

Abstract
The striatum plays critical roles in action control and cognition, and activity of striatal neurons is driven by glutamatergic input. Inhibition of glutamatergic inputs to projection neurons and interneurons of the striatum by presynaptic G protein-coupled receptors (GPCRs) stands to modulate striatal output and striatum-dependent behaviors. Despite knowledge that a substantial number of glutamatergic inputs to striatal neurons originate in the thalamus, most electrophysiological studies assessing GPCR modulation do not differentiate between effects on corticostriatal and thalamostriatal transmission, and synaptic inhibition is frequently assumed to be mediated by activation of GPCRs on corticostriatal terminals. We used optogenetic techniques and recently-discovered pharmacological tools to dissect the effects of a prominent presynaptic GPCR, metabotropic glutamate receptor 2 (mGlu2), on corticostriatal vs. thalamostriatal transmission. We found that an agonist of mGlu2 and mGlu3 induces long-term depression (LTD) at synapses onto MSNs from both the cortex and the thalamus. Thalamostriatal LTD is selectively blocked by an mGlu2-selective negative allosteric modulator and reversed by application of an antagonist following LTD induction. Activation of mGlu2/3 also induces LTD of thalamostriatal transmission in striatal cholinergic interneurons (CINs), and pharmacological activation of mGlu2/3 or selective activation of mGlu2 inhibits CIN-mediated dopamine release evoked by selective stimulation of thalamostriatal inputs. Thus, mGlu2 activation exerts effects on striatal physiology that extend beyond modulation of corticostriatal synapses, and has the potential to influence cognition and striatum-related disorders via inhibition of thalamus-derived glutamate and dopamine release.
Keywords: striatum, metabotropic glutamate receptor, dopamine, cholinergic interneuron, corticostriatal, thalamostriatal
1. Introduction
The basal ganglia are an interconnected group of subcortical nuclei that play important roles in action control and cognition (Graybiel and Grafton, 2015). The striatum serves as the major input nucleus of the basal ganglia, and striatal activity is driven by glutamatergic inputs to medium spiny projection neurons (MSNs, the principal cell type in the striatum) from various cortical regions as well as the thalamus (Alexander et al., 1986; Hintiryan et al., 2016; Smith et al., 2014). This glutamatergic transmission is the main stimulus that drives action potentials in the otherwise passive MSNs, and thus glutamatergic synapses are key points for modulation of striatal function. The striatum is also densely innervated by midbrain dopamine neurons, which modulate a variety of physiological processes and are critical for behaviors including movement, acquisition of motor skills, and instrumental learning (Baik, 2013; Beeler et al., 2014). Substantial effort has been made to understand the physiology of corticostriatal circuits and how they impact behavior. However, despite the abundance of thalamic synapses in the striatum, our understanding of the thalamostriatal system remains limited. Thalamostriatal projections are thought to relay information about salient sensory stimuli and environmental context to regulate behaviors such as attentional shifting, behavioral switching, and reinforcement of instrumental learning (Bradfield et al., 2013; Smith et al., 2011). In addition to regulating striatal output by directly exciting MSNs and indirectly gating corticostriatal transmission (Ding et al., 2010; Smith et al., 2014), thalamostriatal inputs also modulate striatal function by facilitating dopamine release via activation of cholinergic interneurons (CINs) (Threlfell et al., 2012). Although modest progress has been made in elucidating functional roles of thalamostriatal circuits in recent years, many questions remain about the physiology of thalamic inputs to the striatum and their roles in striatal function. For example, little is known about how G protein-coupled receptors (GPCRs) modulate thalamostriatal transmission to influence striatal physiology.
Among the GPCRs that modulate striatal glutamatergic transmission are the metabotropic glutamate (mGlu) receptors. The eight subtypes of mGlu receptors represent three distinct groups (Niswender and Conn, 2010), each of which is known to impact excitatory transmission in the striatum. Group I mGlu receptor (mGlu1 and mGlu5) are expressed postsynaptically in the striatum and contribute to long-term regulation of excitatory transmission by promoting retrograde endocannabinoid signaling (Kreitzer and Malenka, 2005; Sung et al., 2001; Wu et al., 2015). Group II mGlu receptors (mGlu2 and mGlu3) and group III mGlu receptors (mGlu4, −7, −8) act as presynaptic autoreceptors in the striatum (Johnson and Conn, 2012). Electrophysiological recordings in rodent striatal slices have revealed that group II mGlu receptor-selective agonists reduce excitatory transmission, with some reports of transient depression (Lovinger and McCool, 1995; Martella et al., 2009) and other demonstrations of long-term depression (LTD) (Kahn et al., 2001; Kupferschmidt and Lovinger, 2015). Until recently, identification of the specific receptor subtypes that regulate striatal excitatory transmission proved challenging due to a lack of subtype-selective ligands for group II and III mGlu receptors, although one recent study demonstrated that inhibition of evoked field potentials by an mGlu2/3 agonist was absent in mice lacking mGlu2 (Zhou et al., 2013), demonstrating a critical role for this receptor. Robust expression of both mGlu2 and mGlu3 has been described in the striatum (Wright et al., 2013), and mGlu2 expression is likely to be restricted primarily to afferents to the striatum, as little evidence for mGlu2 mRNA has been found within the striatum (Ohishi et al., 1993; Testa et al., 1994). Interestingly, mGlu2 mRNA is detected throughout the cortex as well as in the intralaminar nuclei of the thalamus (Allen Brain Atlas; www.brain-map.org), suggesting that mGlu2 activation could modulate transmission in both striatal inputs. mGlu3 mRNA is also present in the cortex and thalamus, and potential contributions of mGlu3 to regulation of excitatory transmission in the striatum have not been directly evaluated.
The majority of previous studies examining modulation of striatal glutamatergic transmission by GPCRs such as mGlu2/3 employed electrical stimulation either within the striatum or in the overlying white matter. Because cortical and thalamic inputs innervate the same cells (Doig et al., 2010; Huerta-Ocampo et al., 2014), the input specificity of GPCR modulation could not be determined using such techniques, yet inhibition of striatal glutamatergic transmission was often assumed to represent regulation of neurotransmitter release at corticostriatal terminals. To circumvent this shortcoming, we implemented both transgenic and viral techniques to express channelrhodopsin (ChR2) in corticostriatal or thalamostriatal projection neurons and evaluated the ability of group II mGlu receptor activation to reduce glutamate release from each input. Using recently discovered subtype-selective pharmacological tools, we now demonstrate that mGlu2 activation induces robust LTD of thalamostriatal transmission in both MSNs and CINs of the dorsal striatum. In addition, an mGlu2/3 agonist reduces phasic dopamine release that is specifically mediated by thalamostriatal activation of CINs. These findings advance our limited understanding of how presynaptic autoreceptors regulate discrete aspects of striatal physiology.
2. Methods
Animals
Animal care and procedures used for these studies were approved by the Animal Care and Use Committee of the National Institute on Alcohol Abuse and Alcoholism and conformed to the guidelines of the US National Institutes of Health Guide for the Care and Use of Animals. For most experiments, 9–18 week old male C57Bl/6J mice (The Jackson Laboratory, stock no. 000664) were used. For experiments evaluating glutamatergic transmission in CINs, hemizygous ChAT-IRES-Cre mice (The Jackson Laboratory, stock no. 006410) were crossed with Ai14 mice (The Jackson Laboratory, stock no. 007908) to drive TdTomato reporter expression in CINs. For some experiments evaluating corticostriatal and thalamostriatal transmission, Ai32 mice (The Jackson Laboratory, stock no. 024109) were crossed with Emx1-IRES-Cre (The Jackson Laboratory, stock no. 005628) or vGlut2-IRES-Cre mice (The Jackson Laboratory, stock no. 016963, backcrossed at NIAAA to C57Bl/6J mice for at least six generations), respectively. This allowed Cre-dependent expression of ChR2 in the striatal inputs of interest. Animals were housed 2–4 per cage in a temperature- and humidity-controlled room with a standard 12 hour light/dark cycle and ad libitum access to food and water.
