

SUMMARY
The incidence of esophageal adenocarcinoma (EAC) has increased in recent decades. Increases in incidence have been attributed to changes in the prevalence of risk factors for EAC; however, the extent to which these changes explain increases in EAC incidence has not been studied in detail. We used age-period-cohort analysis to estimate changes in the incidence of EAC among white males by age, time period, and birth cohort. Incidence rates per 100,000 individuals were analyzed from 1973 to 2012. Hierarchical Poisson models were used to estimate age, period, and cohort effects, whereby age-specific incidence rates were nested within periods and cohorts. The prevalence of obesity for each time period and birth cohort was included in the model as a fixed-effect. Incidence increased with advancing age (β = 0.12, P < 0.01). There were significant period and birth cohort effects, although the period effect was much larger than the cohort effect. The period effect decreased dramatically when obesity was included as a fixed effect, while the small cohort effect remained unchanged. Results suggest much of the increase in the incidence of EAC can be attributed to a period effect, which may be due to changes in the prevalence of obesity over time.
Keywords: body mass index, esophageal neoplasms, incidence, male, SEER program, time factors
INTRODUCTION
The incidence of esophageal adenocarcinoma (EAC) has rapidly increased since the 1970s,1–4 with annual increases of up to 8% between 1973 and 1996 and 2% since 1996.5 Changes in the incidence patterns of EAC have been largely attributed to changes in the prevalence of risk factors. Obesity and gastroesophageal reflux disease (GERD), both well-established risk factors of EAC,6,7 have increased since the 1970s.8,9 More than one-third of persons living in the U.S. are obese,8 while the prevalence of GERD symptoms (e.g. heartburn, acid reflux) has more than doubled in the last 20 years.9 Both the increases in obesity and GERD coincide with the rise in EAC. Although the increase in EAC has slowed in recent years,5,10 it remains one of the few cancers in the U.S. with a rising incidence.11
Although changes in risk factors across time have been extensively documented, the extent to which these changes explain increases in the incidence of EAC is poorly understood. Further, relatively little attention has been given to cohort effects (i.e. changes across groups of individuals who experience events at the same age during any given time period). Because birth cohorts often have different exposures to behavioral and environmental risk factors, cohort effects are evident in many cancers and chronic diseases.12 Cohort effects are distinct from period effects, which represent variations over time periods that influence all age groups simultaneously.12 Several studies have analyzed secular trends in EAC incidence rates,2,3,5,10,13–15 but by comparison, few have quantified period or cohort effects.
We used age-period-cohort (APC) analysis to estimate changes in the incidence of EAC among white males by age, period, and cohort. APC analysis, widely used in sociology and demography research, combines information on respondent age, time period of observation, and birth cohort to track the prevalence of a health outcome over time.12 These models estimate independent age effects (i.e. distribution of the outcome across the life course due to the biological process of aging), period effects (i.e. secular trends in the prevalence of an outcome that occur in all ages), and cohort effects (i.e. variation in the outcome among those born in or around the same year).
The purpose of this study was to (1) estimate the differential contributions of age, period, and birth cohort to the increasing incidence of EAC, and (2) determine how changes in the prevalence of obesity account for incidence patterns by period and cohort.
MATERIALS AND METHODS
EAC incidence
Incidence of EAC among white males was derived from the National Cancer Institute's (NCI) Surveillance, Epidemiology, and End Results (SEER) program. We limited the study population to white males because EAC is far more common among men than women (4.8 per 100,000 vs. 0.6 per 100,000) and whites than blacks (2.6 per 100,000 vs. 0.6 per 100,000). SEER 9 registries include Atlanta, Connecticut, Detroit, Hawaii, Iowa, New Mexico, San Francisco—Oakland, Seattle—Puget Sound, and Utah, approximately 9.5% of the U.S. population. Incidence rates per 100,000 individuals within the population were analyzed from 1973 to 2012. EAC was defined anatomically as located in the esophagus (International Classification of Disease for Oncology, Third Edition [ICD-O-3] codes 150-159) and histologically as adenocarcinoma (ICD-O-3 codes 8140-8141, 8143-8145, 8190-8231, 8260-8263, 8310, 8401, 8480-8490, 8550-8551, 8570-8574, 8576).
