

Abstract
Regulation of dendritic spines is an important component of synaptic function and plasticity whereas dendritic spine dysregulation is related to several psychiatric and neurological diseases. In the present study, we tested the hypothesis that serotonin (5-HT)2A/2C receptor-induced Rho family transamidation and activation regulates dendritic spine morphology and that activation of multiple types of receptors can induce transglutaminase(TGase)-catalyzed transamidation of small G proteins. We previously reported a novel 5-HT2A receptor downstream effector, TGase-catalyzed serotonylation of the small G protein Rac1 in A1A1v cells, a rat embryonic cortical cell line. We now extend these findings to rat primary cortical cultures which develop dendritic spines; stimulation of 5-HT2A/2C receptors increased transamidation of Rac1 and Cdc42, but not RhoA. Inhibition of TGases significantly decreased transamidation and activation of Rac1 and Cdc42, suggesting that transamidation led to their activation. In primary cortical cultures, stimulation of 5-HT2A/2C receptors by 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane(DOI) caused a transient dendritic spine enlargement, which was blocked by TGase inhibition. Stimulation of both 5-HT2A and 5-HT2C receptors contributed to DOI-induced Rac1 transamidation in primary cortical cultures as demonstrated by selective antagonists. Furthermore, stimulation of muscarinic acetylcholine receptors and NMDA receptors also increased TGase-catalyzed Rac1 activation in SH-SY5Y cells and N2a cells, respectively. Receptor-stimulated TGase-catalyzed transamidation of Rac1 occurs at Q61, a site previously reported to be important in the inactivation of Rac1. These studies demonstrate that TGase-catalyzed transamidation and activation of small G proteins results from stimulation of multiple types of receptors and this novel signaling pathway can regulate dendritic spine morphology and plasticity.
Keywords: transglutaminase, serotonylation, serotonin 2A/2C receptors, NMDA receptors, muscarinic receptors
Graphical abstract
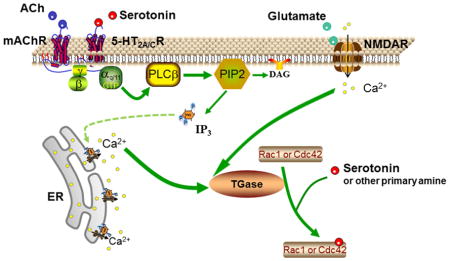
1.0 Introduction
In the central nervous system, the majority of the excitatory synapses are composed of postsynaptic terminals located on dendritic spines (Phillips and Pozzo-Miller, 2015). Changes in size, number and morphology of dendritic spines are tightly coordinated with synaptic function and plasticity, underlying the establishment and remodeling of neuronal circuits, learning and memory, and behavior (Penzes et al., 2011; Kennedy, 2016). Notably, malfunction of dendritic spines accompanies a large number of brain disorders, including bipolar disorder, autism spectrum disorder, schizophrenia and Alzheimer’s disease, suggesting that dendritic spines can serve as a common target for those complex diseases (Penzes et al., 2011; Penzes et al., 2013; Konopaske et al., 2014; Phillips and Pozzo-Miller, 2015). Understanding the molecular underpinnings of dendritic spine regulation may provide essential insights into the etiologies of those disorders and may reveal new drug targets.
Morphological changes of dendritic spines are driven by actin dynamics, which can be regulated by small G proteins of the Rho-family. At the synapse, Rac1, Cdc42 and RhoA play a pivotal role in spine formation and morphogenesis, and synaptic plasticity (Martino et al., 2013). Activation of Rac1 and Cdc42 promotes spine formation, growth and stabilization; conversely, RhoA activation leads to spine pruning. Perturbations in Rho family signaling are implicated in various brain disorders, particularly those associated with cognitive deficits, such as mental retardation, schizophrenia and Alzheimer’s diseases (Ba et al., 2013; Bolognin et al., 2014; Datta et al., 2015).
Numerous studies have demonstrated that the activity of small G proteins including those of the Rho family is regulated by monoaminylation (Muma and Mi, 2015). Monoaminylation is a post-translational modification of proteins in which transglutaminases (TGases) catalyze the transamidation of a primary amine molecule such as serotonin (5-HT) or dopamine to a protein-bound glutamine residue (Muma and Mi, 2015). Serotonylation is a term for the specific transamidation of 5-HT to a protein (Walther et al., 2003). Stimulation of serotonin 2A (5-HT2A) receptors induces serotonylation of Rac1, resulting in Rac1 activation (Dai et al., 2008). An increase in intracellular Ca+2subsequent to receptor stimulation was both necessary and sufficient to stimulate serotonylation and activation of Rac1 (Dai et al., 2011). Together, these findings lead us to hypothesize that multiple receptor subtypes increase TGase-catalyzed transamidation and activation of small G proteins which can alter dendritic spine morphology.
5-HT2A receptors are widely distributed in most forebrain regions. Disrupted function of 5-HT2A receptors has been identified in various neurological and psychiatric disorders such as schizophrenia, Alzheimer’s disease (Fehér et al., 2013), autism, depression and anxiety (Gray and Roth, 2007; Berg et al., 2008; Hervás et al., 2014). 5-HT2A receptors are also the target for several antidepressants, anxiolytics, atypical antipsychotics and hallucinogens (González-Maeso et al., 2007; Mestre et al., 2013; Amodeo et al., 2014). 5-HT2A receptors localize to dendrites, dendritic shafts, and dendritic spines (Cornea-Hebert et al., 2002; Peddie et al., 2008). Initiation of 5-HT2A receptor expression coincides with the period of synaptogenesis (Roth et al., 1991). 5-HT2A receptor activation alters dendritic spine area via a kalirin-7 dependent pathway (Jones et al., 2009). Stimulation of 5-HT2A receptors also changes the density of specific subtypes of dendritic spines (Yoshida et al., 2011). Those studies suggest that 5-HT2A receptors play a role in the regulation of dendritic spine architecture and actin cytoskeleton. However, the underlying mechanisms by which the 5-HT2A receptor signaling regulates dendritic spines and the role of serotonylation of Rac1 and possibly other members of the Rho family in the process are not clear.
NMDA receptors can mediate synaptic plasticity such as long-term potentiation (Collingridge et al., 1983). NMDA receptor activation causes an influx of Ca2+and activation of calmodulin-dependent kinase II. The Ca2+influx and activation of calmodulin-dependent kinase II result in recruitment of AMPA receptors to the synapse and dendritic spine enlargement both of which are determinants of synaptic strength (Matsuzaki et al., 2004). The Ca2+influx is necessary for activation of small G proteins in the dendritic spines and subsequent increase in spine volume (Murakoshi et al., 2011). We hypothesize that the NMDA receptor-dependent influx of Ca2+increases TGase-catalyzed transamidation and activation of small G proteins. Muscarinic receptor signaling has also been shown to be involved in synaptic plasticity and can produce long-term potentiation (Dennis et al., 2015), perhaps also by a mechanism involving TGase-catalyzed transamidation and activation of small G proteins.
In the present study, we used rat primary cortical cultures which develop dendritic spines to test whether activation of TGase via stimulation of 5-HT2A/C receptors induces transamidation and activation of Rac1, Cdc42 and RhoA, and whether the transamidation results in changes of dendritic spine architecture. SH-SY5Y and N2A cell lines were used for biochemical studies to examine muscarinic, NMDA and AMPA receptors. Our results indicate that both G protein-coupled and ligand-gated ion channel receptors can stimulate transamidation and activation of Rac1 and that transamidation can regulate dendritic spine size.
2.0 Materials and Methods
2.1 Reagents
1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane HCl (DOI), N-Methyl-D-aspartic acid (NMDA), (S)-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and (2-hydroxyethyl)trimethylammonium chloride carbamate (carbachol) (Sigma-Aldrich, St. Louis, MO) and 2-aminoethyl disulfide dihydrochloride (cystamine) (MP Biomedicals, Solon, OH) were dissolved in saline or water and further diluted before application to cell cultures. SB 242084 (Sigma-Aldrich, St. Louis, MO) was dissolved in ethanol (the final concentration of ethanol exposure to cells was 0.1%). MDL100907 was kindly provided by Sanofi Aventis (Bridgewater, NJ) and dissolved in DMSO (the final concentration of DMSO exposure to cells was 0.01%).
2.2 Cell culture and transfection
A1A1v cells, SH-SY5Y and N2a cells were cultured and transfected as previously described for all of the biochemical experiments (Yu et al., 1990; Dai et al., 2008; Coleman et al., 2013). In addition, A1A1v cells were transfected with either of the following mammalian expression plasmid constructs: TGase1 (TGM1, clone ID: OHu23436, Genscript, NJ, USA) or TGase3 (TGM3, clone ID: OHu31856, Genscript, NJ, USA) with a common pcDNA3.1+-DYK vector backbone. Before each experiment, cells were maintained for 48h in Dulbecco’s modified Eagle medium (Fisher Scientific, Pittsburgh, PA) containing 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA) treated with charcoal to remove > 99% of endogenous 5-HT (Unsworth and Molinoff, 1992). Cells used as non-transfected control cells were incubated in charcoal filtered FBS but were otherwise untreated.