Viral injections for optogenetics experiments
Male C57Bl/6J mice and male or female ChAT-IRES-Cre;Ai14 mice (5–8 weeks old) were anesthetized with isoflurane (5% induction, ~2% maintenance) and placed into a stereotaxic frame (David Kopf Instruments). Mice were injected with 250–300 nL AAV1.CamKIIα.hChR2(H134R)-eYFP (University of Pennsylvania Vector Core) at a rate of 75 nL/min using a Hamilton syringe. For recordings of corticostriatal transmission, injections were targeted to M1 motor cortex (coordinates relative to bregma: 1.1 anterior; ±1.6 lateral; 0.8 ventral from brain surface). For recordings of thalamostriatal transmission, injections were targeted to the parafascicular nucleus (coordinates relative to bregma: −2.1 posterior; ±0.6 lateral, 3.8 ventral from brain surface). Brain slices were prepared for electrophysiology or FSCV recording 4–10 weeks following surgery. Verification of injection sites and imaging of eYFP fluorescence were performed on an Olympus MVX10 microscope (Olympus Corporation of America).
Brain slice preparation for electrophysiology and fast-scan cyclic voltammetry
Coronal brain slices (250 μm thick) were prepared using a vibratome (Leica Microsystems) as previously described (Atwood et al., 2014a; Crowley et al., 2014; Mathur et al., 2011). Mice were anesthetized with isoflurane, decapitated, and brains were rapidly removed and submerged in ice-cold cutting solution containing (in mM): 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, 10 glucose, and 194 sucrose, continuously bubbled with 95% O2/5% CO2. Slices were immediately removed to a 32°C holding chamber containing artificial cerebrospinal fluid (aCSF) containing (in mM): 124 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 glucose, 305–310 mOsm, continuously bubbled with 95% O2/5% CO2. Slices were allowed to recover for 30–45 minutes at 32°C, and then were incubated at room temperature for at least 30 minutes prior to beginning experiments.
Whole-cell voltage clamp recordings
Whole-cell voltage-clamp recordings were conducted as previously described (Atwood et al., 2014a). Individual hemisected slices were placed in a diamond-shaped recording chamber (Warner Instruments) and were submerged in, and continuously perfused with 30–32°C aCSF at a rate of ~1.5 mL/min. Recording pipettes (2.0–4.0 MΩ resistance in bath) were filled with Cs-based internal solution (295–300 mOsm) containing (in mM): 120 CsMeSO3, 5 NaCl, 10 TEA-Cl, 10 HEPES, 5 QX-314, 1.1 EGTA, 0.3 Na-GTP, and 4 Mg-ATP; pH was adjusted to 7.3 using CsOH. Slices were visualized on a Zeiss Axioskop 2 microscope. Cells for whole-cell recordings were visualized using a 40×/0.8 NA water-immersion objective. Putative MSNs were identified based on size, capacitance, and membrane resistance. CINs were identified by online visualization of the fluorescent reporter. Recordings were performed using a Multiclamp 700A amplifier (Axon Instruments). Cells were voltage-clamped at −60 mV throughout the experiment. For electrically-evoked excitatory postsynaptic current (eEPSC) recordings, a parallel bipolar stimulating electrode (~100 μm tip separation) was positioned at the border of the external capsule and the dorsolateral striatum. Cells were recorded within the dorsolateral striatum. For optically-evoked EPSC (oEPSC) recordings, 470 nm, 2–5 ms pulse-width field illumination was delivered via a High-Power LED Source (Thor Labs). For both electrically-evoked and optically-evoked EPSC recordings, stimulation intensity was typically adjusted to elicit 150–500 pA current amplitudes. Electrically-evoked EPSCs were evoked every 20 seconds, and optically-evoked EPSCs were evoked once per minute. For paired-pulse EPSC recordings, a 50 ms interpulse interval was used. All recordings were filtered at 2 kHz and digitized at 10 kHz (Digidata 1322A, Axon Instruments). For all EPSC recordings, 50 μM picrotoxin was included in the aCSF to block fast GABAergic transmission. Drugs were prepared as stock solutions in water or dimethylsulfoxide (DMSO) and diluted into aCSF prior to each experiment. For experiments using drugs prepared in DMSO, the DMSO concentration was held constant throughout the experiment, with a maximum of 0.1% final DMSO concentration. Drugs were administered via bath application for the designated period of time; antagonists were present throughout the recording. Acquisition was performed using Clamplex 10.3 (Molecular Devices). Access resistance was monitored during recordings and only cells with a stable access resistance (less than 15% change from baseline) were included for analysis.
Fast-scan cyclic voltammetry (FSCV) recordings
A glass-encased cylindrical carbon fiber (Goodfellow, PA; 7 μm diameter, 100–130 μm exposed length) was placed into the DLS at a location expressing eYFP. Optical stimulation was delivered by placing an optical fiber (105 μm core diameter, 0.22 NA, Thorlabs, NJ) in apposition to the brain slice. Extracellular dopamine release was monitored by FSCV by applying a triangular input waveform from −0.4V to +1.2V and back to −0.4V (versus an Ag/AgCl reference electrode immersed in the bath solution) through the carbon fiber electrode. Cyclic voltammograms were collected at 10 Hz using a Chem-Clamp (Dagan Corporation) and DEMON Voltammetry and Analysis software (Yorgason et al., 2011). Once five consecutive stable responses were collected (<10% variation in transient peak), experiments would begin. Four baseline measurements were collected before bath application for 10 minutes of LY379268 (100 nM). Following drug application, four extra drug-washout measurements were collected.
Drugs
Picrotoxin and QX-314 were purchased from Sigma. LY379268, LY395756, and LY341495 were purchased from Tocris Bioscience. VU6001192 and VU0469942 were generously provided by the Vanderbilt Center for Neuroscience Drug Discovery (Vanderbilt University, Nashville, TN).
Data analysis and statistics
EPSC amplitudes were measured using Clampfit 10 software (Molecular Devices). GraphPad Prism 6.0 was used to prepare graphs and perform statistical analyses. All data are reported as mean ± SEM. For electrically-evoked EPSC recordings, amplitudes were averaged each minute. EPSC data were normalized to the average EPSC amplitudes during a 10 minute baseline recording. PPRs were calculated by dividing the amplitude of the second EPSC by the amplitude of the first EPSC. PPR comparisons were made for a 5 minute window before and after drug treatment (6–10 minutes post-drug, during peak drug effect) using a paired t test. Peak drug effects were measured at 6–10 minutes after the onset of drug application unless otherwise indicated. LTD was measured at 25–30 minutes after onset of drug application unless otherwise indicated. Sample traces and % baseline values reported in the text correspond to these time windows. N values represent the number of recorded cells. One experiment was performed on each slice, and slices were always drug-naïve prior to experiments. Each dataset contains experiments obtained from at least 2 animals. Within-group comparisons of drug effects vs. baseline were made using paired t tests. Comparisons of peak depression or LTD magnitude between groups were made using an unpaired t test or a one-way ANOVA followed by post-hoc Dunnett’s comparison with control. Alpha was set at 0.05.