Obesity prevalence
Obesity prevalence was determined using data from the National Health and Nutrition Examination Survey (NHANES), phases I (1971–1975), II (1976–1980), and III (1988–1994) and the continuous cycles (1999–2012). NHANES is the only study that provides estimates on anthropometric measures for the U.S. population. The survey examines a nationally representative sample of about 5,000 persons each year. These persons are located in counties across the country, 15 of which are visited in each data collection cycle. Data collection procedures include a standardized physical examination, where a trained health technologist and recorder work together to collect a complete set of anthropometric measures (e.g. weight, height, upper leg and arm length) from survey participants. Obesity is measured as a body mass index (BMI) ≥30 kg/m2.
To estimate the prevalence of period-specific obesity, we pooled obesity prevalence across all age groups for white males (age ≥21 years) during the corresponding NHANES periods of data collection. Similarly, we estimated the prevalence of cohort-specific obesity by averaging obesity prevalence for white males (age ≥21 years) in each birth cohort across the same NHANES time periods. Because NHANES studies were not conducted in certain years (e.g. 1981–1987), we interpolated obesity prevalence by averaging the prevalence for the previous and subsequent periods or cohorts. To account for the possibility that EAC incidence increases linearly with BMI (vs. a threshold effect of BMI ≥30), we also estimated period- and cohort-specific mean and median BMI using a similar approach.
Statistical analysis
Age, period, and cohort effects can be estimated with a variety of statistical techniques. We used hierarchical Poisson models to nest age-specific incidence rates within levels of period and birth cohort. Specifically, the models estimate fixed effects of age and random effects of period and birth cohort.12 Hierarchical APC analysis avoids the identification problem of linear APC regression models because age, period, and cohort are not assumed to be linear and additive at the same level of analysis. These models enhance the ability to assess the independent effect of each by estimating variance components. Variance components are interpreted as the period or cohort effect. The effect of each cohort is averaged over all periods, and the contribution of each period is averaged over all cohorts. Model components and parameters are defined in detail as Supporting Information.
Hierarchical APC models also provide a framework to incorporate covariates that may explain period and/or cohort effects. To determine the extent to which obesity accounts for the observed variance by period and cohort, we included in the model the prevalence of period- and cohort-specific obesity as covariates with fixed effects. A change in the variance components (in both size and statistical significance) provides evidence for a period and/or cohort effect attributable to obesity. We also specified models with period- and cohort- specific mean and median BMI to account for the possibility that incidence of EAC increases linearly with BMI (vs. a threshold effect of BMI ≥30). Lastly, we developed anthropometry-lagged models that included a lag period of 5, 10, and 15 years between period-obesity and incidence rates.
Cohort and period were divided into approximate 5-year categories for 21 birth cohorts (1885–1991) and 9 time periods (1973–2012). Continuous age (21–85+ years) was centered at the mean to facilitate interpretation of the random effects and reduce the association between the linear and quadratic age terms.
All statistical analyses were conducted using SAS version 9.3 (SAS Institute, Cary, North Carolina, USA).
RESULTS
Period-obesity ranged from 12.1% (1973–1974, 1975–1979) to 40.3% (2010–2012), and cohort-obesity ranged from 10.0% (1885–1890) to 36.8% (1985–1991) (Fig. 1).
Fig. 1.

Estimates of fixed level covariates: obesity prevalence by time period, 1973–2010; B) Mean and median BMI by time period, 1973–2012; C) Obesity prevalence by birth cohort, 1885–1991; D) Mean and median BMI by birth cohort, 1885–1991. Period– and cohort–obesity were dervied from NHANES phases I (1971–1975), II (1976–1980), and III (1988–1994) and the continuous cycles (1999–2012). All estimates were calculated with sampling weights that account for nonresponse and oversampling in NHANES.