2.3 Primary cortical culture
Animals were used in accordance with the National Institute for Health Guide for the Care and Use of Laboratory Animals as approved by the University of Kansas Institutional Animal Care and Use Committee. Primary cortical neurons were isolated from E18 Sprague-Dawley rat embryos as described previously with minor modifications (Srivastava et al., 2011; Beaudoin III et al., 2012). The cortical tissues from feti from each pregnant rat were harvested and the cells from each litter were then pooled. Neurons were plated at a density of 5 × 105 cells/ml on 22mm diameter round cover glass (Neuvitro Corporation, Vancouver, WA) or at a density of 2.7 × 106 cells/ml on T25 cell culture flasks (Fisher Scientific, Lenexa, KS) coated with poly-L-lysine (Sigma-Aldrich, St. Louis, MO). At 21 days in vitro (DIV), cultures were randomly chosen for treatment with 3 μM DOI or vehicle and in some experiments pretreatment with either 1 mM cystamine or a selective 5-HT receptor antagonist or vehicle.
2.4 Immunocytochemistry
Primary neurons were double-labeled with Alexa Fluor® 568 Phalloidin (Life Technologies, Grand Island, NY) for labeling F-actin, antibodies against microtubule-associated protein 2 (Map2) clone HM-2 (Sigma-Aldrich, St. Louis, MO) for labeling dendrites, antibodies against 5-HT2A receptors (Singh et al., 2007), and antibodies against PSD95 (6G6-1C9) (Life Technologies, Grand Island, NY). Alexa Fluor® 488 donkey anti-rabbit IgG (H+L) antibody and Alexa Fluor® 647 conjugated goat anti-mouse IgG (H+L) secondary antibody (Life Technologies, Grand Island, NY) were used to target primary antibodies. Coverslips were mounted onto slides using ProLong anti-fade reagent (Invitrogen, Grand Island, NY).
2.5 Quantitative Analysis of Spine Morphology
Neurons were visualized with an Olympus/3I Spinning Disk confocal microscope using a 100X TIRF oil immersion objective. Z-series of twenty to thirty images were taken at 0.2μm intervals, with 1024 × 1024 or 500 × 500 pixel resolution. Three-dimensional maximum projection reconstructions and deconvolution were performed using Slidebook 5.5 or 6 (Intelligent Imaging Innovations, Inc). To examine the morphology of dendritic spines, dendrites (labeled with MAP2) and dendritic spines were first chosen based on expression of 5-HT2A receptors, and then phalloidin labeling of F-actin was used for measurements of individual spines. Dendrites and individual spines on dendrites were traced, and dendritic area was measured using CellProfiler (Broad Institute, Cambridge, MA). Length of dendrites was measured using Image J (National Institutes of Health, Bethesda, Maryland). Spine density was measured using Neuron Studio (Icahn School of Medicine at Mount Sinai, New York, NY). Spines on one or two primary dendrites of each neuron, and 5–10 neurons for each condition were analyzed from three separate experiments. The length of dendrites used for the analysis was at least 30 microns. The experimenter was blinded to the treatment group during dendritic spine measurements.
2.6 Site-directed Mutagenesis
Site-directed mutagenesis was performed with Rac1 DNA in TOPO® PCR Cloning vector using the Quikchange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). Oligonucleotides containing amino acid substitutions for glutamine 61, 74,161 and 174 of Rac1 were designed using QuikChange® Primer Design Program (Agilent Technologies, Santa Clara, CA) and synthesized by Life Technologies. Wild type and Rac1 mutants were cloned into pcDNA™4/HisMax C Mammalian Expression Vectors (Life Technologies, Grand Island, NY) and transfected into A1A1v cells as described above.
2.7 Small Interfering RNA (siRNA)
To reduce TGase2 protein expression, siRNA duplex targeting the coding sequence of rat TGase2 mRNA was used as previously reported with minor modification (Dai et al., 2008). The target sequence 5-AAGAGCGAGATGATCTGGAAT-3 was synthesized by QIAGEN (Germantown, MD). At 19 DIV, primary neurons were transfected with siRNA at a final concentration of 90 nM using Lipofectamine 3000 without P3000 reagent according to the manufacture’s instruction. 72 hours after transfection, cells were treated with DOI or saline. Cells incubated with Lipofectamine 3000 alone were used as a mock-transfected control.
2.7 Immunoblot
Protein samples were separated on 12% or 14% SDS-polyacrylamide gels as previously described (Dai et al., 2008). The primary antibodies used are as follows: mouse anti-Rac1 antibody, clone 23A8, 1:700 (Millipore Corporation, Billerica, MA); mouse anti-Cdc42 antibody, clone 44, 1:500 (BD Biosciences, BD Biosciences); rat anti-flag antibody, 1:500 (Agilent Technologies, USA); mouse anti-TGase1 antibody (E-6),1:200 (Santa Cruz Biotechnology, Inc, USA), rabbit anti-5-HT2A receptor antibody, 1:20,000 (Singh et al., 2007), mouse anti-5-HT2C receptor antibody (D-12), 1:200 (Santa Cruz Biotechnology, Inc, Dallas, TX), mouse anti-actin antibody, 1:50,000 (MP Biomedicals, Solon, OH). Goat-anti-mouse or goat-anti-rabbit secondary antibodies conjugated to HRP (Jackson ImmunoResearch, West Grove, PA) were used to bind to the primary antibodies and were detected using enhanced chemiluminescence western blotting detection reagents (Amersham Biosciences, Piscataway, NJ). The signals were detected using ChemiDoc™ XRS+ System (Bio-Rad, Hercules, CA) and quantified by calculating the integrated optical density (IOD) of each protein band using Image Lab™ Software (Bio-Rad, Hercules, CA).
2.8 Immunoprecipitation of TGase-Modified Protein
Immunoprecipitation of TGase-modified protein was performed as described previously with minor modifications (Dai et al., 2008). Briefly, cells were harvested and lysed and the protein concentration in the lysates was determined using a BCA Protein Assay kit (Pierce, Rockford, IL). 200μg and 600μg protein were used for detecting Rac1 and Cdc42 transamidation respectively. Isolation of proteins containing TGase-catalyzed bonds was performed by immunoprecipitation using 81D4 mAb (mouse IgM) prebound to Sepharose beads (Covalab, Lyon, France) and the protocol developed by Covalab as described previously (Dai et al. 2008). The 81D4 antibody is selective for the Nε-(γ-glutamyl) lysine isopeptide, which is the TGase-catalyzed bond between a protein-bound lysine residue and protein-bound glutamine residue (el Alaoui et al. 1991). Several other potential epitopes similar to the Nε-(γ-glutamyl) lysine isopeptide were found not to cross-react with the 81D4 antibody (el Alaoui et al. 1991), with the exception of transamidation of a peptide bound lysine to serotonin and possibly other small amines (Dai et al. 2008).
2.9 Activity Assay for Small G Proteins
Active Rac1 and Cdc42 bind to PAK1 and can be separated for measurement on immunoblots. The activity of Rac1 and Cdc42 was measured using Glutathione Transferase (GST) -PAK1 bound Sepharose 4B beads as described previously (Dai et al., 2008). PAK1-bound protein samples were loaded on 14% SDS PAGE and Rac1 or Cdc42 detected on immunoblots as described above.
2.10 Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). All data are analyzed by Student’s t-test, one, or two-way ANOVA, or Kruskal-Wallis one-way ANOVA on Ranks. Post hoc tests were conducted using Newman-Keuls multiple comparison test, Dunn’s method or Tukey’s multiple comparison test. Sigmaplot 12 (Systat Software, Inc., San Jose, CA) Origin 8.0 (OriginLab Corporation, Northampton, MA) and GraphPad Prism version 6.02 (GraphPad Software, Inc., La Jolla, CA) were used for statistical analysis.
3.0 Results
3.1 5-HT2A/2C receptor-stimulated Rac1 activation in primary cortical culture
To determine whether Rac1 activation by 5-HT2A/2C receptor stimulation alters dendritic spine morphology, we used primary cortical neurons from E18 rat pups cultured for DIV21. At this stage, dendritic spines display a relatively mature morphology and form connections with presynaptic partners.
To determine whether Rac1 activity is increased following 5-HT2A/2C receptor stimulation in this primary culture model, we used a PAK1 pull down assay, in which only activated GTP-bound Rac1 can bind to PAK1 (Parrini et al., 2002). We treated the primary neurons with 3μM DOI, a 5-HT2A/2C receptor-selective agonist, for 5min, the period previously used to detect Rac1 activation in A1A1v cells (Dai et al., 2008). As shown in Figure 1A, the ratio of GTP-bound Rac1 to the total Rac1 is significantly increased by 100% after DOI treatment, indicating that Rac1 becomes activated following DOI treatment in primary cultures as in A1A1v cells.