3. Results
3.1 Presynaptic mGlu2 activation induces LTD of electrically-evoked glutamate transmission in the DLS
Previous studies have demonstrated that various mGlu2/3 agonists depress excitatory transmission evoked by electrical stimulation in the dorsal striatum. However, discrepancies exist regrading whether this depression represents short- or long-term changes in neurotransmitter release (for example, see Kahn et al., 2001; Kupferschmidt and Lovinger, 2015; Lovinger and McCool, 1995; Martella et al., 2009). We performed whole-cell voltage clamp recordings of electrically-evoked EPSCs from MSNs in the DLS to determine the effect of the mGlu2/3-selective agonist LY379268 on glutamatergic transmission. After recording stable EPSC amplitudes for at least 10 minutes, LY379268 (100 nM) was bath-applied to slices for 5 minutes. This produced a substantial, long-lasting depression of eEPSC amplitudes (52.8 ± 5.8% of baseline; n = 10; p <0.0001) (Fig. 1a) that began during agonist application. The depression persisted for at least 30 minutes after the termination of drug application in 9/10 cells. Reduction of eEPSC amplitudes was associated with a significant increase in paired-pulse ratios (mean EPSC2/EPSC1 amplitude: baseline, 1.19 ± 0.09 vs. post-drug 1.55 ± 0.11; n = 9; p = 0.037) (Fig. 1b,c), indicating that LY379268 treatment is associated with a decreased probability of glutamate release. Although LY379268-mediated depression of electrically-evoked field potentials was previously shown to require mGlu2 (Zhou et al., 2013), pharmacological analysis of the individual contributions of mGlu2 and mGlu3 has not been reported. To directly evaluate the role of each receptor in non-transgenic mice, we used two recently reported negative allosteric modulators (NAMs), VU6001192 and VU0469942, which are selective for mGlu2 and mGlu3, respectively (Felts et al., 2015; Wenthur et al., 2013). Incubation of slices with VU6001192 (10 μM, present throughout recording) completely prevented the LY379268-mediated depression of eEPSC amplitude (control 50.0 ± 8.2% of baseline, n = 10, vs. VU6001192 99.7 ± 11.0% of baseline, n = 6, at 6–10 minutes following onset of LY379268 application), whereas VU0469942 (10 μM, present throughout recording) did not block the effect of LY379268 (56.6 ± 4.6% of baseline; n = 6). (Fig. 1d–f). One-way ANOVA revealed a main effect of antagonist treatment (F(2, 19) = 8.761; p = 0.002) (Fig. 1f). Post hoc Dunnett’s comparison showed a significant blockade by VU6001192, but not VU0469942, when compared with the LY379268-mediated depression of eEPSC amplitude in control slices (Fig. 1f). These results indicate that mGlu2 mediates the depression of excitatory transmission that is broadly evoked by electrical stimulation, and that mGlu3 is unlikely to play a role.
Figure 1.
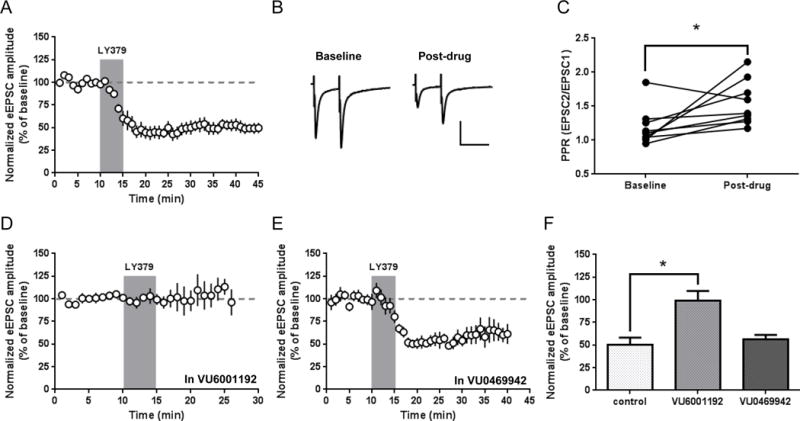
Activation of presynaptic mGlu2 induces LTD of electrically-evoked excitatory transmission. (a) Bath application of LY379268 (100 nM, 5 minutes) produces a long-lasting depression of EPSC amplitudes. (b) Sample traces from paired-pulse recordings (50 ms inter-pulse interval). Traces were averaged over 5 minutes of baseline recording prior to drug application and over the 5 minutes immediately termination of drug application. Scale bars: 200 pA, 50 ms. (c) Paired-pulse ratios (PPR, calculated as EPSC2/EPSC1 amplitude) for individual experiments before and after LY379268 application (p = 0.037, paired t test). (d) Effect of the mGlu2-selective NAM VU6001192 (10 μM, present throughout recording) on LY379268 inhibition of EPSCs. (e) Effect of the mGlu3-selective NAM VU0469942 (10 μM, present throughout recording) on LY379268 inhibition of EPSCs. (f) Summary of LY379268 effect from 6–10 minutes after onset of LY379268 application (mean ± SEM). VU6001192 significant blocked the effect of LY379268 (control vs. VU6001192, p < 0.05, Dunnett’s test). EPSC amplitude time course data are normalized to the average baseline EPSC amplitude and shown as mean ± SEM.
3.2 mGlu2/3 activation depresses corticostriatal transmission
Previous studies of mGlu2/3 agonist effects on electrically-evoked excitatory transmission in the dorsal striatum have generated the prevailing idea that mGlu2 activation produces a depression of corticostriatal transmission. However, none of these studies used techniques that could distinguish modulation of corticostriatal vs. thalamostriatal transmission. To directly evaluate this, we injected an adeno-associated viral (AAV) vector encoding ChR2-EYFP under the control of the CamKIIα promoter into the M1 motor cortex region of C57Bl/6J mice. Four to ten weeks after injection, EYFP-labeled axon terminals were visible in the dorsolateral striatum (Fig. 2a). We recorded optically-evoked EPSCs from MSNs within the DLS and found that 5 minute bath application of LY379268 (100 nM) produced robust LTD (33.3 ± 6.2% of baseline; n = 6; p < 0.001) (Fig. 2b). To evaluate the effect of mGlu2/3 activation on a broader set of cortical inputs, we crossed Ai32 mice, which express ChR2 in a Cre recombinase-dependent manner (Madisen et al., 2012), with Emx1-Cre mice, which express Cre recombinase in glutamatergic projection neurons of the cortex and hippocampus (Gorski et al., 2002; Kupferschmidt and Lovinger, 2015). This produced Emx1-Cre;Ai32 mice with ChR2 specifically expressed in cortical inputs to the striatum. Recordings of oEPSCs from MSNs of Emx1-Cre;Ai32 mice revealed that 5 minute bath application of LY379268 (100 nM) produced a significant reduction of oEPSC amplitude (69.1 ± 6.4% of baseline; n = 11; p = 0.002) (Fig. 2c). Interestingly, the magnitude of LY379268-mediated depression was significantly greater in M1 virus-injected mice than in Emx1-Cre;Ai32 mice (p = 0.0024) (Fig. 2d). To ensure that this result was not due to a general reduction in mGlu2/3 function in Emx1-Cre;Ai32 mice, we performed several recordings in which we interleaved electrical and optical stimulation. In these recordings, LY379268 routinely produced a greater inhibition of electrically-evoked stimulation than optically-evoked stimulation, suggesting that the lower magnitude of the LY379268 effect in the Emx1-Cre;Ai32 mice was specific to corticostriatal inputs activated by optical stimulation (data not shown). Together, these results provide direct evidence that an mGlu2/3 agonist induces LTD at corticostriatal synapses.
Figure 2.
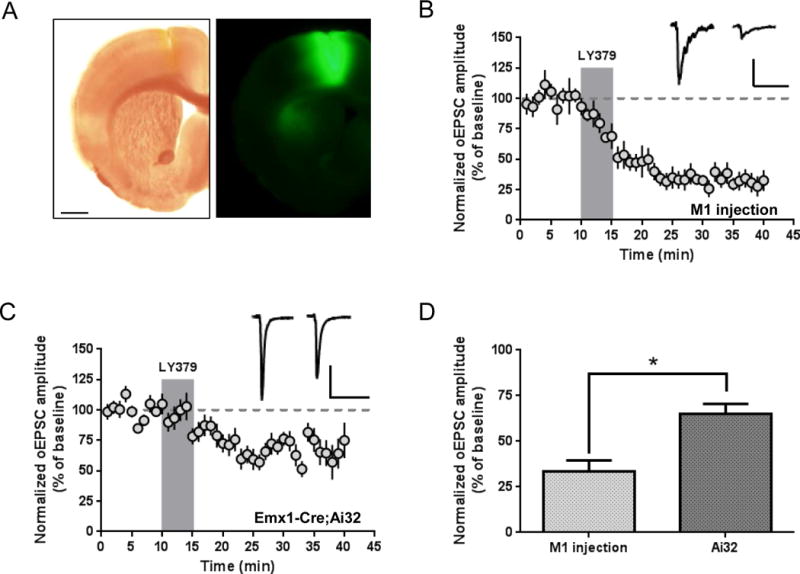
Cortically-evoked EPSCs are inhibited by group II mGlu receptor activation. (a) Expression of ChR2-EYFP in the cortex (primarily M1 motor cortex) following viral expression. Cortical projections are visible in a lateral region of the dorsal striatum. Image is representative of typical fluorescence patterns observed in three recorded mice. Scale bars: 1 mm. (b) LY379268 (100 nM, 5 minutes) induces LTD of optically-evoked corticostriatal EPSCs recorded from MSNs of animals injected with AAV-ChR2. Scale bars: 100 pA, 50 ms. (c) LY379268 (100 nM, 5 minutes) inhibits optically-evoked corticostriatal EPSCs recorded from MSNs in slices from Emx1-Cre;Ai32 mice. Scale bars: 200 pA, 50 ms. (d) Summary of LY379268 effect 26–30 minutes after drug onset (p = 0.0024, unpaired t test). Sample traces are averaged over 5 minutes prior to drug application and from 26–30 minutes following onset of drug application. Data are normalized to the average baseline EPSC amplitude and shown as mean ± SEM.