Age-period incidence rates are shown in Figure 2. Incidence rates generally increased from age 40 through 70 and slightly decreased in the oldest age groups (≥80 years). Incidence rates for all age groups were relatively low through the early 1980s, with marked increases starting in 1990. Age-birth cohort incidence rates (Fig. 3) increased across successive birth cohorts for older age groups (≥60 years) but remained low among the youngest age groups. Period-birth cohort incidence (Fig. 4) increased across the 1900–1930 birth cohorts and decreased successively across more recent birth cohorts.
Fig. 2.
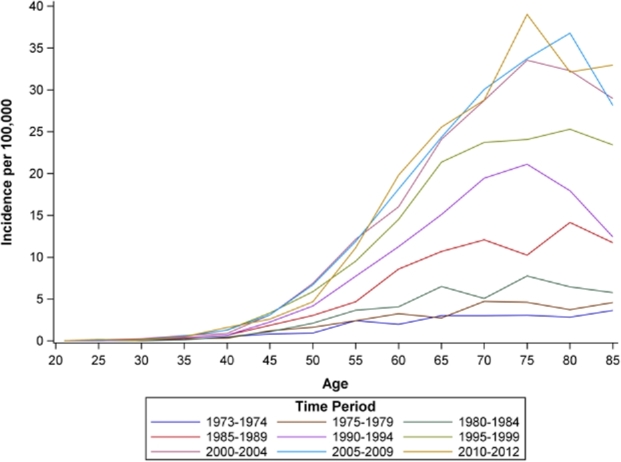
Age by time period incidence rates of esophageal adenocarcinoma for white males, SEER 9, 1973-2012. Note: In the above figure, age (in years) is along the x-axis, and each line represents incidence rates across age during an approximate 5-year time period. Incidence of EAC increased with advancing age, and rates were highest in more recent time periods (e.g. 2010–2012).
Fig. 3.
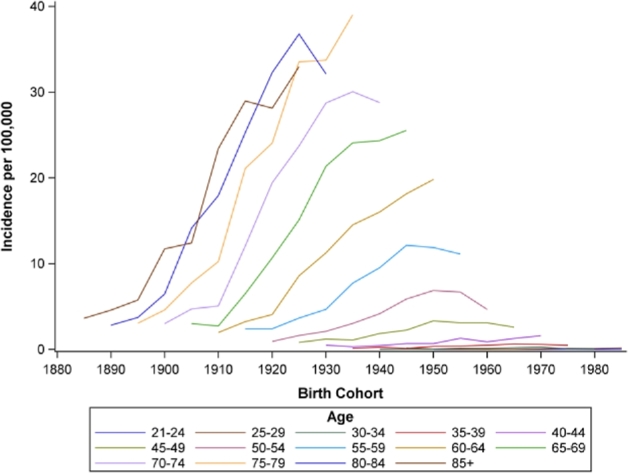
Birth cohort by age incidence rates of esophageal adenocarcinoma for white males, SEER 9, 1973–2012. Note: In the above figure, birth cohort is along the x-axis, and each line represents incidence rates across birth cohort for approximate 5-year age groups. Incidence of EAC increased across successive birth cohorts for older age groups (>=60 years) but remained low among the youngest age groups.
Fig. 4.
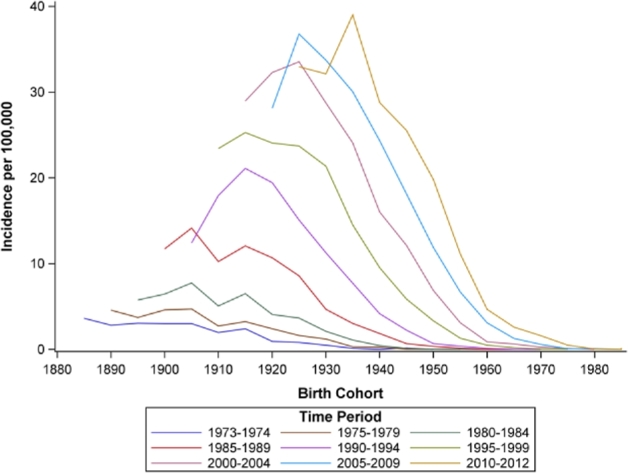
Birth cohort by time period incidence rates of esophageal adenocarcinoma for white males, SEER 9, 1973–2012. Note: In the above figure, birth cohort is along the x-axis, and each line represents incidence rates across birth cohort for approximate 5-year time periods. Incidence of EAC increased across the 1900-1930 birth cohorts and decreased successively across more recent birth cohorts.