Figure 1. 5-HT2A/2C receptor-stimulated Rac1 activation in primary cortical culture.

A. DOI increased of the activity of Rac1 in primary cortical neurons. After 5 min of 3μM DOI treatment, active Rac1 (~21 kDa) was pulled down using GST-PAK1 coupled to glutathione-sepharose beads and measured on immunoblots. Bands at 37 kDa and 27 kDa are non-specific binding to components of the PAK1 pull-down assay as demonstrated by their presence in the negative control lane. The negative control lane was loaded with a sample in which the cell lysate (used in the other samples shown in lanes 1–4) was replaced with lysis buffer but was otherwise treated in an identical manner. The presence of these non-specific bands varied with the batch of sepharose beads. IOD ratio between active Rac1 and total Rac1 was normalized to vehicle-treated control levels. DOI significantly increased activated Rac1 in DOI-treated neurons. Student’s t-test indicates * p≤0.001, n=3.
B. DOI increased Rac1 transamidation in primary cortical neurons. Primary cortical neurons were treated with 3μM DOI, the TGase-modified proteins were immunoprecipitated with 81D4 antibody bound Sepharose beads and detected on immunoblots. Total amount of Rac1 (21kDa band in all immunoblots) in cell lysates was also detected on immunoblots using an anti-Rac1 antibody. Data are quantified by calculating the IOD of each protein band and the IOD ratio between TGase-modified Rac1 and total Rac1 was normalized to vehicle-treated control levels. Rac1 transamidation was significantly increased in DOI-treated neurons. Student’s t-test indicates * p<0.001, n=3.
C. TGase2 protein levels were significantly reduced 72 hours after transfection of primary neurons with 90 nM TGase2-specific siRNA (Student’s t-test indicates * p=0.001, n=3). The reduction in TGase2 is accompanied by an increase in TGase1 protein levels. Student’s t-test indicates * p<0.05, n=3.
D. Transamidation of Rac1 is reduced by inhibition of TGase activity by cystamine or knockdown of TGase2 by siRNA. Cells were incubated with TGase2-specific siRNA for 72 h or with 1mM cystamine for 1 h, and then they were treated with 3μM DOI or vehicle for 15 min, and TGase-modified Rac1 was detected on immunoblots at approximately 21 kDa . The bands at 27 kDa are non-specific binding due to components of the immunoprecipitation assay as demonstrated by the presence of this band in the negative control lane. The negative control lane was loaded with a sample in which the cell lysate was replaced with only lysis buffer but was otherwise treated in an identical manner. Two-way ANOVA analysis of three separate experiments shows significant effects of transfection or pretreatment [F(2,12)=13.011, p<0.001], significant effect of DOI stimulation [F(1,12)=18.343, p=0.001], and a significant interaction between transfection or cystamine with DOI treatment on TGase-modified Rac1 [F(2,12)=7.198, p<0.01] Newman-Keuls multiple comparison test indicates ** p<0.001, n=3 compared with vehicle treatment in mock-transfected cells; * p< 0.05, n=3 compared with vehicle treatment in TGase2 siRNA transfected cells; # p< 0.01, ## p<0.001, n=3 compared with DOI treatment in mock-transfected cells; & p<0.05, n=3 compared with DOI treatment in cystamine-pretreated cells.
E. The increase in Rac1 activation is abolished by inhibition of TGase activity by cystamine or knockdown of TGase2 by siRNA. Two-way ANOVA analysis of three separate experiments shows significant effects of transfection or cystamine pretreatment on Rac1 activation [F(2,12)=5.223, p<0.05], but the effects of DOI stimulation [F(1,12)=4.374, p=0.058] and the interaction between transfection or cystamine with DOI treatment [F(2,12)=3.051, p=0.085] on Rac1 activation are not significant. Newman-Keuls multiple comparison test indicates * p<0.05, n=3 compared with vehicle treatment in mock-transfected cells; # p< 0.05, ## p<0.01, n=3 compared with DOI treatment in mock-transfected cells.
F. Left: DOI-induced Rac 1 transamidation is not catalyzed by TGase1 in A1A1v cells. Cells were transfected with TGase1 constructs. After 5 min of 3μM DOI treatment, active Rac1 was pulled down using GST-PAK1 coupled to glutathione-Sepharose beads and measured on immunoblots. IOD ratio between active Rac1 and total Rac1 was normalized to vehicle-treated control levels. Two-way ANOVA analysis of three separate experiments shows significant effect of DOI stimulation [F(1,8)=8.355, p<0.05] but no effect of TGase1 transfection [F(1,8)=0.3155, p=0.5897] on Rac1 activation. The interaction between TGase1 transfection and DOI stimulation [F(1,8)=0.0089, p=0.9268] is not significant. Tukey’s multiple comparison test shows no significant effect between the comparison groups.
Right: DOI-induced Rac 1 transamidation is not catalyzed by TGase3 in A1A1v cells. Cells were transfected with TGase3 constructs as demonstrated by the anti-flag antibody labeling. After 5 min of 3μM DOI treatment, active Rac1 was pulled down using GST-PAK1 coupled to glutathione-Sepharose beads and measured on immunoblots. IOD ratio between active Rac1 and total Rac1 was normalized to vehicle-treated control levels. Two-way ANOVA analysis of three separate experiments shows no significant effect of DOI stimulation [F(1,8)=0.05524, p=0.8201] or TGase3 transfection [F(1,8)= 0.05541, p= 0.8198] on Rac1 activation in A1A1v cells. There is no significant interaction between TGase3 transfection and DOI stimulation [F(1,8)=1.018, p=0.3425].
G. DOI-induced Rac1 transamidation is mediated by both 5-HT2A and 5-HT2C receptors in rat primary cortical neurons. Vehicle-treated rat primary cortical neurons express 5-HT2A (~45kDa) and 5-HT2C receptors (~48kDa) as demonstrated on immunoblots. Cells were treated with MDL 100907, SB24084 or vehicle followed by DOI stimulation. TGase-modified Rac1 was detected on immunoblots. The percent increase in transamidation following treatment with DOI varies between experiments from approximately a 50% increase, as in figure 1B, to a nearly 300% increase in figure 1G. Also note that there was large variability in the data presented in figure 1G in the group treated with vehicle/DOI as indicated by the large error bar. Two-way ANOVA analysis of three separate experiments shows significant effects of pretreatment [F(2,12)=7.827, p<0.01], significant effect of DOI stimulation [F(1,12)=9.051, p<0.05]. The interaction between pre-treated and DOI treatment [F(2,12)=2.915, p=0.093] is not significant. Newman-Keuls multiple comparison test indicates * p<0.01, n=3 compared with vehicle treatment in vehicle-pretreated cells; # p< 0.01, n=3 compared with DOI treatment in vehicle-pretreated cells.
Next, to determine whether Rac1 transamidation is increased in rat primary culture, we treated the neurons with 3μM DOI for 15min, the time point at which we observed the most abundant Rac1 transamidation in A1A1v cells (Dai et al, 2008). The ratio of TGase-modified Rac1 to total Rac1 is significantly increased following DOI treatment (Figure 1B), suggesting Rac1 is transamidated after stimulation of 5-HT2A/2C receptors in rat primary cortical cultures.
To exam whether the DOI-stimulated increase of active Rac1 is due to TGase-catalyzed transamidation, we inhibited the activity or expression of TGase by treating the primary neurons with 1mM cystamine, a TGase inhibitor, or transfecting a siRNA targeting TGase2 into the primary neurons, followed by DOI treatment. We also examined the expression of other TGase proteins to determine the selectivity and possible off-target effects of the siRNA. As shown in Figure 1C, siRNA transfection resulted in a 45% decrease in TGase2 protein levels accompanied by an 85% increase in TGase1 protein levels. TGase3 was not detected on Western blot using a TGase3 specific antibody (data not shown). Compared with DOI-stimulated mock-transfected cells, cystamine treatment caused a 70% decrease and siRNA transfection caused 40% decrease in TGase-modified Rac1 following DOI treatment, suggesting that 5-HT2A/2C receptor-mediated Rac1 transamidation is dependent on the activity of TGases (Figure 1D). The reduction in DOI-induced Rac1 transamidation caused by cystamine is significantly higher than TGase2 siRNA. We also found that treatment with cystamine and siRNA transfection decreases the amount of DOI-induced Rac1 activation by 56% and 38%, respectively, compared with mock-transfected cells. These results indicate that TGase-catalyzed transamidation is necessary for the increase in Rac1 activity upon 5-HT2A/2C receptor stimulation in rat primary cortical neurons (Figure 1E).