3.3 mGlu2 activation produces a reversible form of LTD at thalamostriatal synapses
Whereas GPCR-mediated modulation of corticostriatal transmission has received a great deal of attention in the literature, few studies have examined modulation of thalamostriatal transmission. To assess the potential regulation of thalamostriatal transmission by group II mGlu receptors, we virally expressed ChR2 via injections targeting the parafascicular nucleus of the thalamus, which provides a major input to the dorsal striatum (Smith et al., 2014). Four to ten weeks after injection, we observed diffuse EYFP-labeled axon terminals throughout the striatum (Fig. 3a), including in the DLS. We recorded oEPSCs from MSNs in the DLS and found that 5 minute bath application of LY379268 (100 nM) produced LTD that persisted for at least 30 minutes following the onset of drug application (39.3 ± 6.2% of baseline; n = 8; p < 0.0001) (Fig. 3b). As an alternative approach, we crossed Ai32 mice with Vglut2-Cre mice to obtain Vglut2-Cre;Ai32 mice. In accordance with previously identified Vglut2 expression patterns, these mice express Cre recombinase in the thalamus and not in striatum-projecting cortical neurons (Fremeau et al., 2004; Vong et al., 2011). As with virus-injected mice, LY379268 (100 nM, 5 minutes) induced LTD in DLS MSNs of Vglut2-Cre;Ai32 mice (42.0 ± 5.4% of baseline; n = 10; p < 0.0001) (Fig. 3c). There was no difference in the magnitude of LTD induced by LY379268 when using the two different methods of ChR2 expression (p = 0.75) (Fig. 3d). Together, these results provide the first direct evidence that in addition to the widely-accepted regulation of corticostriatal transmission, activation of group II mGlu receptors strongly modulates striatal glutamatergic transmission arising from the thalamus.
Figure 3.
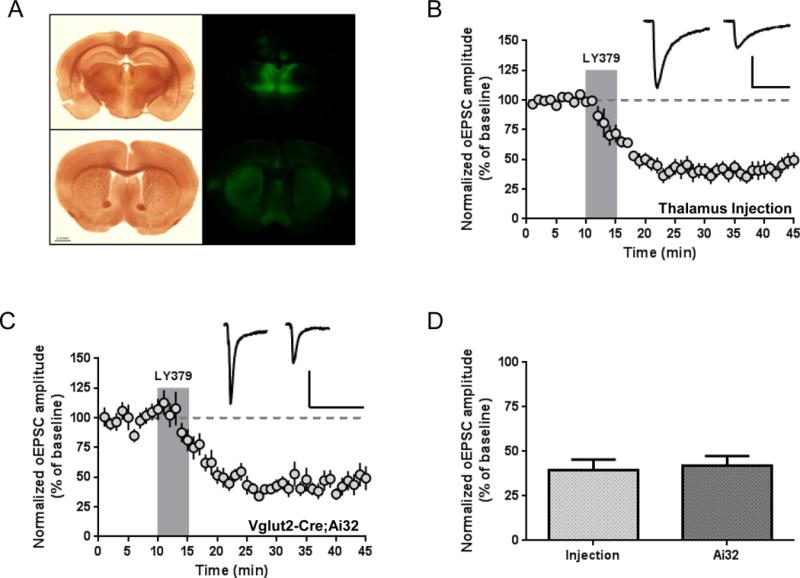
Activation of group II mGlu receptors induces LTD of thalamically-evoked glutamatergic transmission. (a) Expression of ChR2-EYFP in the intralaminar nuclei of the thalamus (including the parafascicular nucleus) following viral expression (top panels). Thalamic projections are visible throughout the striatum (bottom panels). Scale bar: 1 mm. (b) LY379268 (100 nM, 5 minutes) induces LTD of optically-evoked thalamostriatal EPSCs recorded from MSNs of animals injected with AAV-ChR2. Scale bars: 200 pA, 50 ms. (c) LY379268 (100 nM, 5 minutes) induces LTD of optically-evoked thalamostriatal EPSCs recorded from MSNs of Vglut2-Cre;Ai32 mice. Scale bars: 200 pA, 50 ms. (d) There is no difference in magnitude of LTD in injected vs. Vglut2-Cre;Ai32 mice (p = 0.75, unpaired t test). Sample traces are averaged over 5 minutes prior to drug application and from 26–30 minutes following onset of drug application. Data are normalized to the average baseline EPSC amplitude and shown as mean ± SEM.
We next used subtype-selective pharmacological tools to explicitly test the roles of mGlu2 and mGlu3 on thalamostriatal transmission in mice virally expressing ChR2 in the parafascicular nucleus. We recorded the effect of LY379268 (100 nM, 5 minutes) in the presence of the group II mGlu receptor-preferring antagonist LY341495 (500 nM) (Fig. 4c), the mGlu2-selective NAM VU6001192 (10 μM, Fig. 4a,c), or the mGlu3-selective NAM VU0469942 (10 μM, Fig. 4b,c). One-way ANOVA revealed a significant effect of antagonist treatment (F(3, 23) = 13.57, p < 0.0001). Post hoc analysis revealed that both LY341495 and VU6001192 significantly reduced the effect of LY379268 when compared with control slices (control 57.5 ± 3.6% of baseline, n = 7; LY341495 107.1 ± 8.9% of baseline, n = 7; VU6001192 85.0 ± 6.1% of baseline, n = 6; 6–10 minutes following onset of drug application) (Fig. 4a,c), whereas VU0469942 had no effect (51.3 ± 8.2% of baseline, n = 7) (Fig. 4b,c). Concordant with our observations of electrically-evoked stimulation, our results suggest that mGlu2 exclusively regulates thalamostriatal transmission.
Figure 4.
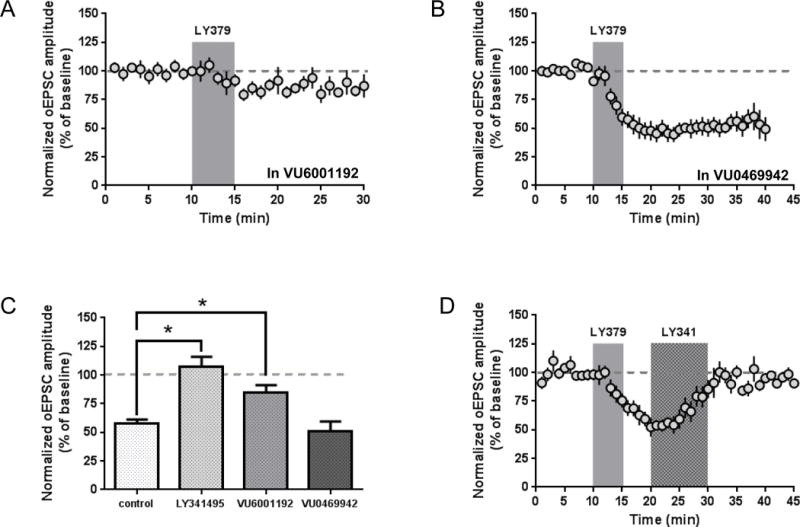
Thalamostriatal LTD requires mGlu2 and is reversed by a competitive antagonist. LY379268-induced LTD is prevented by incubation of slices with the mGlu2-selective NAM VU6001192 (10 μM, present throughout recording) (a), but is not impaired by the mGlu3-selective NAM VU0469942 (10 μM, present throughout recording) (b). (c) Summary of LY379268 inhibition of thalamostriatal transmission in the presence of the mGlu2/3-preferring competitive antagonist LY341495 (500 nM), VU6001192, or VU0469942 (control vs. LY341495 and control vs. VU6001192, p < 0.05, Dunnett’s test). (d) Application of LY341495 (500 nM, 10 minutes) ten minutes after onset of LY379268 (100 nM, 5 minutes) restores EPSC amplitudes to baseline levels. Antagonist experiments were performed in animals injected with AAV-ChR2 in the parafascicular nucleus. Data are normalized to the average baseline EPSC amplitude and shown as mean ± SEM.
