

Abstract
This study tries to find detectable signals of gene flow of Sub-Saharan origin into the Mediterranean in four genomic regions previously associated with coronary artery disease. A total of 366 single-nucleotide polymorphisms were genotyped in 772 individuals from 10 Mediterranean countries. Population structure analyses were performed, in which a noticeable Sub-Saharan component was found in the studied samples. The overall percentage of this Sub-Saharan component presents differences between the two Mediterranean coasts. D-statistics suggest possible Sub-Saharan introgression into one of the studied genomic regions (10q11). We also found differences in linkage disequilibrium patterns between the two Mediterranean coasts, possibly attributable to differential Sub-Saharan admixture. Our results confirm the potentially important role of human demographic history when performing epidemiological studies.
Introduction
Cardiovascular diseases are the leading cause of morbidity and mortality in Western societies.1 According to the World Health Organisation, 17.3 million people died of cardiovascular diseases in 2008, representing 30% of all deaths worldwide (http://www.who.int/cardiovascular_diseases/about_cvd/en/). One of the most common types of cardiovascular disease is coronary artery disease (CAD), which has increased by 44% during the last 20 years in North Africa and the Middle East (http://www.who.int/whr/2013/report/en/), and is manifested as stable and unstable angina, myocardial infarction or sudden cardiac death. In all these heart conditions, genetic and environmental factors interact to determine the clinical phenotype.2 Recently, several genetic variants have been robustly associated with CAD through genome-wide association studies (GWASs).3
Genotype and phenotype variation can present geographic patterns, as is for instance the case of height across Europe.4 Such patterns can be shaped by selection and/or various demographic phenomena (population expansions, subdivisions, gene flow and/or bottlenecks). As an example, a recent study reported that genetic differences in CAD risk among worldwide populations are due to demographic processes.5 In some occasions, allele frequencies at specific genomic regions increase through introgression from other populations with which there was admixing. These processes can also affect linkage disequilibrium (LD) patterns across populations. As a result, allele frequencies and LD patterns in introgressed regions for a given population tend to be more similar to distantly related populations than to others otherwise closer. An example of this is the Neanderthal introgression into Eurasian populations.6
In this study, we carried out a fine-scale genetic analysis on a broad collection of European populations using variation from four genomic regions putatively or provenly2 associated with CAD risk. These four regions have shown disparity in the association of specific markers with CAD risk between Southern European and North African samples.7 To see if this disparity could be partially attributed to introgression, we looked for signals of gene flow of Sub-Saharan origin into the Mediterranean at the four CAD-related genomic regions mentioned above.
Materials and methods
Populations
Our analysis was based on genetic data from 772 individuals of Southern European and North African ancestry. The studied populations encompass 10 Mediterranean countries: Spain (Basque Country, Girona, Valencia and Las Alpujarras); France (Toulouse); Italy (Sardinia and Sicily); Bosnia-Herzegovina; Greece (Crete); Turkey; Morocco (Khenifra and Chouala); Tunisia; Algeria (M'zab); Libya; and Jordan (Bedouin Jordanians and non-Bedouin Jordanians).7, 8 In addition, we considered the Yoruba (YRI) and Han Chinese (CHB) populations from the phase III 1000 Genomes Project in order to have genetic representation of Sub-Saharan Africa and East Asia. The geographic location of the samples is shown in Figure 1.
Figure 1.

Populations studied. 1, Chouala; 2, Khenifra; 3, Las Alpujarras; 4, Valencia; 5, Basque Country; 6, Girona; 7, Toulouse; 8, M'zab; 9, Tunisia; 10, Sardinia; 11, Sicily; 12, Libya; 13, Bosnia-Herzegovina; 14, Crete; 15, Turkey; 16, General Jordan; 17, Bedouin.