3.2 TGase 1 and 3 do not activate Rac1 due to 5-HT2A receptor-stimulation in A1A1v cells
Since TGase2 siRNA increased expression of TGase1 in rat primary cells, we sought to determine if TGase1 and 3, which are both expressed in brain, are able to catalyze the activation of Rac1. To examine whether the DOI-stimulated increase of active Rac1 can be catalyzed by TGase1 and TGase3, we overexpressed TGase1 and TGase3 constructs in A1A1v cells and treated the cells with 3μM DOI. In cells transfected with the TGase1 construct, there was a significant effect of DOI treatment but no effect of TGase1 transfection on Rac1 activation (Figure 1F, left). In cells transfected with the TGase3 construct, there was no effect of either treatment or TGase3 transfection on Rac1 activation (Figure 1F, right). An anti-flag antibody was used to detect expression of TGase1 and 3. We further verified expression of TGase1 using a TGase1-specific antibody since only low levels of TGase1 expression were detected using the anti-flag antibody. Overall, our results suggest that neither TGase1 nor TGase3 catalyze Rac1 activation due to 5-HT2A/C receptor-stimulation in A1A1v cells.
3.3 DOI-induced Rac1 transamidation is mediated by both 5-HT2A and 5-HT2C receptors in primary cortical neurons
DOI is a selective 5-HT2A/2C receptor agonist. We determined whether both 5-HT2A and 5-HT2C receptors are expressed in the primary cortical cultures using western blots and found that both receptors are expressed as shown in Figure 1G. In order to test whether 5-HT2A or 5-HT2C, or both receptors mediate the DOI-stimulated Rac1 transamidation in rat primary cortical cells, we pretreated the rat primary neurons with 3.6nM MDL100907, a selective 5-HT2A receptor antagonist, 10nM SB242084, a selective antagonist for the 5-HT2C receptors, or DMSO, for one hour. We then stimulated cells with DOI or saline for 15min and examined Rac1 transamidation as described above. As shown in Figure 1G, the ratio of TGase-modified Rac1 to total Rac1 is significantly increased following DOI treatment, and both MDL100907 and SB24084 suppressed DOI-induced Rac1 transamidation. These results suggest that both 5-HT2A and 5-HT2C receptors contribute to DOI-stimulated Rac1 transamidation mediated by TGases.
3.4 Stimulation of 5-HT2A/2C receptors increases activation of Cdc42
We examined TGase-modified Cdc42 in A1A1v cells after treatment with 3 μM DOI for 30min, 1 hour, 2 hours or 3 hours. The Cdc42 transamidation is significantly elevated after 2 hour and 3 hour of DOI treatment (Figure 2A).
Figure 2. Stimulation of 5-HT2A/2C receptors increases activation of Cdc42.

A. DOI increased transamidation of Cdc42 in A1A1v cells. One-way ANOVA indicates a significant difference among groups [F(4,10)=11.828, p<0.001]. Newman-Keuls multiple comparison test indicates * p<0.01, n=3.
B. DOI increased the activity of Cdc42 in A1A1v cells. Cdc42 activity was transiently increased at 30min, 1hr and 2hr after DOI treatment and was reduced back to control levels at 3hr after DOI treatment. One-way ANOVA indicates a significant difference among groups [F(4,10)=11.631, p<0.001]. Newman-Keuls multiple comparison test indicates * p< 0.05, ** p< 0.01, *** p< 0.001, n=3.
C. Inhibition of TGase activity by cystamine prevents Cdc42 activation in A1A1v cells. Two-way ANOVA analysis of three separate experiments shows a significant effect of cystamine pretreatment [F(1,16)=37.072, p<0.001], a significant effect of DOI stimulation [F(3,16)=4.595, p<0.05], and a significant interaction between cystamine pretreatment with DOI treatment on Cdc42 activity [F(3,16)=3.924, p<0.05]. Newman-Keuls multiple comparison test indicates *, p< 0.05, **, p<0.01, n=3 compared with non-stimulated in vehicle pretreated cells; #, p≤0.01, ##, p<0.001, n=3 compared with vehicle-pretreated control cells stimulated with DOI for the same length of time.
D. Inhibition of TGase activity by cystamine or knockdown of TGase2 by siRNA prevents Cdc42 transamidation in rat primary cells. Two-way ANOVA analysis of three separate experiments shows significant effects of transfection or cystamine pretreatment [F(2,12)=12.374, p<0.001]. The effects of DOI stimulation [F(1,12)=4.003, p=0.069] and interaction between transfection or cystamine with DOI treatment on Cdc42 transamidation [F(2,12)=1.456, p=0.272] are not significant. Newman-Keuls multiple comparison test indicates * p<0.05, n=3 compared with vehicle treatment in mock-transfected cells; # p< 0.01, n=3 compared with DOI treatment in mock-transfected cells; & p<0.05, n=3 compared with DOI treatment in cystamine-pretreated cells.
E. Inhibition of TGase activity by cystamine or knockdown of TGase2 by siRNA prevents Cdc42 activation in rat primary cells. Two-way ANOVA analysis shows significant effects of transfection or cystamine pretreatment [F(2,12)=17.503, p<0.001], significant effect of DOI stimulation [F(1,12)=16.065, p=0.002], and a significant interaction between transfection or cystamine with DOI treatment on Cdc42 activation [F(2,12)=10.895, p=0.002]. Newman-Keuls multiple comparison test indicates * p<0.001, n=3 compared with vehicle treatment in mock-transfected cells; # p<0.01, n=3 compared with DOI treatment in mock-transfected cells.
F. RhoA is not transamidated following DOI treatment in primary cortical neurons. Primary cortical neurons were treated with 3μM DOI treatment for 15 min, 30 min, 1 hour, 3 hour and 24 hour. Then the TGase-modified proteins were immunoprecipitated with 81D4 antibody bound Sepharose beads and detected on immunoblots using anti-RhoA antibody. Total amount of RhoA (molecular mass ~22kDa) in cell lysates was also detected on western blots using an anti-RhoA antibody. The experiment was repeated three times with similar results. “Neg” indicates 81D4 beads were only incubated with IP buffer but not protein lysate.
We observed an increase in Cdc42 activity at 5min, 15min and 30min post-DOI treatment in A1A1v cells (data not shown). To further determine the duration of DOI-induced Cdc42 activation, we examined a more extended time-course (0.5, 1, 2, and 3 hr) of DOI-treatment in A1A1v cells. As shown in Figure 2B, the activity of Cdc42 is significantly increased following 30 min, 1 hour and 2 hour DOI treatment but not 3 hours compared with vehicle-treated cells, suggesting that DOI-induced Cdc42 activation lasts up to 2 hour with a peak at one hour in A1A1v cells.
We found that pretreatment with cystamine decreases the amount of activated Cdc42 at all three time points, 30 min, 1 hour or 3 hours, suggesting the DOI-induced Cdc42 activation is due to the TGase-catalyzed transamidation of Cdc42 in A1A1v cells (Figure 2C).
Next, we found that Cdc42 is activated and transamidated after DOI treatment for 15 minutes in primary cortical neurons (Figure 2D&E). A significant reduction in TGase-modified Cdc42 by cystamine pre-treatment and TGase2 siRNA transfection was shown in Figure 2D, suggesting 5-HT2A/2C receptor-mediated Cdc42 transamidation is dependent on the activity of TGases in primary culture. Compared to DOI-stimulated mock-transfected cells, cystamine treatment caused an 84% decrease and siRNA transfection caused a 34% decrease in TGase-modified Cdc42 following DOI treatment. As shown in Figure 2E, DOI-induced increases in the ratio of GTP-bound Cdc42 to the total Cdc42 were attenuated 75% and 54% by pretreatment with 1mM cystamine and TGase2 siRNA, respectively. These results suggest that DOI-stimulated TGase-catalyzed Cdc42 transamidation leads to Cdc42 activation in rat primary cortical neurons.
3.5 RhoA is not transamidated following DOI treatment in primary cortical neurons
We also tested whether RhoA is modified by TGases following DOI treatment. We were not able to detect TGase-catalyzed modification of RhoA in rat primary cortical cells at any of the time points examined, from 15 minutes up to 24 hours (Figure 2F). These results suggest that stimulation of 5HT2A/2C receptors does not induce significant levels of TGase-catalyzed RhoA transamidation in rat primary cortical cells.
3.6 TGase-catalyzed transamidation of Rac1 occurs at Q61 of Rac1 sequence in neuronal cells
We searched for GTP/Mg2+ binding sites, and GTPase activating protein (GAP), guanine nucleotide exchange factor (GEF), and guanine nucleotide dissociation inhibitor (GDI) interaction sites in the Rac1 sequence (accession number: NP_008839) using the Conserved Domain Database (available on http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) since TGase-catalyzed modifications at these sites would more likely lead to functional changes that would impact Rac1 activity. Four glutamine residues (Q61, Q74, Q141, and Q162) are located in the activity-related domains and were targeted for site-directed mutagenesis. We generated two plasmid constructs containing double mutations, one with a Q61/74N and the other with Q141/162N. We transfected wild type (WT) Rac1 and Rac1 mutants into A1A1v cells, and 48 hours later stimulated 5-HT2A/2C receptors with DOI treatment. As shown in Figure 3A, DOI significantly increases TGase-modified WT Rac1. Transamidation of Q61/74N Rac1 was not increased following DOI treatment compared with vehicle-treated control. To determine whether Q61 or Q74 or both are modified by TGase, we generated another two Rac1 mutants bearing a single mutation at Q61 or Q74, and transfected them into A1A1v cells. DOI stimulation significantly increased TGase-modified Q74N Rac1 and WT Rac1, but not Q61N Rac1, suggesting that Q61 is the site that is transamidated following stimulation of 5-HT2A/C receptors (Figure 3B). Transfection of Q61N Rac1 resulted in an increase in Rac1 activation independent of DOI stimulation (Figure 3C). These results suggest that the Q61N Rac1 mutant is constitutively active.