A common feature of mGlu receptor-mediated LTD is that antagonist application following induction of LTD restores baseline levels of synaptic transmission (Lodge et al., 2013). To test the reversibility of mGlu2-LTD of thalamostriatal transmission, we induced LTD with LY379268 (100 nM, 5 minutes), then applied the mGlu2/3-preferring antagonist LY341495 (500 nM) to slices from 10 to 20 minutes following the onset of LY379268 application. Similar to previous reports (Kupferschmidt and Lovinger, 2015), we found that mGlu2-LTD was indeed reversible upon subsequent antagonist exposure (Fig. 4d). This is unlikely to be caused by incomplete washout of LY379268, as this drug is known to produce reversible effects in similar preparations (for example, (Walker et al., 2015). Unlike several reports of “reversibly reversible” antagonist effects on mGlu receptor-mediated LTD (Lodge et al., 2013), we did not observe restoration of depressed transmission after washout of LY341495 (Fig. 4d).
To further assess the ability of mGlu2 to modulate excitatory transmission, we examined the effect of LY395756 (10 μM), an mGlu2 partial agonist that also blocks mGlu3 responses at higher concentrations (Dominguez et al., 2005; Hanna et al., 2013; Sanger et al., 2013), on both electrically-evoked EPSCs and optically-evoked thalamostriatal EPSCs. Surprisingly, five minute bath application of LY395756 produced a striking but reversible inhibition of both electrically-evoked (Fig. 5a) and optically-evoked (Fig. 5b) EPSCs (electrically-evoked: 33.4 ± 5.9% of baseline at 4–6 minutes following onset of drug application, n = 6, p < 0.001; optically-evoked: 28.0 ± 4.2% of baseline, n = 6, p < 0.0001). It is unlikely that the concentration of LY395756 used was insufficient to induce LTD given the remarkable magnitude of the acute depression and previous reports that this drug induces LTD at other synapses at lower concentrations (Lucas et al., 2013). These findings further support the role of mGlu2 in modulating thalamostriatal transmission, but also demonstrate that ligands with distinct pharmacological profiles (i.e. full vs. partial agonist) can produce different forms of synaptic modulation.
Figure 5.
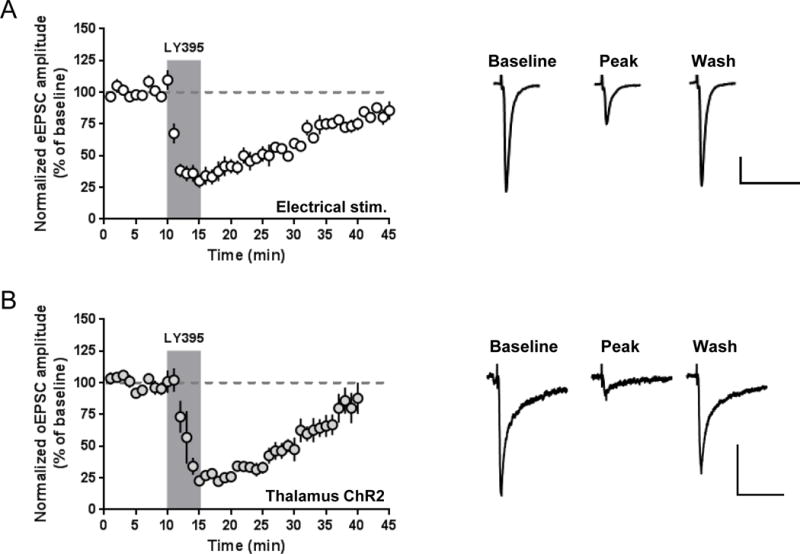
The mGlu2 agonist/mGlu3 antagonist LY395756 reversibly inhibits excitatory transmission. (a) LY395756 (10 μM, 5 minutes) transiently reduces amplitudes of electrically-evoked EPSCs recorded from MSNs. (b) LY395756 (10 μM, 5 minutes) reversibly reduces amplitudes of optically-evoked thalamostriatal EPSCs recorded from MSNs of animals injected with AAV-ChR2. Scale bars: 100 pA, 50 ms. Sample traces are averaged for 5 minutes prior to drug application, from 4–6 minutes following onset of drug application, and for the last 3 minutes of data shown in the time courses. All data are normalized to the average baseline EPSC amplitude and shown as mean ± SEM.
3.4 mGlu2 modulates thalamically-driven dopamine release mediated by cholinergic interneuron activation
Because thalamostriatal inputs to CINs are important regulators of striatum-dependent learning (Bradfield et al., 2013), we proceeded to test the impact of mGlu2/3 activation on thalamic inputs to CINs. We performed whole-cell recordings of optically-evoked EPSCs from CINs of ChAT-Cre;Ai14 mice virally expressing ChR2 in thalamic inputs to the striatum. ChAT-Cre;Ai14 mice express the fluorescent reporter TdTomato in CINs, allowing online visual identification of CINs. Five minute bath application of LY379268 (100 nM) induced robust LTD of thalamostriatal transmission (46.8 ± 5.6% of baseline; n = 6; p < 0.0001) (Fig. 6a). Thalamostriatal activation of CINs is known to drive dopamine release in the dorsal striatum (Threlfell et al., 2012), so we next assessed the ability of mGlu2/3 activation to reduce dopamine release evoked by optical stimulation of thalamostriatal inputs using FSCV in brain slices. Comparable to effects on excitatory transmission in CINs, we found that LY379268 (100 nM, 10 minutes) produced a substantial reduction of dopamine release (43.2 ± 10.4% of baseline upon termination of drug application; baseline [DA] 0.96 ± 0.20 μM vs. LY379268 0.37 ± 0.13 μM; n = 6; p = 0.0275) (Fig. 6b,c). To directly evaluate the role of mGlu2 in the inhibition of dopamine release, we applied LY395756 (10 μM, 10 minutes) to slices and found that this drug also robustly reduced dopamine release (61.0 ± 3.2% of baseline upon termination of drug application; baseline [DA] 0.49 ± 0.08 μM vs. LY395756 0.29 ± 0.05 μM; n = 6; p = 0.0035) (Fig. 6d), indicating a specific role for mGlu2 in the regulation of thalamically-driven dopamine release.
Figure 6.
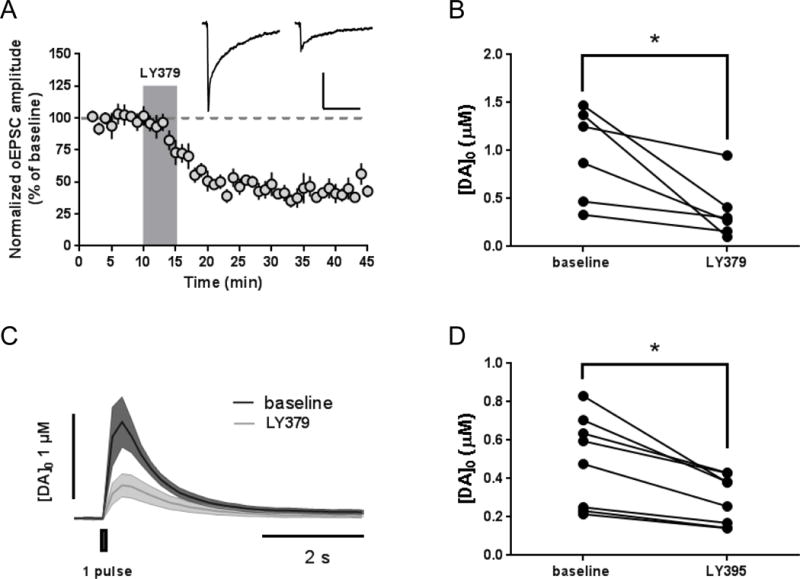
mGlu2 activation reduces thalamostriatal transmission in CINs and thalamically-driven dopamine release. (a) LY379268 application (100 nM, 5 minutes) induces LTD of optically-evoked thalamostriatal EPSCs recorded from CINs of ChAT-Cre;Ai14 mice injected with AAV-ChR2. Sample traces are averaged for 5 minutes prior to drug application and from 26–30 minutes following onset of drug application. Scale bars: 50 pA, 100 ms. Data are normalized to the average baseline EPSC amplitude and shown as mean ± SEM. (b) FSCV experiments in ChAT-Cre;Ai14 mice reveal that LY379268 (100 nM, 10 minutes) reduces dopamine release evoked by optical stimulation of thalamostriatal inputs to the dorsal striatum in slices obtained from ChAT-Cre;Ai14 mice injected with AAV-ChR2 (p = 0.0275, paired t test). (c) FSCV traces are averaged from all recordings. Baseline values and traces are averaged for 4 sweeps prior to drug application. LY379 values and traces represent the [DA] amplitude after 10 minutes of drug application. Shaded areas of sample traces represent SEM. (d) FSCV experiments in C57Bl/6J mice injected in the thalamus with AAV-ChR2 reveal that LY395756 (10 μM, 10 minutes) reduces dopamine release evoked by optical stimulation (p = 0.0035, paired t test).