Results of the hierarchical APC analysis (Table 1, Model A) demonstrate significant age effects (β = 0.116, P < 0.001). Incidence rates increased with increasing age and decreased slightly in the oldest ages (β = −0.002, P < 0.001). For example, predicted incidence at age 80 years across all periods and cohorts was 15.3/100,000 (95% confidence interval [CI]: 9.5/100,000, 24.6/100,000) and 14.3/100,000 (95% CI: 8.9/100,000, 23.0/100,000) at age 85 years. There was also a period effect (σ = 0.498, P = 0.025), suggesting, independent of both age and birth cohort, incidence rates increased monotonically over time. The observed cohort effect (σ = 0.027, P = 0.046) was smaller by comparison.
Table 1.
Hierarchical Poisson model estimates of incidence rates of esophageal adenocarcinoma for white males, SEER 9, 1973–2012
| Model A | Model B | Model C | Model D | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fixed effects | Coefficient | SE | P | Coefficient | SE | P | Coefficient | SE | P | Coefficient | SE | P |
| Model for mean | ||||||||||||
| Intercept | −10.317 | 0.240 | <0.001 | −12.643 | 0.312 | <0.001 | −29.762 | 2.385 | <0.001 | −31.590 | 3.262 | <0.001 |
| Age (linear, centered) | 0.116 | 0.002 | <0.001 | 0.125 | 0.004 | <0.001 | 0.123 | 0.003 | <0.001 | 0.120 | 0.003 | <0.001 |
| Age (quadratic) | −0.002 | <0.001 | <0.001 | −0.002 | <0.001 | <0.001 | −0.002 | <0.001 | <0.001 | −0.002 | <0.001 | <0.001 |
| Period Obesity (%) | 6.101 | 1.082 | <0.001 | |||||||||
| Cohort obesity (%) | 4.582 | 1.455 | 0.002 | |||||||||
| Period mean BMI | 0.453 | 0.070 | <0.001 | |||||||||
| Cohort Mean BMI | 0.267 | 0.087 | 0.002 | |||||||||
| Period median BMI | 0.553 | 0.101 | <0.001 | |||||||||
| Cohort median BMI | 0.252 | 0.095 | 0.008 | |||||||||
| Variance Components | Variance | SE | P | Variance | SE | P | Variance | SE | P | Variance | SE | P |
| Period | ||||||||||||
| Intercept | 0.498 | 0.255 | 0.025 | 0.052 | 0.030 | 0.038 | 0.045 | 0.026 | 0.039 | 0.080 | 0.045 | 0.039 |
| Cohort | ||||||||||||
| Intercept | 0.027 | 0.016 | 0.046 | 0.034 | 0.018 | 0.028 | 0.032 | 0.017 | 0.032 | 0.033 | 0.019 | 0.036 |
BMI, body mass index; SE, standard error.
When the prevalence of period- and cohort- obesity were added as fixed covariates to the model (Table 1, Model B), linear and quadratic age remained significant (linear: β = 0.125, P < 0.001; quadratic: β = −0.002, P < 0.001). Period-obesity (β = 6.101, P < 0.001) and cohort-obesity (β = 4.582, P = 0.008) were also significant fixed effects. The period effect decreased (σ = 0.052, P = 0.038), but the cohort effect did not change substantively (σ = 0.034, P = 0.028). Similar results were observed when mean (Model C) and median (Model D) BMI were included as fixed covariates.
Models with 5-, 10-, and 15-year lag periods are shown in Table 2. The magnitude of period and cohort effects were similar to that observed in models without lag periods. However, the cohort effect decreased as lag time increased, from 0.032 in the 5-year model to 0.016 in the 15-year model. The period effect decreased from 0.072 in the 10-year model to 0.035 in the 15-year model.