Figure 3. TGase-catalyzed transamidation of Rac1 occurs at Q61 of Rac1 sequence in neuronal cells.

A. DOI-stimulated transamidation was prevented in Q61/74N Rac1 mutants. A1A1v cells were transfected with Rac1 constructs containing amino acid substitutions for glutamine 61& 74, 141&162 and wild type (WT) Rac1 and 48 hours later, stimulated with DOI for 15min. Transfected Rac1 constructs migrate on immunoblots at ~25kDa versus endogenous Rac1 at 21kDa. Two-way ANOVA: main effects of Rac1 constructs [F(2,12) = 5.442, p<0.05], main effects of DOI stimulation [F(1,12)= 3.601, p=0.082], and interaction between Rac1 constructs and DOI treatment [F(2,12)=1.579, p=0.246]. Newman-Keuls multiple comparison test indicates * p<0.05 compared with vehicle treatment in WT Rac1-transfected cells; # p<0.05 compared with DOI treatment in WT Rac1-transfected cells, n=3; & p<0.05 compared with DOI treatment in Q61/74N Rac1-transfected cells.
B. DOI-stimulated transamidation was prevented in Q61N Rac1 mutants. Two-way ANOVA: main effects of Rac1 constructs [F(2,12) = 26.132, p<0.001], main effects of DOI stimulation [F(1,12)=33.442, p<0.001], and interaction between Rac1 constructs and DOI treatment [F(2,12)=7.107, p<0.01]. *: p<0.001 compared with vehicle treatment in the same Rac1 construct groups; #: p< 0.001 compared with DOI treatment in WT Rac1-transfected cells, by Newman-Keuls post-hoc test indicates. n=3.
C. Q61N Rac1 is constitutively activated independent of DOI stimulation. PAK1 pull-down and detection of Rac1 on immunoblots demonstrate high constitutive activity for Q61N Rac1 mutants with and without DOI stimulation. Higher exposure demonstrates that activity of both wild-type and Q74N Rac1 mutants are stimulated with DOI. The immunoblot for Rac1 verifies transfection of the Rac1 constructs in cell lysates used for PAK1 pull-down. The experiment was repeated three times with similar results. We did not quantitate the blots for these experiments because the extremely large differences in the intensity of the bands are outside of the linear range of the assay.
3.7 Stimulation of muscarinic receptors in SH-SY5Y cells causes TGase-dependent Rac1 transamidation and activation
As previously reported, 5-HT2A receptor-induced PLC activation is necessary for TGase-catalyzed Rac1 transamidation, and an increase in intracellular Ca2+ is sufficient to cause Rac1 transamidation in A1A1v cells (Dai et al., 2011). Therefore, we hypothesized that other Gαq/11 linked-receptors that activate PLC and increase in intracellular Ca2+ also result in Rac1 transamidation. To test this hypothesis, we used a human neuroblastoma cell line SH-SY5Y cells, which express M1, M2 and M3 receptors (Kukkonen et al., 1992). In this cell line, carbachol treatment can activate muscarinic receptors and induce PLC activation and subsequently an increase in cytosolic Ca2+ (Wojcikiewicz et al., 1994). As shown in Figure 4A, carbachol increases Rac1 activity significantly at 10min but not at 5min. To test whether carbachol-increased Rac1 activity is due to transamidation, we pretreated SH-SY5Y cells with 1mM cystamine to inhibit TGases and then stimulated muscarinic receptors with carbachol. As shown in Figure 4B, carbachol increased Rac1 transamidation by 150% in vehicle-pretreated cells. Cystamine reduced the carbachol-stimulated Rac1 transamidation by 78% compared to vehicle-pretreated cells. As shown in Figure 4C, cystamine also decreased carbachol-induced Rac1 activity by 78% compared to vehicle-pretreated controls.
Figure 4. Stimulation of muscarinic receptors in SH-SY5Y cells and NMDA receptors in N2a cells causes TGase-dependent Rac1 activation.

A. Carbachol treatment increased Rac1 activity in SH-SY5Y cells. SH-SY5Y cells were treated with vehicle or 1mM carbachol, a muscarinic receptor agonist, for 5min and 10min, and Rac1 activity was detected. One-way ANOVA shows a significant difference among groups [F(2,6)=13.85, p<0.01]. Newman-Keuls multiple comparison test indicates * p<0.01, n=3.
B. Carbachol-stimulated Rac1 transamidation is TGase dependent. Pretreatment with cystamine prevents carbachol-stimulated Rac1 transamidation. Two-way ANOVA analysis shows a significant effect of cystamine pretreatment, F(1,8)=90.978, p<0.001], significant effect of carbachol stimulation [F(1,8)=31.28, p<0.001], and a significant interaction between cystamine and carbachol treatment on Rac1 transamidation [F(1,8)=59.003, p<0.001]. Newman-Keuls multiple comparison test indicates * p< 0.001, n=3 compared with non-stimulated in vehicle pretreated cells; # p<0.001, n=3 compared with carbachol-stimulated controls in vehicle-pretreated cells.
C. Carbachol-stimulated Rac1 activation is TGase dependent. Cystamine pretreatment inhibited carbachol-stimulated activity of Rac1. Two-way ANOVA: main effects of cystamine pretreatment [F(1,8) = 9.405, p<0.05], main effects of carbachol stimulation [F(1,8)=3.155, p=0.114], and interaction between cystamine pretreatment and carbachol treatment [F(1,8)=5.134, p=0.053]. Newman-Keuls multiple comparison test indicates * p< 0.05, n=3 compared with non-stimulated in vehicle pretreated cells; # p<0.01, n=3 compared with carbachol-stimulated controls in vehicle-pretreated cells.
D. NMDA stimulates Rac1 activation in N2A cells. N2a cells were treated with 50μM NMDA or 100μM AMPA or vehicle for 5 minutes. NMDA increased Rac1 activation and pretreatment with the TGase inhibitor cystamine prevented this increase. Stimulation of AMPA receptors did not increase Rac1 activity at either 5 (data not shown) or 15 minutes post-stimulation. One-way ANOVA: F(3,8) = 6.234, p= 0.017, Newman-Keuls multiple comparison test indicates that NMDA-treated cells were significantly different from vehicle-treated cells (shown by *) and # indicates cells treated with both cystamine and NMDA were significantly different cells treated with NMDA alone at p < 0.05.
3.8 Stimulation of NMDA receptors in N2a cells causes TGase-dependent Rac1 activation
Ca2+ is a co-factor for the enzyme TGase and since an increase in intracellular Ca2+was sufficient to increase TGase-catalyzed Rac1 transamidation and activation (Dai et al., 2011), we reasoned that stimulation of other types of receptors that increase free intracellular Ca2+, such as ligand-gated ion channels NMDA and AMPA might also be able to increase TGase-catalyzed activation of Rac1. To test this hypothesis, N2a cells, which express NMDA and AMPA receptors, were stimulated with 50μM NMDA or 100μM AMPA for 5 or 15 minutes. As shown in Figure 4D, NMDA increased Rac1 activation and pretreatment with the TGase inhibitor cystamine prevented this increase. Stimulation of AMPA receptors did not increase Rac1 activity at 5 and 15 minutes post-stimulation, time points which previously resulted in activation following stimulation of the Gαq/11 linked receptors.
3.9 DOI-induced dendritic spine enlargement is dependent on TGase activity
In our cultured rat primary neurons, phalloidin labeled f-actin in dendritic spines (Figure 5A) and MAP2 immunolabeled the dendrites. To test whether stimulation of 5-HT2A receptors influences dendritic spine morphology, we performed a time course study in which primary neurons were treated with 3μM DOI for up to 60min. The area of dendritic spines increased by DOI treatment at 30min, and recovered to vehicle-treated control levels at 60min (Figure 5B). To determine whether TGase-catalyzed transamidation of small G proteins including Rac1 and Cdc42 mediates the effect of DOI on dendritic spine morphology, we inhibited TGase by pretreatment with 1mM cystamine for 1 hour, prior to DOI treatment for 30min. DOI-induced spine enlargement is prevented by cystamine pretreatment (Figure 5A & C), suggesting the 5-HT2A and/or 5-HT2C receptor stimulation induced increases in spine area is dependent on TGase activity, likely transamidation of small G proteins Rac1 and Cdc42. Two-way ANOVA test indicates that neither cystamine (1.5 hours post-treatment) nor DOI (30 min post-treatment) has a significant effect on dendritic spine density (Figure 5D).