4. Discussion
We employed optogenetic techniques to dissect circuit-specific modulation of striatal excitatory transmission by mGlu2. Recently, our laboratory and others have used similar approaches to distinguish roles of opioid, cannabinoid, and histamine receptors in regulating corticostriatal vs. thalamostriatal transmission (Atwood et al., 2014a; Ellender et al., 2011; Wu et al., 2015). Indeed, for each of these receptor classes, differential modulation of corticostriatal vs. thalamostriatal transmission has been reported. Interestingly, mGlu2 appears to be the first example of a GPCR that modulates both synapses to a similar extent. However, because cortical and thalamic synapses have distinct properties, such as differences in short-term plasticity, basal probability of glutamate release, and postsynaptic ionotropic glutamate receptor composition (Ding et al., 2008; Smeal et al., 2008), it is possible that activation of mGlu2 in each pathway yields differential effects on striatal output. Intriguingly, we observed a substantial divergence in the magnitude of LTD when recording inputs from the motor cortex vs. broadly activating cortical inputs in the Emx1-Cre;Ai32 mouse (Fig. 2b–d). This finding suggests that striatal inputs from distinct cortical regions are differentially regulated by mGlu2 activation. Further studies evaluating modulation of discrete corticostriatal inputs will be necessary to elucidate the role of mGlu2 in various circuits that impact different aspects of basal ganglia-related behaviors.
Both cortical and thalamic inputs to the striatum drive phasic dopamine release by activating CINs and releasing acetylcholine, which evokes dopamine release by activating presynaptic nicotinic receptors on dopamine terminals (Cachope et al., 2012; Kosillo et al., 2016; Threlfell et al., 2012). Thus, it stands to reason that mGlu2 activation could regulate dopamine release. We report that activation of mGlu2 inhibits dopamine release evoked by optogenetic activation of thalamic inputs to the striatum (Fig. 6b,c). This finding is in agreement with results of microdialyisis studies in the ventral striatum which suggest that group II mGlu receptors tonically reduce extracellular dopamine levels (Baker et al., 2002; Hu et al., 1999). However, more recent microdialysis studies failed to observe a decrease in dorsal striatal dopamine release evoked by in vivo electrical stimulation of midbrain dopamine neurons (Pehrson and Moghaddam, 2010), suggesting that mGlu2 does not modulate dopamine release by acting as a heteroreceptor on dopaminergic axon terminals. The ability of LY379268 to robustly depress thalamostriatal transmission in CINs (Fig. 6a) suggests that mGlu2 on thalamostriatal terminals mediates the inhibition of thalamically-driven dopamine release, although an additional role for mGlu2/3 activation on CIN terminals cannot be ruled out (Marti et al., 2001).
We found that mGlu2-mediated depression of thalamostriatal transmission is both long-lasting and reversible upon application of a competitive antagonist following LTD induction (Fig. 3, Fig. 4d). Thus, this represents a “labile” form of GPCR-mediated LTD, a phenomenon which is commonly observed across all groups of mGlu receptors (Atwood et al., 2014b; Lodge et al., 2013). Discrepancies exist in the literature regarding whether or not the depression of electrically-evoked excitatory transmission by mGlu2/3 activation is transient or represents a form of agonist-induced LTD. These inconsistent findings may result from technical differences, the agonist used in the study, or the species, as reversible inhibition has been reported in rats (Lovinger and McCool, 1995; Picconi et al., 2002), whereas LTD has commonly been observed in mice (Kahn et al., 2001; Kupferschmidt and Lovinger, 2015; Zhou et al., 2013). In addition, previous data suggested that mGlu2-mediated LTD in the striatum was not affected by subsequent antagonist application (Kahn et al., 2001), whereas this and other reports from our laboratory indicate that presynaptic mGlu2 effects in the mouse striatum represent labile forms of LTD (Kupferschmidt and Lovinger, 2015). Although it remains unclear how mGlu receptors continue to signal long after agonist washout, the prevailing hypothesis is that the receptors adopt a constitutively active conformation that allows agonist-independent activity but remains sensitive to antagonist treatment (Lodge et al., 2013; Young et al., 2013). The continued development of techniques to examine nuanced changes in receptor structure during and following ligand-binding could provide critical insight into the mechanisms by which mGlu receptors mediate flexible forms of synaptic plasticity.
A particularly intriguing finding of our study was that the mGlu2 partial agonist/mGlu3 antagonist LY395756 produces a transient rather than a long-term form of synaptic inhibition (Fig. 5). Several explanations might account for the differential efficacy of LY379268 vs. LY395756. One interpretation of this finding is that mGlu3 activation is required for the long-term component of the LY379268 effect, and in fact this has been demonstrated in the medial prefrontal cortex (Walker et al., 2015). However, our finding that the mGlu3 NAM VU0469942 fails to convert the LY379268 effect from LTD to a reversible form of synaptic depression is inconsistent with this interpretation (Fig. 1e, Fig. 4c). Another possibility is that the efficacy of LY395756 is insufficient to induce LTD; this notion is supported by a recent report that the active enantiomer, LY541850, is a partial agonist in primary neuron cultures, although the efficacy of this drug may be context dependent (i.e. dependent on levels of receptor reserve) (Hanna et al., 2013; Sanger et al., 2013). Given the strikingly robust acute inhibition produced by LY395756 (Fig. 5), this explanation also seems unlikely. A third possibility is that LY395756 acts as a functionally biased agonist such that mechanisms that mediate short-term reductions in glutamate release probability are preferentially engaged over the signaling pathways required for induction of LTD. Additional insight into the mechanistic underpinnings of presynaptic LTD will be necessary to further explore this possibility. Our demonstration of the inconsistent effects of structurally similar agonists highlights the importance of considering nuanced characteristics of pharmacological tools when interrogating GPCR function in native systems.
Modulation of glutamatergic transmission by striatal group II mGlu receptors is altered in a number of animal models of CNS disorders. For example, striatal dopamine denervation by 6-hydroxydopamine lesion produces an increase in the potency with which LY379268 depresses electrically-evoked excitatory transmission (Picconi et al., 2002). Similarly, enhanced sensitivity to LY379268 was reported in multiple genetic models of Parkinson’s disease (Martella et al., 2009). Conversely, reduced mGlu2/3 function has been observed in models of alcohol dependence and cocaine self-administration (Baker et al., 2003; Meinhardt et al., 2013). The potential involvement of these receptors in behavioral alterations associated with pathological conditions provides compelling reasons to elucidate their input-specific functions in both normal and pathological states. Identification of discrete circuits that experience changes in neuromodulation will advance our understanding of the neurobiology of CNS disorders and provide insight into how aberrant circuit function produces pathological behaviors. However, a thorough understanding of receptor function under normal conditions is necessary to interpret effects of disease-relevant manipulations. In addition, knowledge of how presynaptic GPCRs such as mGlu2 modulate circuits that are involved in specific CNS disorders facilitates identification of therapeutic targets. Indeed, agonists and positive allosteric modulators of mGlu2 have entered clinical trials in recent years for treatment of striatum-related disorders including addiction and schizophrenia (Cross, 2013; Patil et al., 2007; Salih et al., 2015), and enhancing our understanding of how these drugs affect relevant circuits within the context of a particular disorder could facilitate predictions regarding their efficacy for particular symptom-related endpoints.
