJ8 by Sanger sequencing as a second line test, or concurrently with sequencing of ABCC9 as part of panel, exome or genome sequencing.
1.7 Analytical validation
Sanger sequencing is predicted to detect >99% of variants in the targeted regions of the two known genes. For other sequencing methods, sensitivity will depend on the characteristics of the test, including coverage of the coding regions and intron-exon boundaries, read depth and sequencing quality.
1.8 Estimated frequency of the disease
Incidence at birth (‘birth prevalence') or population prevalence. If known to be variable between ethnic groups, please report):
Unknown. There is no known variation in incidence or prevalence among ethnic groups.
1.9 Diagnostic setting

Comment: Testing is useful for confirming or making the diagnosis of Cantú syndrome in an affected individual. Testing of relatives allows assessment of reproductive risks – for example, in the parents of an affected child. Prenatal diagnosis is an option where one parent is affected and a pathogenic variant has been identified. Targeted non-invasive prenatal diagnosis (NIPD) is an emerging option for families in which the father is affected. NIPD may be possible where a previous affected child has a de novo variant, but the suitability of this will depend on the healthcare setting.
2. Test characteristics
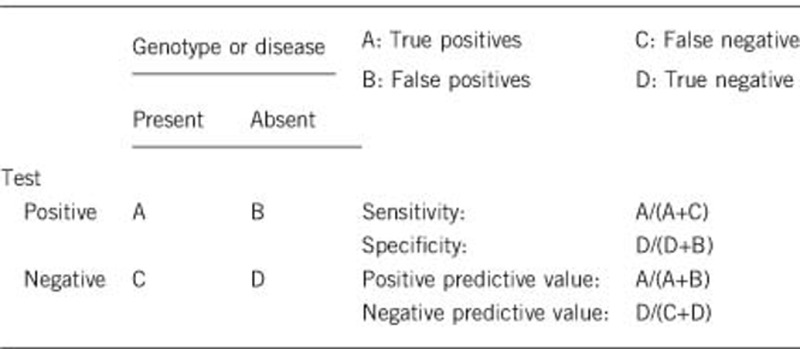
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Nearly 100%.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
Nearly 100%.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The sensitivity of a clinical diagnosis can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Of 30 patients with a clinical diagnosis of Cantú syndrome included in two papers,1, 2 28 had variants identified in ABCC9. One of the two with no identified variant was subsequently found to have a different disorder, with a duplication of the 17q24.2-q24.3 region. This suggests that the clinical sensitivity of testing for variants in ABCC9 is >95% addition of KCNJ8 sequencing would be expected to improve this to close to 100%. However, this assumes accurate clinical phenotyping. Depending on the specific features in an affected individual, there are a number of disorders which can present with similar features. In addition to 17q24.2-q24.3 copy number variants, these include lysosomal storage disorders, Beckwith-Wiedemann syndrome, endocrine disorders such as congenital hypothyroidism and acromegaly, Wiedemann-Steiner syndrome, Zimmerman-Laband and Temple-Baraitser syndromes and Berardinelli-Seip congenital lipodystrophy. Treatment with minoxidil can also produce very similar clinical features.6 The clinical sensitivity of testing will depend at least in part on the extent to which alternative diagnoses have been excluded, on clinical grounds or by specific testing, prior to testing for variants in ABCC9 and KCNJ8.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The specificity of a clinical diagnosis can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
If the disease is not present, no pathogenic variants should be found in either ABCC9 or KCNJ8, and thus clinical specificity should be 100%.
2.5 Positive clinical predictive value (life-time risk of developing the disease if the test is positive)
Clinical features are usually present from childhood. Thus, positive clinical predictive value is mainly relevant in the context of prenatal diagnosis, in the setting of a known familial pathogenic variant. In that setting, positive clinical predictive value is likely to be close to 100%, with only the small risk of laboratory error or misinterpretation of a variant as pathogenic likely to lead to a genotype-positive fetus being clinically unaffected at birth.
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
100% if there is a proven pathogenic variant in the index case.
Index case in that family had not been tested:
This depends on whether the clinical diagnosis in the proband is correct. If a confident clinical diagnosis has been made in the proband, negative clinical predictive value would be expected to be high (but difficult to quantify) in this scenario as well.