Figure 5. DOI-induced dendritic spine enlargement is dependent on TGase activity.

A. F-actin in dendritic spines is labeled with phalloidin in primary cortical cultures treated with vehicle (Veh) and Veh, Veh and DOI, cystamine (Cys) and Veh, or Cys and DOI. Scale bar represents 5μm.
B. DOI causes an increase in dendritic spine area after 30min treatment. Bars represent absolute value of dendritic spine area. Each dendrite had 11–41 dendritic spines, 8–10 dendrites (from 5 – 10 neurons/group, n = 39 neurons) were measured for each treatment, resulting in 150–238 dendritic spines/treatment group for a total of 1,007 dendritic spines measured. Log transformation was performed on original data to achieved normality. One-way repeated measures ANOVA suggests a significant difference between five treatment groups [F(4,34)= 9.342, p<0.001]. Newman-Keuls multiple comparison test indicates * p ≤ 0.001 compared with vehicle treatment group, and groups treated with DOI for 5min, 15min and 60min.
C. Cystamine inhibits the DOI-induced dendritic spine enlargement. Bars represent absolute value of dendritic spine area. The number of spines measured in each group was 154 for vehicle, vehicle treated, 223 for vehicle, DOI treated, 122 for cystamine, vehicle treated and 164 for cystamine, DOI treated cells. Since log transformation did not result in a normal distribution of the data, analysis was performed on the original data using a non-parametric test. Kruskal-Wallis one-way ANOVA on Ranks indicates a significant difference between four treatment groups (p<0.001, n = 21 neurons). Post hoc Dunn’s test suggests * p<0.05 compared with vehicle-vehicle treatment; # p<0.05 compared with vehicle-DOI treatment.
D. DOI and cystamine have no significant effects on dendritic spine density. Bars represent absolute value of dendritic spine density. Log transformation was performed on original data to achieved normality. Two-way ANOVA test indicates that neither cystamine nor DOI has a significant effect on dendritic spine density.
4.0 Discussion
We previously reported that stimulation of 5-HT2A receptors induced Rac1 transamidation and activation, both of which can be blocked by inhibiting TGase (Dai et al., 2008). However, as a novel effector and second messenger of the 5-HT2A receptor signaling pathway, the functional impact of Rac1 transamidation in neuronal cells was unknown. Small G proteins of the Rho family are well known regulators of the actin cytoskeleton, neurite outgrowth and dendritic spine formation, morphogenesis and plasticity (Ba et al., 2013; Martino et al., 2013). Specifically, activation of Rac1 and Cdc42 leads to spine formation and enlargement. Stimulation of 5-HT2A receptors produces a transient increase in dendritic spine size that is dependent on Pak1 activation, a downstream effector of Rac1 and Cdc42 activation (Jones et al., 2009). Therefore, we hypothesized that the transient spine enlargement caused by stimulation of 5-HT2A receptors is dependent on TGase-catalyzed transamidation of Rac1 and Cdc42. Our time course experiment demonstrated that the area of dendritic spines is significantly increased 30 minutes after treatment with DOI, consistent with previous results reported by Jones et al, (2009). Furthermore, the present study demonstrated, for the first time, that regulation of dendritic spines via 5-HT2A/2C receptors is dependent on TGase activation. We found that cystamine is able to inhibit the DOI-induced transient spine enlargement, suggesting that TGase-catalyzed Rac1 and/or Cdc42 transamidation plays a vital role in mediating the regulation of dendritic spines via the 5-HT2A/2C receptor signaling pathway.
In the present study, we first demonstrated that stimulation of 5-HT2A/C receptors causes TGase-mediated transamidation and activation of small G proteins Rac1 and Cdc42 in primary culture as previously shown for Rac1 in A1A1v cells. We used two approaches to suppress TGase activity in primary culture, the competitive TGase inhibitor cystamine and TGase2 siRNA. Cystamine produced a more robust reduction in DOI-induced Rac1 transamidation which correlated with a more robust reduction in Rac1 activity than with TGase2 siRNA. TGase2 siRNA resulted in a 95% reduction of TGases2 protein level in A1A1v cells (Dai et al., 2008), however, in rat primary cortical culture only a 45% reduction of TGase2 was achieved. As a substrate for TGase, cystamine acts as a competitive inhibitor for all TGase subtypes, while the siRNA only targets TGase2. We found that the TGase2 siRNA also increased TGase1 protein expression in primary cortical culture, which may be a compensatory effect of TGase2 reduction. TGase2 is ubiquitously expressed in neuronal tissues, in contrast to TGase1 and 3 which are differentially expressed in various brain regions (Kim et al., 1999; Zainelli et al., 2005; Wilhelmus et al., 2009). TGase3 was not detectable in the primary neuronal cultures by western blots. Neither TGase1 nor TGase3 over-expression increased activation of Rac1 following DOI stimulation of 5-HT2A receptors in A1A1v cells. These results suggest that the limited reduction in TGase2 protein with the siRNA treatment caused the less robust effect of TGase siRNA on DOI-induced Rac1 transamidation and activity compared to the cystamine rather than the increase in TGase1 protein.
In the present study, we demonstrated for the first time that Cdc42 is transamidated and activated following stimulation with DOI. First using A1A1v cells, we found that Cdc42 is transamidated and activated in a more delayed time course than Rac1 following treatment with DOI. Cdc42 transamidation and activation in A1A1v cells was significantly increased at two hours and 30 minutes after DOI treatment, respectively. It is unclear why there is a discordant time course for the effects of transamidation and activation, possibly due to the differences in the sensitivity of each assay or to additional mechanisms beyond transamidation playing a role in the activation of Cdc42. However in primary cortical cell cultures, both transamidation and activation of Cdc42 occurred rapidly, 15 minutes after DOI treatment.
We identified Q61 as the site on Rac1 that is transamidated; we found that the Rac1 Q61N mutant cannot be transamidated following 5-HT2A/2C receptor stimulation and is constitutively active. Previous studies found that transamidation or deamidation of Rac1 and Cdc42 at Q61 and RhoA at Q63 could inhibit both intrinsic and GAP-catalyzed hydrolysis of those GTPases, thereby rendering them constitutively active (Lerm et al., 1999; Schmidt et al., 1999; Flatau et al., 2000). Together, these results suggest that transamidation of Rac1 at Q61 inhibits GTP hydrolysis of Rac1, thereby inhibiting Rac1 inactivation. Although further studies are needed to confirm that Rac1 and Cdc42 are indeed the TGase substrates mediating the increase in dendritic spine size, the Q61 mutant constructs cannot be used to address this issue. Since the Q61Rac1 mutant is constitutively active, it unfortunately cannot be used to prevent Rac1 activation via transamidation and thereby assess the effects on functional outcomes such as dendritic spine morphology.
Several studies indicate that stimulation of Gαq/11-coupled receptors, such as bradykinin or endothelin-1 receptor, can cause small G protein activation (van Leeuwen et al., 1999; Clerk et al., 2001). However, the underlying mechanisms still remain unclear. Our previous study indicated that 5-HT2A receptor-coupled PLC activation, as demonstrated by the PLC inhibitor U73122, and the subsequent increase in intracellular Ca2+, as demonstrated by chelation, are necessary for TGase-catalyzed Rac1 transamidation and activation (Dai et al., 2011). Furthermore, an increase in intracellular Ca2+ induced by the calcium ionophore ionomycin, is sufficient to cause Rac1 transamidation and activation in A1A1 cells, suggesting that receptor systems that increase in intracellular Ca2+may activate TGases to induce monoaminylation (Dai et al., 2011). In present study, we extended these findings to 5-HT2C receptors and another family of Gαq/11-protein-coupled receptors, muscarinic acetylcholine receptors. Pretreatment with either the 5-HT2A or 5-HT2C receptor selective antagonist blocked the DOI-induced increase in Rac1 transamidation. These results suggest that both 5-HT2A and 5-HT2C receptors are able to cause an increase in Rac1 transamidation and that both receptors are necessary for the full effect of DOI on transamidation. In fact, pretreatment with a selective antagonist alone had a significant effect on Rac1 transamidation (main effect for pretreatment, p < 0.01), although there was no significant difference between the vehicle/vehicle and vehicle/MDL100907 or vehicle/SB242084 treated groups. Further studies are needed to determine if there is a constitutive effect of 5-HT2A or 5-HT2C receptor signaling on Rac1 activity. We found that stimulation of muscarinic receptors with carbachol increased the transamidation and activity of Rac1, both of which were suppressed by the TGase inhibitor cystamine. Furthermore, stimulation of NMDA receptors increased TGase-catalyzed activation of Rac1. Our results suggest that TGase-mediated monoaminylation of small G proteins can serve as a common mechanism underlying the Gαq/11-coupled and ligand gated ion channel receptor-induced small G protein activation.