Table 2.
Hierarchical Poisson model estimates of incidence rates of esophageal adenocarcinoma, including lag periods, of white males, SEER, 1973–2012
| Model | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 5-Year Time Lag* | 10-Year Time Lag† | 15-Year Time Lag‡ | |||||||
| Fixed effects | Coefficient | SE | P | Coefficient | SE | P | Coefficient | SE | P |
| Model for mean | |||||||||
| Intercept | −12.254 | 0.364 | <0.001 | −11.727 | 0.395 | <0.001 | −11.072 | 0.353 | <0.001 |
| Age (linear, centered) | 0.126 | 0.004 | <0.001 | 0.126 | 0.003 | <0.001 | 0.125 | 0.003 | <0.001 |
| Age (quadratic) | −0.002 | <0.001 | <0.001 | −0.002 | <0.001 | <0.001 | −0.002 | <0.001 | <0.001 |
| Period obesity (%) | 5.588 | 1.418 | <0.001 | 4.929 | 1.741 | 0.005 | 3.812 | 1.655 | 0.021 |
| Cohort obesity (%) | 4.268 | 1.400 | 0.002 | 3.523 | 1.297 | 0.007 | 2.481 | 1.220 | 0.042 |
| Variance Components | Variance | SE | P | Variance | SE | P | Variance | SE | P |
| Period | |||||||||
| Intercept | 0.075 | 0.044 | 0.046 | 0.072 | 0.047 | 0.062 | 0.035 | 0.026 | 0.086 |
| Cohort | |||||||||
| Intercept | 0.029 | 0.016 | 0.032 | 0.021 | 0.012 | 0.038 | 0.016 | 0.010 | 0.052 |
SE, standard error.
*Limited to incidence rates from SEER, 1975–2012.
†Limited to incidence rates from SEER, 1980–2012.
‡Limited to incidence rates from SEER, 1985–2012.
DISCUSSION
The results of our study suggest the increasing incidence of EAC is a complex function of age, time period, and birth cohort. We observed that much of the rise in incidence is driven by a period effect. Others have similarly reported large increases in incidence over time, with annual increases of 2–8% since the 1970s.5,11 Our study extends the findings of previous research by demonstrating that, not only has incidence increased over time, but that the increase is likely attributable to the growing prevalence of obesity. Many have hypothesized the increase in incidence is related to trends in obesity prevalence because obesity is independently associated with a risk of both EAC16 and Barrett's esophagus.17,18 Although its role in EAC has been recognized for many years, the extent to which trends in the prevalence of obesity might account for changes in incidence has not been extensively studied. Incorporating obesity as a fixed effect in our model allowed us to quantify its contribution to changes in incidence patterns.
Considerable evidence links obesity to EAC risk. The relationship between obesity and EAC was first reported in case-control studies in the 1990s, which showed an elevated risk of cancer for the heaviest level of BMI compared to normal weight.6,19–21 These findings have since been validated by large, population-based cohort studies22–24 that also suggest higher BMI increases risk. To date, most research has focused on the relationship between obesity and GERD as the mechanical pathway linking BMI with EAC. Obesity predisposes patients to reflux through a variety of mechanisms, including increased intra-abdominal pressure, delayed gastric emptying, relaxation of the lower esophageal sphincter, and loss of the angle of His. These mechanisms are supported by the dose-dependent relationship between BMI and GERD, whereby the prevalence of GERD increases with increasing levels of BMI.25,26 More recent evidence suggests there may be an indirect metabolic effect of abdominal obesity on risk of EAC.27,28 Although the exact mechanism by which obesity affects EAC pathogenesis has not been completely elucidated, substantial evidence provides support for the association between obesity and EAC.