Genetic data
Samples were genotyped for a total of 366 single-nucleotide polymorphisms (SNPs) distributed along four CAD-associated chromosomal regions: 1p13.3, 1q41, 9p21 and 10q112 (Table 1). Genotyping was carried out with the Custom GoldenGate Panel (Illumina Inc., San Diego, CA, USA) genotyping platform. SNPs were previously selected as representatives of common variation in each of the four genomic regions according to the following criteria: (i) coverage of one SNP every ~1.5 Kb; (ii) minor allele frequency (MAF)>0.05 in European populations; (iii) avoiding markers in strong LD in European populations (r2>0.8); and (iv) giving priority to tag SNPs as well as markers previously reported to be associated with CAD.7 All of the participants signed an informed consent form before sample donation, and the Ethics Committee of the University of Barcelona approved the study. Individual genotypes and SNP information is available through the EMBL-EBI ArrayExpress public repository (accession number: E-MTAB-5265).
Table 1. Chromosomal regions studied.
| Chromosomal region | Number of SNPs | Span (kbp) | Known genes |
|---|---|---|---|
| 1p13.3 | 59 | 150 | CELSR2, PSRC1, MYBPHL, SORT1 |
| 1q41 | 37 | 100 | TAF1A, MIA3, AIDA |
| 9p21 | 155 | 300 | CDKN2A, CDKN2B |
| 10q11 | 115 | 200 | CXCL12 |
Abbreviation: SNP, single-nucleotide polymorphism.
Statistical analysis
Standard quality control was performed with Plink v1.9.9 Only one marker was excluded for not fitting Hardy–Weinberg proportions (P-value <10−5). Per-individual and per-locus genotype missingness was zero, whereas none of the SNPs was monomorphic. In all subsequent analyses, we used all Mediterranean populations together with YRI and CHB. We first explored population structure in our samples with PCA and ancestry component analysis, using Plink and Admixture v1.3.0,10 respectively.
We further used qpdstat from AdmixTools11 to calculate D-statistics. We applied qpdstat to each of the four genomic regions separately, setting jackknife block size to 0.02–0.06 cm depending on the number of markers in each genomic region. We set block size to a smaller than the default value in order to compensate for the relatively low number of SNPs analysed. Each Mediterranean population was compared with CHB, YRI, and a dummy individual representing a hypothetical outgroup. This dummy individual was homozygous for the ancestral allele for each of the studied SNPs as defined by comparison with six primate species. The Z-scores reported by AdmixTools indicate whether there has been gene flow between two out of four given populations when one of these populations is an outgroup (in our case the dummy individual). In particular, for the topology (((X, CHB); YRI); Outgroup) shown in Figure 2a, where X is a given Mediterranean population, each of the four genomic regions is tested to define whether allele frequencies are more similar between X and CHB (which would denote lack of Sub-Saharan gene flow) or between X and YRI (which could be interpreted as suggestive of Sub-Saharan gene flow). Positive Z-scores indicate more similarity of a Mediterranean population with YRI, whereas negative Z-scores indicate more similarity of CHB with YRI. We set a threshold of |Z-score|>2 necessary for a D-statistic to be significant.
Figure 2.

(a) Topology assumed for the D-statistics, where X represents a Mediterranean population. (b) Boxplots showing the results of the D-statistics. The points represent the Z-score values (abscissas axis) obtained for each genomic region in a given Mediterranean population X (ordinates axis). As the topology assumed is (((X, CHB), YRI), Ancestral), significant Z-score-positive values indicate a gene flow event between YRI and X. The significance threshold is set at ±2. See legend at Figure 3 for clarification of the population names.
In order to check whether there were significant differences in the Z-scores between North African and Southern European populations for each genomic region, we ran a Mann–Whitney rank sum test. Furthermore, for the genomic regions for which the Mann–Whitney test was significant (P<0.0125 after Bonferroni correction for multiple testing), populations were further grouped into four geographic categories (Southwest Europe, Southeast Europe, Northwest Africa and Northeast Africa) and a follow-up Kruskal–Wallis test was carried out to see if the structuring pattern could be further explained by an East–West axis.
We also analysed the differences in LD patterns between the Southern European and North African groups using varLD v1.012 with 1000 iterations per test. Finally, we obtained phased haplotypes with SHAPEIT13 for nine LD blocks that we identified with Haploview14 and explored haplotype sharing between Southern Europe, North Africa and Sub-Saharan groups with Venn diagrams.