3. Clinical utility
3.1 (Differential) diagnostics: The tested person is clinically affected
(to be answered if in 1.9 ‘A' was marked)
3.11 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
The diagnosis of Cantú syndrome can be made on clinical grounds alone (hypertrichosis, distinctive facial features and the presence of other common features such as polyhydramnios and large birthweight).7 However, diagnostic imaging can make a contribution to the diagnosis. This may include skeletal survey looking for the variable mild osteochondrodysplasia, as well as cardiomegaly. The burden of this to the patient relates mainly to radiation exposure. Echocardiography may also be useful. Patent ductus arteriosus is common. There may be cardiac enlargement, with high cardiac output probably due to reduced peripheral vascular resistance (not a cardiomyopathy, although the heart is often enlarged).8 Blood pressure measurement (standing and prone) may be helpful. Pericardial effusion has also been observed, and some patients have pulmonary hypertension. Biochemistry is not diagnostic, but investigations such as urinary glycosaminoglycans and leucocyte enzymology for storage disorders may be helpful in excluding differential diagnoses (also low burden to the patient). Similarly, chromosome microarray could be considered to exclude 17q24.2-q24.3 copy number variants. Echocardiography, biochemistry and microarray testing all have a low burden for the patient.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
The investigations described above are relatively inexpensive individually. If there is strong clinical suspicion of Cantú syndrome, the combined expense of radiological investigations, biochemical testing and chromosome microarray are comparable to or exceed the cost of proceeding directly to sequencing of ABCC9/KCNJ8. However, echocardiography is indicated even in a patient with a confirmed diagnosis, because it is required for clinical management.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe)Yes, see 3.1.4.
If the test result is negative (please describe)Depends on clinical manifestations in the individual who has been tested.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no test has been done (please describe)?
There are no specific options in relation to this disorder.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes—if a pathogenic variant is identified in the proband, testing can be offered to other family members as appropriate. Risks to offspring of family members can be accurately assessed and options such as prenatal diagnosis offered.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Clinical features can be highly variable. In particular, hypertrichosis may be much less prominent in fair-haired individuals. Testing of a mildly affected family member in whom the diagnosis is in doubt may confirm or exclude the diagnosis, making additional investigations (as discussed in 3.1.2) unnecessary. In the proband, if there is early suspicion of the diagnosis, extensive investigations for alternative diagnoses (such as skeletal survey, testing for storage disorders and endocrine testing for hypertrichosis) may be obviated by confirmation of the diagnosis of Cantú syndrome.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Penetrance appears to be close to 100%, so this situation is unlikely to arise.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes. Children of affected individuals are at 1 in 2 risk of being affected. The risk to sibs where parents are unaffected is low, but recurrences have been reported in this setting,9 presumably due to somatic or germline mosaicism. In either case, prenatal diagnosis using molecular testing is enabled by a positive genetic test result in the index patient. Targeted non-invasive prenatal diagnosis could also be used in the case of a de novo variant or when the father is affected.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
The main application of testing (other than the medical issues discussed in 3.1.4) is for reproductive purposes. In addition to the option of prenatal diagnosis (see 3.4.1) identification of a pathogenic variant in an adult makes it possible to consider preimplantation genetic diagnosis.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics, 6th Joint call for European Research Projects on Rare Diseases (JTC 2014, project CantuTreat), and by a grant from the Children's Discovery Institute at Washington University (CH-MI-II-2015-488).
Footnotes
The authors declare no conflict of interest.
References
- Harakalova M, van Harssel JJT, Terhal PA et al: Dominant missense mutations in ABCC9 cause Cantú syndrome. Nat Genet 2012; 44: 793–796. [DOI] [PubMed] [Google Scholar]
- van Bon BWM, Gilissen C, Grange DK et al: Cantú Syndrome is Caused by Mutations in ABCC9. Am J Hum Genet 2012; 90: 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstein CA, Towne MC, Luquette LJ et al: Mutation of KCNJ8 in a patient with Cantú syndrome with unique vascular abnormalities – Support for the role of K(ATP) channels in this condition. Eur J Hum Genet 2013; S6: 678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper PE, Reutter H, Woelfle J et al: Cantú Syndrome Resulting from Activating Mutation in the KCNJ8 Gene. Hum Mutat 2014; 35: 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange DK, Nichols CG, Singh GK. Cantú syndrome and related disorders. In: Pagon RA, Adam MP, Ardinger HH et al (eds): GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle, 2014; 1993–2016. Available from https://www.ncbi.nlm.nih.gov/books/NBK246980/. [PubMed] [Google Scholar]
- Nguyen KH, Marks JG: Pseudoacromegaly induced by the long-term use of minoxidil. J Am Acad Derm 2003; 48: 962–965. [DOI] [PubMed] [Google Scholar]
- Scurr I, Wilson L, Lees M et al: Cantú Syndrome: Report of Nine New Cases and Expansion of the Clinical Phenotype. Am J Med Genet 2011; 155: 508–518. [DOI] [PubMed] [Google Scholar]
- Levin MD, Singh GK, Zhang HX et al: KATP channel gain-of-function leads to increased myocardial L-type Ca2+ current and contractility in Cantu syndrome. Proc Natl Acad Sci USA 2016; 113: 6773–6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantú JM, García-Cruz D, Sánchez-Corona J, Hernández A, Nazará Z: A Distinct Osteochondrodysplasia with Hypertrichosis – Individualization of a Probable Autosomal Recessive Entity. Hum Genet 1982; 60: 36–41. [DOI] [PubMed] [Google Scholar]