We also observed a birth cohort effect on incidence patterns, which was not explained when obesity was included as a fixed covariate in the model. Some have suggested the decline in Helicobacter pylori colonization, which is inversely associated with risk of EAC,29 has contributed to rises in incidence.30 The prevalence of H. pylori in western countries has decreased in the last 30 years, and several studies have shown that much of this decrease is due to a birth cohort effect.31 There are many proposed mechanisms by which H. pylori may decrease risk of EAC,30 but most have examined the role of H. pylori in suppressing gastric acid production and preventing damage to the distal esophagus when reflux arises.32 Others have argued that H. pylori only plays a minor role in the sharp increase in incidence rates or is merely a maker of some other risk factor (e.g. changes in microbiota). Our study underscores the need for a better understanding of the critical factors involved with cohort-mechanisms, including H. pylori or microbiota, which may be related to risk of EAC.
Our findings are similar to other APC studies of EAC, both in the U.S. and Europe, which have also shown independent period and cohort effects. The methods used across these studies vary; many use log-linear regression analysis,4,33–35 where coefficients are constrained to be equal, while others have developed multistage models that incorporate features of the carcinogenic process in estimating effects by age, period, and cohort.36–38 Despite different methodological approaches, the findings are consistent and support the conclusions reached in this study. Increases in incidence can be attributed to both a period and cohort effect. Efforts to include explanatory variables (e.g. symptomatic GERD, obesity) in models, however, have had less consistent results. Some research suggests trends in EAC incidence do not match corresponding trends in lifestyle-related risk factors, including obesity.37 Yet, a different study reports BMI is the predominant driver of disease progression.38 Differences in our results and those of others may be explained by differences in model assumptions, measurement of explanatory variables (e.g. self-reported BMI), and outcome (i.e. absolute vs. relative increases in incidence).
There are several strengths of this study. We used a new and innovative form of APC analysis, which has not been previously applied to cancer incidence rates. By developing a mixed effects model, we were able to avoid the identification problem of linear APC models and independently estimate the contribution of age, period, and birth cohort to incidence rates. The mixed effects model also allowed us to quantify the impact of obesity on increasing incidence rates. Data for the study were derived from two large, population-based sources (SEER, NHANES), which have a similar age and racial distribution of the entire U.S. population.
An important limitation of the models developed for our study is that they are not models of etiology and cannot provide information on the specific mechanism by which obesity is related to carcinogenesis. Our models do not distinguish between the many factors that have led to increases in obesity prevalence (e.g. diet, physical inactivity) that could also be related to risk. There may be other etiologic factors or exposures that cannot be accounted for in the model, either because their relationship with risk is still unknown or the prevalence is not monitored in population-based sources with sufficiently long follow-up. This limitation highlights the importance of robust data sources to fully understand the evolution of the disease over time. There is also some evidence to suggest increases in EAC predated SEER data,39 which may result in an underestimate of the period effect. Finally, our analysis was also limited to white men. Incidence of EAC is lower in other race-sex groups, and there may be different contributions of age, period, birth cohort, and explanatory covariates in these population subgroups.
EAC is one of the few cancers in the U.S. with a rising incidence, and incidence is expected to increase in the coming years. Our results tell an unambiguous story regarding the role of obesity in EAC: changes in incidence have been largely driven by increases in the prevalence of obesity over time. Although we observed a small cohort effect, the magnitude of the period effect and contribution of obesity was much larger by comparison. This has important implications for when and where to intervene along the EAC continuum. Specifically, efforts related to the prevention of EAC may be best spent reducing the prevalence of obesity. Given the low absolute risk of EAC in the general population, it may be worth considering whether directing obesity prevention at populations with an increased risk profile, including obese persons with GERD or Barrett's esophagus, is appropriate in preventing EAC. Causal mechanisms and pathways by which obesity increases cancer risk are still debated, but targeting efforts at weight reduction to these high risk groups may be an effective strategy to reduce the burden of EAC.
Acknowledgments
This work was supported by the National Institutes of Health (Grant T32 DK07634).
SUPPORTING INFORMATION
Supplementary data are available at DOTESO online.