Results
Population structure
Figure 3a shows the centroid coordinates for each population along the first two principal components. We can see a North–South separation along PC1, with PC2 defining an East–West gradient. In general, populations were visually separated in four groups (North African, Southern European, Sub-Saharan and East Asian) with the Mediterranean populations naturally showing the closest proximity. The centroids match roughly the geography – with the exception of the Basques who appear as an outlier in the Southern European cluster.
Figure 3.

(a) PCA based on allele frequencies. Big green and red points are the centroids of YRI and CHB populations, respectively. Triangles represent the centroids of North African populations, whereas small circles represent the centroids of the Southern European populations. (b) Admixture analysis for K=3 ancestral components represented as pie charts. The pie chart of each population is located on its geographic position. ALP, Las Alpujarras; BED, Bedouin; BOS, Bosnia-Herzegovina; BSC, Basque Country; CHB, Han Chinese from Beijing; CHO, Chouala; CRT, Crete; GIR, Girona; JOR, Jordan; KHE, Khenifra; LIB, Libya; MZB, M'zab;SAR, Sardinia; SIC, Sicily; TOU, Toulouse; TUN, Tunisia; TUR, Turkey; VAL, Valencia; YRI, Yoruba. A full colour version of this figure is available at the European Journal of Human Genetics journal online.
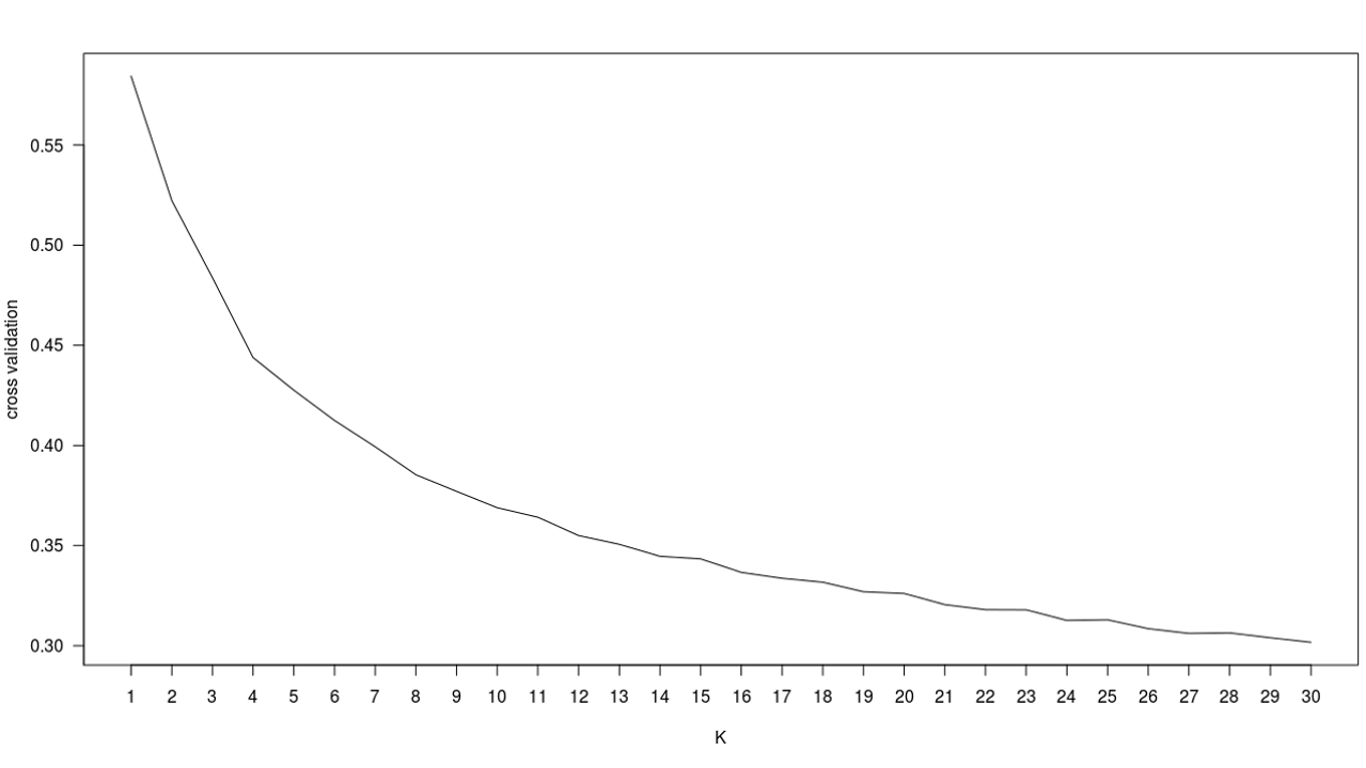
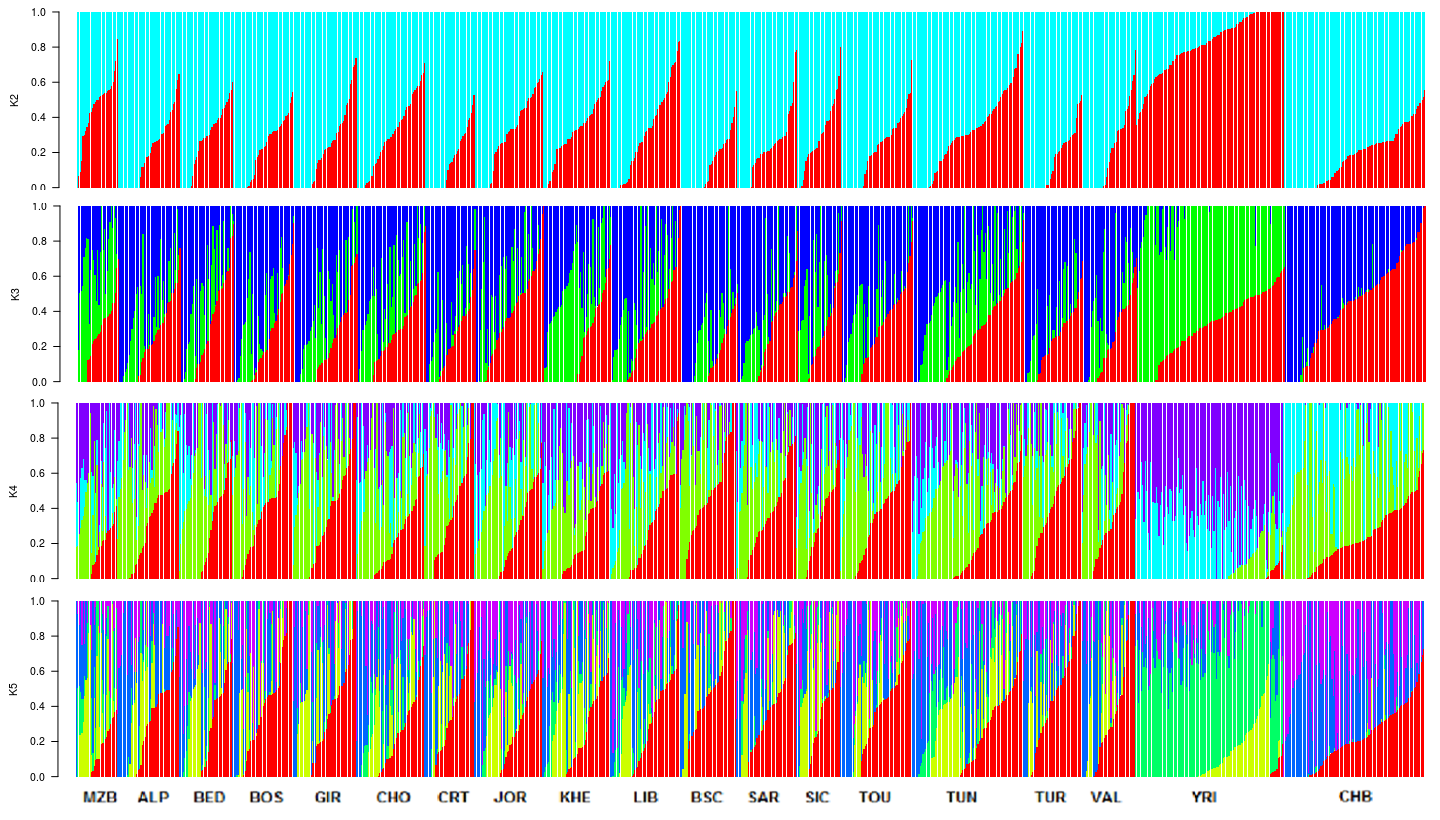
Regarding the Admixture analysis, due to the lack of an optimal cross-validation value for the tested numbers of ancestral components (K={1, 2, …, 30} Supplementary Information 1), we focused our analysis on K=3 ancestral components, roughly matching the three major geographic regions of