In conclusion, we have used optogenetic techniques to demonstrate that mGlu2-mediated inhibition of striatal excitatory transmission, a phenomenon which has long been attributed to activation of presynaptic receptors on cortical afferents, occurs similarly at both corticostriatal and thalamostriatal inputs. Use of this approach will likely be advantageous when considering the routine functions and therapeutic applications of specific presynaptic GPCRs, as it provides refined information about how discrete circuits are modulated. As our understanding of how individual circuits contribute to normal and disease-related behaviors increases, we will likely see a substantial improvement in our ability to predict how pharmacological manipulation of GPCRs will impact behavior and alleviate symptoms of CNS disorders. This is especially important given the fact that drugs targeted at GPCRs, including presynaptic GPCRs, are among the most widely used therapeutic agents.
The mGlu2/3 agonist LY379268 induces long-term depression (LTD) of both corticostriatal and thalamostriatal transmission in the dorsal striatum.
LTD induction is selectively blocked by a negative allosteric modulator of mGlu2.
Thalamostriatal LTD is observed at synapses on both medium spiny neurons and cholinergic interneurons.
mGlu2 activation reduces phasic dopamine release driven by thalamostriatal excitation of cholinergic interneurons.
Acknowledgments
This work was supported by the National Institute on Alcohol Abuse and Alcoholism Division of Intramural Clinical and Biological Research and a National Institute of General Medical Sciences PRAT Fellowship to K.A.J. We acknowledge Guoxiang Luo for assistance with mouse genotyping. The authors thank Drs. P. Jeffrey Conn and Craig Lindsley of the Vanderbilt Center for Neuroscience Drug Discovery (Vanderbilt University) for generously providing VU6001192 and VU0469942, and Dr. Karl Deisseroth (Stanford University) for permission to use the ChR2 construct.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–381. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- Atwood BK, Kupferschmidt DA, Lovinger DM. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat Neurosci. 2014a;17:540–548. doi: 10.1038/nn.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Lovinger DM, Mathur BN. Presynaptic long-term depression mediated by Gi/o-coupled receptors. Trends Neurosci. 2014b;37:663–673. doi: 10.1016/j.tins.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baik JH. Dopamine signaling in reward-related behaviors. Front Neural Circuits. 2013;7:152. doi: 10.3389/fncir.2013.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA, Cools R, Luciana M, Ostlund SB, Petzinger G. A kinder, gentler dopamine… highlighting dopamine’s role in behavioral flexibility. Front Neurosci. 2014;8:4. doi: 10.3389/fnins.2014.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradfield LA, Bertran-Gonzalez J, Chieng B, Balleine BW. The thalamostriatal pathway and cholinergic control of goal-directed action: interlacing new with existing learning in the striatum. Neuron. 2013;79:153–166. doi: 10.1016/j.neuron.2013.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, Lovinger DM, Cheer JF. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2012;2:33–41. doi: 10.1016/j.celrep.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AJ. AZD8529-an mGluR2 positive allosteric modulator for the treatment of schizophrenia. Neuropsychopharmacology. 2013;38 [Google Scholar]
- Crowley NA, Cody PA, Davis MI, Lovinger DM, Mateo Y. Chronic methylphenidate exposure during adolescence reduces striatal synaptic responses to ethanol. Eur J Neurosci. 2014;39:548–556. doi: 10.1111/ejn.12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Peterson JD, Surmeier DJ. Corticostriatal and thalamostriatal synapses have distinctive properties. J Neurosci. 2008;28:6483–6492. doi: 10.1523/JNEUROSCI.0435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JB, Guzman JN, Peterson JD, Goldberg JA, Surmeier DJ. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron. 2010;67:294–307. doi: 10.1016/j.neuron.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig NM, Moss J, Bolam JP. Cortical and thalamic innervation of direct and indirect pathway medium-sized spiny neurons in mouse striatum. J Neurosci. 2010;30:14610–14618. doi: 10.1523/JNEUROSCI.1623-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C, Prieto L, Valli MJ, Massey SM, Bures M, Wright RA, Johnson BG, Andis SL, Kingston A, Schoepp DD, Monn JA. Methyl substitution of 2-aminobicyclo[3.1.0]hexane 2,6-dicarboxylate (LY354740) determines functional activity at metabotropic glutamate receptors: identification of a subtype selective mGlu2 receptor agonist. J Med Chem. 2005;48:3605–3612. doi: 10.1021/jm040222y. [DOI] [PubMed] [Google Scholar]
- Ellender TJ, Huerta-Ocampo I, Deisseroth K, Capogna M, Bolam JP. Differential modulation of excitatory and inhibitory striatal synaptic transmission by histamine. J Neurosci. 2011;31:15340–15351. doi: 10.1523/JNEUROSCI.3144-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts AS, Rodriguez AL, Smith KA, Engers JL, Morrison RD, Byers FW, Blobaum AL, Locuson CW, Chang S, Venable DF, Niswender CM, Daniels JS, Conn PJ, Lindsley CW, Emmitte KA. Design of 4-Oxo-1-aryl-1,4-dihydroquinoline-3-carboxamides as Selective Negative Allosteric Modulators of Metabotropic Glutamate Receptor Subtype 2. J Med Chem. 2015;58:9027–9040. doi: 10.1021/acs.jmedchem.5b01371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Grafton ST. The striatum: where skills and habits meet. Cold Spring Harb Perspect Biol. 2015;7:a021691. doi: 10.1101/cshperspect.a021691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna L, Ceolin L, Lucas S, Monn J, Johnson B, Collingridge G, Bortolotto Z, Lodge D. Differentiating the roles of mGlu2 and mGlu3 receptors using LY541850, an mGlu2 agonist/mGlu3 antagonist. Neuropharmacology. 2013;66:114–121. doi: 10.1016/j.neuropharm.2012.02.023. [DOI] [PubMed] [Google Scholar]
- Hintiryan H, Foster NN, Bowman I, Bay M, Song MY, Gou L, Yamashita S, Bienkowski MS, Zingg B, Zhu M, Yang XW, Shih JC, Toga AW, Dong HW. The mouse cortico-striatal projectome. Nat Neurosci. 2016;19:1100–1114. doi: 10.1038/nn.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Duffy P, Swanson C, Ghasemzadeh MB, Kalivas PW. The regulation of dopamine transmission by metabotropic glutamate receptors. J Pharmacol Exp Ther. 1999;289:412–416. [PubMed] [Google Scholar]
- Huerta-Ocampo I, Mena-Segovia J, Bolam JP. Convergence of cortical and thalamic input to direct and indirect pathway medium spiny neurons in the striatum. Brain Struct Funct. 2014;219:1787–1800. doi: 10.1007/s00429-013-0601-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Conn PJ. Metabotropic Glutamate Receptor-Dopamine Interactions in the Basal Ganglia Motor Circuit. In: Jones S, editor. Dopamine-Glutamate Interactions in the Basal Ganglia. CRC Press; Boca Raton, FL: 2012. pp. 1–30. [Google Scholar]
- Kahn L, Alonso G, Robbe D, Bockaert J, Manzoni OJ. Group 2 metabotropic glutamate receptors induced long term depression in mouse striatal slices. Neurosci Lett. 2001;316:178–182. doi: 10.1016/s0304-3940(01)02397-7. [DOI] [PubMed] [Google Scholar]