NMDA receptor stimulation resulted in TGase-dependent activation of Rac1 suggesting that transamidation of small G proteins could play a role in NMDA-dependent long term potentiation and synaptic plasticity. NMDA receptors are involved in the induction of synaptic plasticity and an increase in dendritic spine volume is a “morphological correlate of plasticity” (Collingridge et al., 1983; Nicoll and Roche, 2013). Persistent activation of the Rho family of small G-proteins is necessary for synaptic plasticity (Murakoshi et al., 2011). The link between NMDA receptor-stimulated increases in intracellular Ca2+and the persistent activation of Rho family G-proteins is unclear but our results suggest that it could involve TGase-catalyzed transamidation of the Rho G-proteins.
Numerous studies have shown that Gαq/11-coupled receptors, such as the 5-HT2A receptor, metabotropic glutamate receptor 5 and muscarinic receptors, as well as PLC, Ca2+ and calmodulin are upstream regulators of synaptogenesis and dendritic spines (Wess, 2004; Spires et al., 2005; Horne and Dell’Acqua, 2007; Wijetunge et al., 2008; Yamasaki et al., 2010; Schätzle et al., 2011). However, little was known about the signaling molecules coordinating these mechanisms. Our results suggest that 5-HT2A/2C receptor signaling pathways regulate dendritic spine morphology in rat cortical neurons via a TGase-mediated mechanism, likely monoaminylation of small G proteins. Furthermore, the results point to a possible role for TGase-mediated transamidation as a common down-stream signaling pathway for several receptor systems, most importantly NMDA receptors, in the regulation of dendritic spines. In conclusion, we report a novel receptor signaling pathway in the regulation of dendritic spines, thus providing further insight into neuropsychiatric and neurological disorders in which those receptors and dendritic spines are involved.
Highlights.
Stimulation of 5-HT2A & 5-HT2C receptors transamidates and activates Rac1 and Cdc42
Stimulation of muscarinic acetylcholine and NMDA receptors activates Rac1
Activation of Rac1 & Cdc42 is reduced by inhibition or knock-down of transglutaminase
Receptor-stimulated transglutaminase-catalyzed transamidation of Rac1 occurs at Q61
5-HT2A/C receptors induce transglutaminase-dependent dendritic spine size increases
Acknowledgments
MDL100907 was kindly provided by Sanofi Aventis (Bridgewater, NJ). Special thanks to Dr. David Moore and Heather Shinogle for their efforts on setting up confocal microscopy configurations and building up CellProfiler pipelines. Thanks to Zak Tazkargy for his contributions to dendritic spine area measuring.
Funding: This work was supported by the National Institutes of Health [MH068612] to N.A.M. and by the National Institutes of Health Kansas INBRE, P20 GM103418.
Abbreviations
- DIV
days in vitro
- DOI
1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane
- 5-HT
serotonin
- TGase
transglutaminase
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5.0 References
- Amodeo DA, Jones JH, Sweeney JA, Ragozzino ME. Risperidone and the 5 - HT2A Receptor Antagonist M100907 Improve Probabilistic Reversal Learning in BTBR T+ tf/J Mice. Autism Research. 2014;7:555–567. doi: 10.1002/aur.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba W, van der Raadt J, Kasri NN. Rho GTPase signaling at the synapse: implications for intellectual disability. Experimental cell research. 2013;319:2368–2374. doi: 10.1016/j.yexcr.2013.05.033. [DOI] [PubMed] [Google Scholar]
- Beaudoin GM, III, Lee S-H, Singh D, Yuan Y, Ng Y-G, Reichardt LF, Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nature protocols. 2012;7:1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- Berg KA, Harvey JA, Spampinato U, Clarke WP. Physiological and therapeutic relevance of constitutive activity of 5-HT 2A and 5-HT 2C receptors for the treatment of depression. Progress in brain research. 2008;172:287–305. doi: 10.1016/S0079-6123(08)00914-X. [DOI] [PubMed] [Google Scholar]
- Bolognin S, Lorenzetto E, Diana G, Buffelli M. The Potential Role of Rho GTPases in Alzheimer’s Disease Pathogenesis. Molecular neurobiology. 2014;50:406–422. doi: 10.1007/s12035-014-8637-5. [DOI] [PubMed] [Google Scholar]
- Clerk A, Pham FH, Fuller SJ, Sahai E, Aktories K, Marais R, Marshall C, Sugden PH. Regulation of mitogen-activated protein kinases in cardiac myocytes through the small G protein Rac1. Molecular and cellular biology. 2001;21:1173–1184. doi: 10.1128/MCB.21.4.1173-1184.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman N, Ates-Alagoz Z, Gaye B, Farbaniec M, Sun S, Adejare A. Toxicity Studies on Novel N-Substituted Bicyclo-Heptan-2-Amines at NMDA Receptors. Pharmaceuticals. 2013;6:536–545. doi: 10.3390/ph6040536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. The Journal of physiology. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornea-Hebert V, Watkins K, Roth B, Kroeze W, Gaudreau P, Leclerc N, Descarries L. Similar ultrastructural distribution of the 5-HT 2A serotonin receptor and microtubule-associated protein MAP1A in cortical dendrites of adult rat. Neuroscience. 2002;113:23–35. doi: 10.1016/s0306-4522(02)00146-x. [DOI] [PubMed] [Google Scholar]
- Dai Y, Dudek NL, Patel TB, Muma NA. Transglutaminase-catalyzed transamidation: a novel mechanism for Rac1 activation by 5-hydroxytryptamine2A receptor stimulation. Journal of Pharmacology and Experimental Therapeutics. 2008;326:153–162. doi: 10.1124/jpet.107.135046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Dudek NL, Li Q, Muma NA. Phospholipase C, Ca2+, and calmodulin signaling are required for 5-HT2A receptor-mediated transamidation of Rac1 by transglutaminase. Psychopharmacology. 2011;213:403–412. doi: 10.1007/s00213-010-1984-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta D, Arion D, Corradi JP, Lewis DA. Altered expression of CDC42 signaling pathway components in cortical layer 3 pyramidal cells in schizophrenia. Biological psychiatry. 2015 doi: 10.1016/j.biopsych.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis SH, Pasqui F, Colvin EM, Sanger H, Mogg AJ, Felder CC, Broad LM, Fitzjohn SM, Isaac JT, Mellor JR. Activation of Muscarinic M1 Acetylcholine Receptors Induces Long-Term Potentiation in the Hippocampus. Cerebral cortex. 2015 doi: 10.1093/cercor/bhv227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Alaoui S, Legastelois S, Roch AM, Chantepie J, Quash G. Transglutaminase activity and N epsilon (gamma glutamyl) lysine isopeptide levels during cell growth: an enzymatic and immunological study. Int J Cancer. 1991;48:221–226. doi: 10.1002/ijc.2910480212. [DOI] [PubMed] [Google Scholar]
- Fehér Á, Juhász A, László A, Pákáski M, Kálmán J, Janka Z. Serotonin transporter and serotonin receptor 2A gene polymorphisms in Alzheimer’s disease. Neuroscience letters. 2013;534:233–236. doi: 10.1016/j.neulet.2012.12.020. [DOI] [PubMed] [Google Scholar]
- Flatau G, Landraud L, Boquet P, Bruzzone M, Munro P. Deamidation of RhoA glutamine 63 by the Escherichia coli CNF1 toxin requires a short sequence of the GTPase switch 2 domain. Biochemical and biophysical research communications. 2000;267:588–592. doi: 10.1006/bbrc.1999.1904. [DOI] [PubMed] [Google Scholar]
- González-Maeso J, Weisstaub NV, Zhou M, Chan P, Ivic L, Ang R, Lira A, Bradley-Moore M, Ge Y, Zhou Q. Hallucinogens recruit specific cortical 5-HT 2A receptor-mediated signaling pathways to affect behavior. Neuron. 2007;53:439–452. doi: 10.1016/j.neuron.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Gray JA, Roth BL. Molecular targets for treating cognitive dysfunction in schizophrenia. Schizophrenia Bulletin. 2007;33:1100–1119. doi: 10.1093/schbul/sbm074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervás A, Toma C, Romarís P, Ribasés M, Salgado M, Bayes M, Balmaña N, Cormand B, Maristany M, Guijarro S. The involvement of serotonin polymorphisms in autistic spectrum symptomatology. Psychiatric genetics. 2014;24:158–163. doi: 10.1097/YPG.0000000000000034. [DOI] [PubMed] [Google Scholar]