our study: Mediterranean, East Asia and Sub-Saharan Africa. Figure 3b shows the mixture profiles of the studied populations. We observed a component that was predominant in Sub-Saharan Africa (green), as well as two other components: one that was present in both the Mediterranean and the Asian samples at varying proportions (blue), and one that was present in all samples (red). Due to the small number of SNPs used, these two components were not representative of continent-level geographic regions. The Mediterranean samples show rather homogeneous proportions for all three components, with slightly higher rates of Sub-Saharan ancestry in the North African populations. For visual comparisons, we provide the Admixture plots for K={2, 3, 4, 5} in Supplementary Information 2.
Signals of Sub-Saharan gene flow
The virtually indistinguishable mixture patterns in all Mediterranean populations motivated the search for signals of differential Sub-Saharan gene flow with more sensitive methods – in particular AdmixTools (Figure 2b). Despite the effort, we did not obtain consistent positive Z-scores for either 1q41 or 9p21, and we even found suggestively negative values for 1p13.3, contrasting a possible gene flow from YRI to the Mediterranean samples for these three regions. However, Z-scores were consistently positive and close to the significance threshold (|Z-score|>2) for the 10q11 region. Moreover, Mann–Whitney comparisons showed significant differences in Z-scores between Southern Europe and North Africa (Table 2), which can be refined in some cases including a West–East differentiation grouping, namely Southwest Europe and Northeast Africa (Mann–Whitney, P=0.02).
Table 2. P-values for the D-statistic comparisons between the two Mediterranean coasts.