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
References
- 1. Blot W, Devesa S, Kneller R et al. Rising incidence of adenocarcinoma of the esophagus and gastric cardia. JAMA 1991; 265: 1287–9. [PubMed] [Google Scholar]
- 2. Devesa S, Blot W, Fraumeni J Jr. Changing patterns in the incidence of esophageal and gastric carcinoma in the United States. Cancer 1998; 83: 2049–53. [PubMed] [Google Scholar]
- 3. Pohl H, Welch H. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst 2005; 97: 142–6. [DOI] [PubMed] [Google Scholar]
- 4. Vizcaino A, Moreno V, Lambert R et al. Time trends incidence of both major histologic types of esophageal carcinomas in selected countries, 1973-1995. Int J Cancer 2002; 99: 860–8. [DOI] [PubMed] [Google Scholar]
- 5. Pohl H, Sirovich B, Welch H. Esophageal adenocarcinoma incidence: are we reaching the peak? Cancer Epidemiol Biomarkers Prev 2010; 19: 1468–70. [DOI] [PubMed] [Google Scholar]
- 6. Lagergren J, Bergstrom R, Nyren O. Association between body mass and adenocarcinoma of the esophagus and gastric cardia. Ann Intern Med 1999; 130: 883–90. [DOI] [PubMed] [Google Scholar]
- 7. Lagergren J, Bergstrom R, Lindgren A et al. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med 1999; 340: 825–31. [DOI] [PubMed] [Google Scholar]
- 8. Flegal K, Carroll M, Ogden C et al. Prevalence and trends in obesity among US adults, 1999-2008. JAMA 2010; 303: 235–41. [DOI] [PubMed] [Google Scholar]
- 9. El-Serag H. Time trends of gastroesophageal reflux disease: a systematic review. Clin Gastroenterol Hepatol 2007; 5: 17–26. [DOI] [PubMed] [Google Scholar]
- 10. Hur C, Miller M, Kong C et al. Trends in esophageal adenocarcinoma incidence and mortality. Cancer 2013; 119: 1149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simard E, Ward E, Siegel R et al. Cancers with increasing incidence trends in the United States: 1999 through 2008. CA Cancer J Clin 2012; 62: 118–28. [DOI] [PubMed] [Google Scholar]
- 12. Yang Y, Land K. Age-period-Cohort Analysis: New Models, Methods, and Empirical Applications. CRC Press, 2013. Boca Raton, FL. [Google Scholar]
- 13. Brown L, Devesa S, Chow W. Incidence of adenocarcinoma of the esophagus among white Americans by sex, stage, and age. J Natl Cancer Inst 2008; 100: 1184–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bollschweiler E, Wolfgarten E, Gutschow C et al. Demographic variations in the rising incidence of esophageal adenocarcinoma in white males. Cancer 2001; 92: 549–55. [DOI] [PubMed] [Google Scholar]
- 15. Dubecz A, Solymosi N, Stadlhuber R et al. Does the incidence of adenocarcinoma of the esophagus and gastric cardia continue to rise in the twenty-first century? a SEER database analysis. J Gastrointest Surg 2013; 18: 124–9. [DOI] [PubMed] [Google Scholar]
- 16. Kubo A, Corley D. Body mass index and adenocarcinomas of the esophagus or gastric cardia: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev 2006; 15: 872–8. [DOI] [PubMed] [Google Scholar]
- 17. Corley D, Kubo A, Levin T et al. Abdominal obesity and body mass index as risk factors for Barrett's esophagus. Gastroenterology 2007; 133: 34–41. [DOI] [PubMed] [Google Scholar]
- 18. El-Serag H, Kvapil P, Hacken-Bitar J et al. Abdominal obesity and the risk of Barrett's esophagus. Am J Gastroenterol 2005; 100: 2151–6. [DOI] [PubMed] [Google Scholar]
- 19. Brown L, Swanson C, Gridley G et al. Adenocarcinoma of the esophagus: role of obesity and diet. J Natl Cancer Inst 1995; 87: 104–9. [DOI] [PubMed] [Google Scholar]
- 20. Vaughan T, Davis S, Kristal A et al. Obesity, alcohol, and tobacco as risk factors for cancers of the esophagus and gastric cardia: adenocarcinoma versus squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 1995; 4: 85–92. [PubMed] [Google Scholar]