- Horne EA, Dell’Acqua ML. Phospholipase C is required for changes in postsynaptic structure and function associated with NMDA receptor-dependent long-term depression. The Journal of neuroscience. 2007;27:3523–3534. doi: 10.1523/JNEUROSCI.4340-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Srivastava DP, Allen JA, Strachan RT, Roth BL, Penzes P. Rapid modulation of spine morphology by the 5-HT2A serotonin receptor through kalirin-7 signaling. Proceedings of the National Academy of Sciences. 2009;106:19575–19580. doi: 10.1073/pnas.0905884106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MB. Synaptic Signaling in Learning and Memory. Cold Spring Harbor perspectives in biology. 2016;8:a016824. doi: 10.1101/cshperspect.a016824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-Y, Grant P, Lee J-H, Pant HC, Steinert PM. Differential expression of multiple transglutaminases in human brain Increased expression and cross-linking by transglutaminases 1 and 2 in Alzheimer’s disease. Journal of Biological Chemistry. 1999;274:30715–30721. doi: 10.1074/jbc.274.43.30715. [DOI] [PubMed] [Google Scholar]
- Konopaske GT, Lange N, Coyle JT, Benes FM. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA psychiatry. 2014;71:1323–1331. doi: 10.1001/jamapsychiatry.2014.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukkonen J, Ojala P, Näsman J, Hämäläinen H, Heikkilä J, Akerman K. Muscarinic receptor subtypes in human neuroblastoma cell lines SH-SY5Y and IMR-32 as determined by receptor binding, Ca++ mobilization and northern blotting. Journal of Pharmacology and Experimental Therapeutics. 1992;263:1487–1493. [PubMed] [Google Scholar]
- Lerm M, Selzer J, Hoffmeyer A, Rapp U, Aktories K, Schmidt G. Deamidation of Cdc42 and Rac by Escherichia colicytotoxic necrotizing factor 1: activation of c-Jun N-terminal kinase in HeLa cells. Infection and immunity. 1999;67:496–503. doi: 10.1128/iai.67.2.496-503.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino A, Ettorre M, Musilli M, Lorenzetto E, Buffelli M, Diana G. Rho GTPase-dependent plasticity of dendritic spines in the adult brain. Frontiers in cellular neuroscience. 2013:7. doi: 10.3389/fncel.2013.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestre TA, Zurowski M, Fox SH. 5-Hydroxytryptamine 2A receptor antagonists as potential treatment for psychiatric disorders. Expert opinion on investigational drugs. 2013;22:411–421. doi: 10.1517/13543784.2013.769957. [DOI] [PubMed] [Google Scholar]
- Muma NA, Mi Z. Serotonylation and transamidation of other monoamines. ACS Chemical Neuroscience. 2015 doi: 10.1021/cn500329r. [DOI] [PubMed] [Google Scholar]
- Murakoshi H, Wang H, Yasuda R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature. 2011;472:100–104. doi: 10.1038/nature09823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Roche KW. Long-term potentiation: peeling the onion. Neuropharmacology. 2013;74:18–22. doi: 10.1016/j.neuropharm.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrini MC, Lei M, Harrison SC, Mayer BJ. Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Molecular cell. 2002;9:73–83. doi: 10.1016/s1097-2765(01)00428-2. [DOI] [PubMed] [Google Scholar]
- Peddie C, Davies H, Colyer F, Stewart M, Rodriguez J. Colocalisation of serotonin 2A receptors with the glutamate receptor subunits NR1 and GluR2 in the dentate gyrus: an ultrastructural study of a modulatory role. Experimental neurology. 2008;211:561–573. doi: 10.1016/j.expneurol.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen J-E, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nature neuroscience. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Buonanno A, Passafaro M, Sala C, Sweet RA. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. Journal of neurochemistry. 2013;126:165–182. doi: 10.1111/jnc.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M, Pozzo-Miller L. Dendritic spine dysgenesis in autism related disorders. Neuroscience letters. 2015 doi: 10.1016/j.neulet.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Hamblin M, Ciaranello R. Developmental regulation of 5-HT 2 and 5-HT 1C mRNA and receptor levels. Developmental brain research. 1991;58:51–58. doi: 10.1016/0165-3806(91)90236-c. [DOI] [PubMed] [Google Scholar]
- Schätzle P, Ster J, Verbich D, McKinney RA, Gerber U, Sonderegger P, María Mateos J. Rapid and reversible formation of spine head filopodia in response to muscarinic receptor activation in CA1 pyramidal cells. The Journal of physiology. 2011;589:4353–4364. doi: 10.1113/jphysiol.2010.204446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt G, Goehring U-M, Schirmer J, Lerm M, Aktories K. Identification of the C-terminal Part of BordetellaDermonecrotic Toxin as a Transglutaminase for Rho GTPases. Journal of Biological Chemistry. 1999;274:31875–31881. doi: 10.1074/jbc.274.45.31875. [DOI] [PubMed] [Google Scholar]
- Singh RK, Shi J, Zemaitaitis BW, Muma NA. Olanzapine increases RGS7 protein expression via stimulation of the Janus tyrosine kinase-signal transducer and activator of transcription signaling cascade. Journal of Pharmacology and Experimental Therapeutics. 2007;322:133–140. doi: 10.1124/jpet.107.120386. [DOI] [PubMed] [Google Scholar]
- Spires TL, Molnár Z, Kind PC, Cordery PM, Upton AL, Blakemore C, Hannan AJ. Activity-dependent regulation of synapse and dendritic spine morphology in developing barrel cortex requires phospholipase C-β1 signalling. Cerebral cortex. 2005;15:385–393. doi: 10.1093/cercor/bhh141. [DOI] [PubMed] [Google Scholar]
- Srivastava DP, Woolfrey KM, Penzes P. Analysis of dendritic spine morphology in cultured CNS neurons. Journal of Visualized Experiments. 2011 doi: 10.3791/2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen FN, van Delft S, Kain HE, van der Kammen RA, Collard JG. Rac regulates phosphorylation of the myosin-II heavy chain, actinomyosin disassembly and cell spreading. Nature Cell Biology. 1999;1:242–248. doi: 10.1038/12068. [DOI] [PubMed] [Google Scholar]
- Walther DJ, Peter J-U, Winter S, Höltje M, Paulmann N, Grohmann M, Vowinckel J, Alamo-Bethencourt V, Wilhelm CS, Ahnert-Hilger G. Serotonylation of small GTPases is a signal transduction pathway that triggers platelet α-granule release. Cell. 2003;115:851–862. doi: 10.1016/s0092-8674(03)01014-6. [DOI] [PubMed] [Google Scholar]
- Wess J. Muscarinic Acetylcholine Receptor Knockout Mice: Novel Phenotypes and Clinical Implications*. Annu Rev Pharmacol Toxicol. 2004;44:423–450. doi: 10.1146/annurev.pharmtox.44.101802.121622. [DOI] [PubMed] [Google Scholar]
- Wijetunge LS, Till SM, Gillingwater TH, Ingham CA, Kind PC. mGluR5 regulates glutamate-dependent development of the mouse somatosensory cortex. The Journal of Neuroscience. 2008;28:13028–13037. doi: 10.1523/JNEUROSCI.2600-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmus MM, Grunberg S, Bol JG, Van Dam AM, Hoozemans JJ, Rozemuller AJ, Drukarch B. Transglutaminases and Transglutaminase - Catalyzed Cross - Links Colocalize with the Pathological Lesions in Alzheimer’s Disease Brain. Brain Pathology. 2009;19:612–622. doi: 10.1111/j.1750-3639.2008.00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcikiewicz RJ, Tobin AB, Nahorski SR. Muscarinic Receptor - Mediated Inositol 1, 4, 5 - Trisphosphate Formation in SH - SY5Y Neuroblastoma Cells Is Regulated Acutely by Cytosolic Ca2+ and by Rapid Desensitization. Journal of neurochemistry. 1994;63:177–185. doi: 10.1046/j.1471-4159.1994.63010177.x. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Matsui M, Watanabe M. Preferential localization of muscarinic M1 receptor on dendritic shaft and spine of cortical pyramidal cells and its anatomical evidence for volume transmission. The Journal of neuroscience. 2010;30:4408–4418. doi: 10.1523/JNEUROSCI.5719-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Kanamaru C, Ohtani A, Li F, Senzaki K, Shiga T. Subtype specific roles of serotonin receptors in the spine formation of cortical neurons in vitro. Neuroscience research. 2011;71:311–314. doi: 10.1016/j.neures.2011.07.1824. [DOI] [PubMed] [Google Scholar]
- Yu VC, Eiger S, Duan DS, Lameh J, Sadée W. Regulation of Cyclic AMP by the μ - Opioid Receptor in Human Neuroblastoma SH - SY5Y Cells. Journal of neurochemistry. 1990;55:1390–1396. doi: 10.1111/j.1471-4159.1990.tb03151.x. [DOI] [PubMed] [Google Scholar]
- Zainelli GM, Dudek NL, Ross CA, Kim S-Y, Muma NA. Mutant huntingtin protein: a substrate for transglutaminase 1, 2, and 3. Journal of Neuropathology & Experimental Neurology. 2005;64:58–65. doi: 10.1093/jnen/64.1.58. [DOI] [PubMed] [Google Scholar]