| 1p13.3 | 1q41 | 9p21 | 10q11 | |
|---|---|---|---|---|
| Southern Europe vs North Africa | 0.16 | 0.23 | 0.06 | 0.01 |
| SW Europe vs SE Europe vs NW Africa vs NE Africa | 0.03 | |||
| SW Europe vs SE Europe | 0.07 | |||
| SW Europe vs NW Africa | 0.38 | |||
| SW Europe vs NE Africa | 0.02 | |||
| SE Europe vs NW Africa | 0.86 | |||
| SE Europe vs NE Africa | 0.06 | |||
| NW Africa vs NE Africa | 0.20 |
Abbreviations: NE, North East; NW, North West; SE, South East; SW, South West. Bold number denotes values below significance (P-value <0.0125 for first two tests, P-value <0.0083 for the last two tests); numbers in italics denote nominal significance.
Differences in LD patterns
All comparisons of LD patterns among the four studied genomic regions were significant (P<0.01). Pairwise score matrices are reported in the Supplementary Information 3–6. The highest LD pattern differences occurred at the 9p21 genomic region, followed by those at the 10q11 genomic region. This could be reflecting the differences in the degree of Sub-Saharan gene flow between the two Mediterranean coasts also seen in the structure analyses, as higher levels of admixture between two relatively non-admixed populations result in higher levels of LD in the admixed individuals.15
Haplotype sharing
We identified a total of nine LD blocks in the four studied genomic regions, which correspond to a total 2372 unique haplotypes. Comparisons between Southern European, North African and Sub-Saharan African groups showed that there is generally higher haplotype sharing between North Africa and Sub-Saharan Africa than between Southern Europe and Sub-Saharan Africa, providing additional independent evidence for greater Sub-Saharan gene flow towards the former (Figure 4).
Figure 4.