- 21. Chow W, Blot W, Vaughan T et al. Body mass index and risk of adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst 1998; 90: 150–5. [DOI] [PubMed] [Google Scholar]
- 22. Merry A, Schouten L, Goldbohm R et al. Body mass index, height and risk of adenocarcinoma of the oesophagus and gastric cardia: a prospective cohort study. Gut 2007; 56: 1503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. MacInnis R, English D, Hopper J et al. Body size and composition and the risk of gastric and oesophageal adenocarcinoma. Int J Cancer 2006; 118: 2628–31. [DOI] [PubMed] [Google Scholar]
- 24. Abnet C, Freedman N, Hollenbeck A et al. A prospective study of BMI and risk of oesophageal and gastric adenocarcinoma. Eur J Cancer 2008; 44: 465–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Corley D, Kubo A. Body mass index and gastroesophageal reflux disease: a systematic review and meta-analysis. Am J Gastroenterol 2006; 101: 2619–28. [DOI] [PubMed] [Google Scholar]
- 26. Hampel H, Abraham N, El-Serag H. Meta-analysis: obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med 2005; 143: 199–211. [DOI] [PubMed] [Google Scholar]
- 27. Ryan A, Duong M, Healy L et al. Obesity, metabolic syndrome and esophageal adenocarcinoma: epidemiology, etiology and new targets. Cancer Epidemiol 2011; 35: 309–19. [DOI] [PubMed] [Google Scholar]
- 28. Howard J, Cathcart M, Healy L et al. Leptin and adiponectin receptor expression in oesophageal cancer. Br J Surg 2014; 101: 643–52. [DOI] [PubMed] [Google Scholar]
- 29. Islami F, Kamangar F. Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer Prev Res 2008; 1: 329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blaser M. Disappearing microbiota: helicobacter pylori protection against esophageal adenocarcinoma. Cancer Prev Res (Phila) 2008; 1: 308–11. [DOI] [PubMed] [Google Scholar]
- 31. Grad Y, Lipsitch M, Aiello A. Secular trends in helicobacter pylori seroprevalence in adults in the United States: evidence for sustained race/ethnic disparities. Am J Epidemiol 2012; 175: 54–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peek R, Blaser M. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer 2002; 2: 28–37. [DOI] [PubMed] [Google Scholar]
- 33. Thrift A, Whiteman D. The incidence of esophageal adenocarcinoma continues to rise: analysis of period and birth cohort effects on recent trends. Ann Oncol 2012; 23: 3155–62. [DOI] [PubMed] [Google Scholar]
- 34. Lepage C, Rachet B, Jooste V et al. Continuing rapid increase in esophageal adenocarcinoma in England and Wales. Am J Gastroenterol 2008; 103: 2694–9. [DOI] [PubMed] [Google Scholar]
- 35. van Blankenstein M, Looman C, Siersema P et al. Trends in the incidence of adenocarcinoma of the oesophagus and cardia in the Netherlands 1989-2003. Br J Cancer 2007; 96: 1767–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jeon J, Luebeck E, Moolgavkar S. Age effects and temporal trends in adenocarcinoma of the esophagus and gastric cardia (United States). Cancer Causes Control 2006; 17: 971–81. [DOI] [PubMed] [Google Scholar]
- 37. Kong C, Kroep S, Curtius K et al. Exploring the recent trend in esophageal adenocarcinoma incidence and mortality using comparative simulation modeling. Cancer Epidemiol Biomarkers Prev 2014; 23: 997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hazelton W, Curtius K, Inadomi J et al. The role of gastroesophageal reflux and other factors during progression to esophageal adenocarcinoma. Cancer Epidemiol Biomarkers Prev 2015; 24: 1012–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Abrams J, Sharaiha R, Gonsalves L et al. Dating the rise of esophageal adenocarcinoma: analysis of Connecticut Tumor Registry Data, 1940-2007. Cancer Epidemiol Biomarkers Prev 2011;20:183–6. [DOI] [PMC free article] [PubMed] [Google Scholar]