- Kosillo P, Zhang YF, Threlfell S, Cragg SJ. Cortical Control of Striatal Dopamine Transmission via Striatal Cholinergic Interneurons. Cereb Cortex. 2016 doi: 10.1093/cercor/bhw252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt DA, Lovinger DM. Inhibition of presynaptic calcium transients in cortical inputs to the dorsolateral striatum by metabotropic GABA(B) and mGlu2/3 receptors. J Physiol. 2015;593:2295–2310. doi: 10.1113/JP270045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge D, Tidball P, Mercier MS, Lucas SJ, Hanna L, Ceolin L, Kritikos M, Fitzjohn SM, Sherwood JL, Bannister N, Volianskis A, Jane DE, Bortolotto ZA, Collingridge GL. Antagonists reversibly reverse chemical LTD induced by group I, group II and group III metabotropic glutamate receptors. Neuropharmacology. 2013;74:135–146. doi: 10.1016/j.neuropharm.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, McCool BA. Metabotropic glutamate receptor-mediated presynaptic depression at corticostriatal synapses involves mGLuR2 or 3. J Neurophysiol. 1995;73:1076–1083. doi: 10.1152/jn.1995.73.3.1076. [DOI] [PubMed] [Google Scholar]
- Lucas SJ, Bortolotto ZA, Collingridge GL, Lodge D. Selective activation of either mGlu2 or mGlu3 receptors can induce LTD in the amygdala. Neuropharmacology. 2013;66:196–201. doi: 10.1016/j.neuropharm.2012.04.006. [DOI] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, Hsu YW, Garcia AJ, 3rd, Gu X, Zanella S, Kidney J, Gu H, Mao Y, Hooks BM, Boyden ES, Buzsaki G, Ramirez JM, Jones AR, Svoboda K, Han X, Turner EE, Zeng H. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci. 2012;15:793–802. doi: 10.1038/nn.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella G, Platania P, Vita D, Sciamanna G, Cuomo D, Tassone A, Tscherter A, Kitada T, Bonsi P, Shen J, Pisani A. Enhanced sensitivity to group II mGlu receptor activation at corticostriatal synapses in mice lacking the familial parkinsonism-linked genes PINK1 or Parkin. Exp Neurol. 2009;215:388–396. doi: 10.1016/j.expneurol.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Paganini F, Stocchi S, Bianchi C, Beani L, Morari M. Presynaptic group I and II metabotropic glutamate receptors oppositely modulate striatal acetylcholine release. Eur J Neurosci. 2001;14:1181–1184. doi: 10.1046/j.0953-816x.2001.01750.x. [DOI] [PubMed] [Google Scholar]
- Mathur BN, Capik NA, Alvarez VA, Lovinger DM. Serotonin induces long-term depression at corticostriatal synapses. J Neurosci. 2011;31:7402–7411. doi: 10.1523/JNEUROSCI.6250-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhardt MW, Hansson AC, Perreau-Lenz S, Bauder-Wenz C, Stahlin O, Heilig M, Harper C, Drescher KU, Spanagel R, Sommer WH. Rescue of infralimbic mGluR2 deficit restores control over drug-seeking behavior in alcohol dependence. J Neurosci. 2013;33:2794–2806. doi: 10.1523/JNEUROSCI.4062-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the messenger RNA for a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat. Neuroscience. 1993;53:1009–1018. doi: 10.1016/0306-4522(93)90485-x. [DOI] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- Pehrson AL, Moghaddam B. Impact of metabotropic glutamate 2/3 receptor stimulation on activated dopamine release and locomotion. Psychopharmacology (Berl) 2010;211:443–455. doi: 10.1007/s00213-010-1914-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picconi B, Pisani A, Centonze D, Battaglia G, Storto M, Nicoletti F, Bernardi G, Calabresi P. Striatal metabotropic glutamate receptor function following experimental parkinsonism and chronic levodopa treatment. Brain. 2002;125:2635–2645. doi: 10.1093/brain/awf269. [DOI] [PubMed] [Google Scholar]
- Salih H, Anghelescu I, Kezic I, Sinha V, Hoeben E, Van Nueten L, De Smedt H, De Boer P. Pharmacokinetic and pharmacodynamic characterisation of JNJ-40411813, a positive allosteric modulator of mGluR2, in two randomised, double-blind phase-I studies. J Psychopharmacol. 2015;29:414–425. doi: 10.1177/0269881115573403. [DOI] [PubMed] [Google Scholar]
- Sanger H, Hanna L, Colvin EM, Grubisha O, Ursu D, Heinz BA, Findlay JD, Vivier RG, Sher E, Lodge D, Monn JA, Broad LM. Pharmacological profiling of native group II metabotropic glutamate receptors in primary cortical neuronal cultures using a FLIPR. Neuropharmacology. 2013;66:264–273. doi: 10.1016/j.neuropharm.2012.05.023. [DOI] [PubMed] [Google Scholar]
- Smeal RM, Keefe KA, Wilcox KS. Differences in excitatory transmission between thalamic and cortical afferents to single spiny efferent neurons of rat dorsal striatum. Eur J Neurosci. 2008;28:2041–2052. doi: 10.1111/j.1460-9568.2008.06505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Y, Galvan A, Ellender TJ, Doig N, Villalba RM, Huerta-Ocampo I, Wichmann T, Bolam JP. The thalamostriatal system in normal and diseased states. Front Syst Neurosci. 2014;8:5. doi: 10.3389/fnsys.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Y, Surmeier DJ, Redgrave P, Kimura M. Thalamic contributions to Basal Ganglia-related behavioral switching and reinforcement. J Neurosci. 2011;31:16102–16106. doi: 10.1523/JNEUROSCI.4634-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung KW, Choi S, Lovinger DM. Activation of group I mGluRs is necessary for induction of long-term depression at striatal synapses. J Neurophysiol. 2001;86:2405–2412. doi: 10.1152/jn.2001.86.5.2405. [DOI] [PubMed] [Google Scholar]
- Testa CM, Standaert DG, Young AB, Penney JB., Jr Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci. 1994;14:3005–3018. doi: 10.1523/JNEUROSCI.14-05-03005.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S, Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71:142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AG, Wenthur CJ, Xiang Z, Rook JM, Emmitte KA, Niswender CM, Lindsley CW, Conn PJ. Metabotropic glutamate receptor 3 activation is required for long-term depression in medial prefrontal cortex and fear extinction. Proc Natl Acad Sci U S A. 2015;112:1196–1201. doi: 10.1073/pnas.1416196112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthur CJ, Morrison R, Felts AS, Smith KA, Engers JL, Byers FW, Daniels JS, Emmitte KA, Conn PJ, Lindsley CW. Discovery of (R)-(2-fluoro-4-((-4-methoxyphenyl)ethynyl)phenyl) (3-hydroxypiperidin-1-yl)methanone (ML337), an mGlu3 selective and CNS penetrant negative allosteric modulator (NAM) J Med Chem. 2013;56:5208–5212. doi: 10.1021/jm400439t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright RA, Johnson BG, Zhang C, Salhoff C, Kingston AE, Calligaro DO, Monn JA, Schoepp DD, Marek GJ. CNS distribution of metabotropic glutamate 2 and 3 receptors: transgenic mice and [(3)H]LY459477 autoradiography. Neuropharmacology. 2013;66:89–98. doi: 10.1016/j.neuropharm.2012.01.019. [DOI] [PubMed] [Google Scholar]
- Wu YW, Kim JI, Tawfik VL, Lalchandani RR, Scherrer G, Ding JB. Input- and cell-type-specific endocannabinoid-dependent LTD in the striatum. Cell Rep. 2015;10:75–87. doi: 10.1016/j.celrep.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason JT, Espana RA, Jones SR. Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J Neurosci Methods. 2011;202:158–164. doi: 10.1016/j.jneumeth.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SR, Chuang SC, Zhao W, Wong RK, Bianchi R. Persistent receptor activity underlies group I mGluR-mediated cellular plasticity in CA3 neuron. J Neurosci. 2013;33:2526–2540. doi: 10.1523/JNEUROSCI.3338-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Karlsson C, Liang T, Xiong W, Kimura M, Tapocik JD, Yuan Q, Barbier E, Feng A, Flanigan M, Augier E, Enoch MA, Hodgkinson CA, Shen PH, Lovinger DM, Edenberg HJ, Heilig M, Goldman D. Loss of metabotropic glutamate receptor 2 escalates alcohol consumption. Proc Natl Acad Sci U S A. 2013;110:16963–16968. doi: 10.1073/pnas.1309839110. [DOI] [PMC free article] [PubMed] [Google Scholar]