Venn diagram containing the haplotypes from all the LD blocks identified in the four genomic regions studied. The population groups are Southern Europe, North Africa and Sub-Saharan Africa (Yoruba).
Discussion
This study provides insights into how demographic events that are stochastic by nature (such as introgression) have the potential of affecting the differential geographic distribution of variants associated with common diseases. Specifically, through the analysis of genotype data from top CAD risk loci, we suggest that gene flow of Sub-Saharan origin may have played a role in the current geographic distribution of variants associated with CAD.
Our Mediterranean samples presented a considerable proportion of Sub-Saharan admixture, suggesting introgression in at least some of the four genomic regions. The Sub-Saharan component was noticeably more prevalent in North Africa than in Southern Europe, a fact also reflected in the elevated proportion of shared haplotypes of YRI with North Africa than with Southern Europe (Supplementary Information 7). These results are in accord with previous studies suggesting a more intense Sub-Saharan gene flow into North Africa than into Southern Europe due to geographic proximity and/or the potential role of the Mediterranean sea as a genetic barrier.8, 16, 17, 18, 19, 20, 21 Among the European populations, the ones that present a higher Sub-Saharan proportion are Sicily, Girona, Valencia and Andalusia (26%, 22%, 19% and 17%, respectively). This observation matches historical of North African influence in these regions during the Middle Ages.22 Likewise, the LD differences found between the two Mediterranean coasts match the observation of North African populations presenting higher levels of Sub-Saharan admixture.
The results from the D-statistics together with those from Admixture also suggest a potential gene flow of Sub-Saharan origin into the Mediterranean at least for the 10q11 region. This region includes CXCL12, a gene that codes for a chemokine ligand linked to cardiovascular disease with protective effects.23 It is also worth noting that potential signals of balancing selection have been identified at 10q11,8 implying that natural selection could have maintained this signal of Sub-Saharan gene flow in this genomic region by favouring admixed individuals against cardiovascular disease. Further research is warranted to shed more light on this hypothesis.
It is important to note that the relatively small size of the studied chromosomal regions and the low number of markers pose a limitation to the robustness of the results obtained, with potential bias also due to fact that the SNP used in the analyses was ascertained primarily in European populations. In addition, the studied loci are not the only ones associated with CAD and therefore an extension to more disease-associated loci and samples would be desirable. The small number of SNPs is probably also the reason behind the lack of an optimal cross-validation value in the Admixture analysis, warrantying caution at interpreting the obtained results. Finally, given that none of the Z-scores for 10q11 passed the established significance threshold, though very close to it, our results are subject to be false positives. Future work could address efficiently the above issues by analysing a higher number of regions and markers.
Our results build on the notion of a differential gene flow from Sub-Saharan Africa into North Africa that, according to recent studies, could have taken place 750–1200 years ago during the trans-Saharan slave trade.24 Sub-Saharan introgression into Europe could have been the result of indirect contact of Europeans with North Africans already admixed with Sub-Saharan populations.25, 26 This two-step Sub-Saharan introgression into Europe could be an interesting subject of future research validated through demographic simulations.
Acknowledgments
This research was supported by the Ministerio de Ciencia e Innovación CGL2011-27866 project. We would like to thank all collaborators who provided samples of general populations: N Harich and M Kandil (Morocco), H Chabaani (Tunisia and Libya), M Sadiq (Jordan), JM Dougoujon (France and Algeria), C Calò (Sardinia), N Pojskic (Bosnia), N Moschonas (Crete, Greece), N Bissar-Tadmouri (Turkey), J Santamaría (Valencia, Spain) and F Luna (Las Alpujarras, Spain). All volunteers are gratefully acknowledged for sample donation.
Footnotes
Supplementary Information accompanies this paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
The authors declare no conflict of interest.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Wilson PWF, Agostino RBD, Levy D, Belanger AM, Silbershatz H, Kannel WB: Prediction of coronary heart disease using risk factor categories. Circulation 1998; 97: 1837–1847. [DOI] [PubMed] [Google Scholar]
- Girelli D, Martinelli N, Peyvandi F, Olivieri O: Genetic architecture of coronary artery disease in the genome-wide era: implications for the emerging ‘golden dozen' loci. Semin Thromb Hemost 2009; 35: 671–682. [DOI] [PubMed] [Google Scholar]
- Lieb W, Vasan RS: Genetics of coronary artery disease. Circulation 2013; 128: 1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MR, Hemani G, Medina-gomez C et al: Population genetic differentiation of height and body mass index across Europe. Nat Genet 2015; 47: 1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona E, Dudley JT, Butte AJ: Extreme evolutionary disparities seen in positive selection across seven complex diseases. PLoS One 2010; 5: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW et al: A draft sequence of the Neandertal genome. Science 2010; 328: 710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti D, Via M, Carreras-Torres R et al: Analysis of genomic regions associated with coronary artery disease reveals continent-specific single nucleotide polymorphisms in North African populations. J Epidemiol 2016; 643: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti D, Carreras-Torres R, Esteban E, Via M, Moral P: Potential signals of natural selection in the top risk loci for coronary artery disease: 9p21 and 10q11. PLoS One 2015; 10: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ: Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015; 4: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DH, Novembre J, Lange K: Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009; 19: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Moorjani P, Luo Y et al: Ancient admixture in human history. Genetics 2012; 192: 1065–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong RT, Teo Y: varLD: a program for quantifying variation in linkage disequilibrium patterns between populations. Bioinformatics 2010; 26: 1269–1270. [DOI] [PubMed] [Google Scholar]
- O'Connell J, Gurdasani D, Delaneau O et al: A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet 2014; 10: e1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ: Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265. [DOI] [PubMed] [Google Scholar]
- Shiheng T, Rongmei Z, Jianhua C: A population genetics model of linkage disequilibrium in admixed populations. Chinese Sci Bull 2001; 46: 193–197. [Google Scholar]
- Athanasiadis G, González-pérez E, Esteban E, Dugoujon J, Stoneking M, Moral P: The Mediterranean Sea as a barrier to gene flow: evidence from variation in and around the F7 and F12 genomic regions. BMC Evol Biol 2010; 10: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanasiadis G, Moral P: Spatial principal component analysis points at global genetic structure in the Western Mediterranean. J Hum Genet 2013; 58: 762–765. [DOI] [PubMed] [Google Scholar]
- Bosch E, Calafell F, Comas D, Oefner PJ, Underhill PA: High-resolution analysis of human Y-chromosome variation shows a sharp discontinuity and limited gene flow between Northwestern Africa and the Iberian peninsula. Am J Hum Genet 2001; 68: 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capelli C, Redhead N, Romano V et al: Population structure in the Mediterranean Basin: a Y chromosome perspective. Ann Hum Genet 2006; 70: 207–225. [DOI] [PubMed] [Google Scholar]
- Comas D, Calafell F, Benchemsi N, Helal A, Lefranc G, Stoneking M: Alu insertion polymorphisms in NW Africa and the Iberian Peninsula: evidence for a strong genetic boundary through the Gibraltar Straits. Hum Genet 2000; 107: 312–319. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez E, Esteban E, Via M et al: Population relationships in the Mediterranean revealed by autosomal genetic data (Alu and Alu/STR Compound Systems). Am J Phys Anthropol 2010; 141: 430–439. [DOI] [PubMed] [Google Scholar]
- Humphreys RS: Islamic History: a Framework for Inquiry. Princeton University Press:Princeton,1991. [Google Scholar]
- Döring Y, Pawig L, Weber C, Noels H: The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular disease. Front Psychol 2014; 5: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn BM, Botigue LR, Gravel S et al: Genomic ancestry of North Africans supports back-to-Africa migrations. PLoS Genet 2012; 8: e1002397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piancatelli D, Canossi A, Aureli A et al: Human leukocyte antigen-A, -B, and -Cw polymorphism in a Berber population from North Morocco using sequence-based typing. Tissue Antigens 2004; 63: 158–172. [DOI] [PubMed] [Google Scholar]
- Coudray C, Olivieri A, Achilli A, Pala M, Melhaoui M, Cherkaoui M: The complex and diversified mitochondrial gene pool of Berber populations. Ann Hum Genet 2009; 73: 196–214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.