

Abstract
Apratoxin E provided the inspiration for the design of apratoxin A/E hybrids under preclinical development. Through total synthesis using two different strategies, it was determined that the originally proposed configuration of the thiazoline at C30 is opposite from that in apratoxin A, in contrast to previous assumptions on biosynthetic grounds. The epimer and true natural apratoxin E were synthesized, and the biological activities were evaluated.
Graphical Abstract
Apratoxins are potent cytotoxins derived from marine cyanobacteria.1 Their cytotoxicity is due to potent inhibition of cotranslational translocation, leading to downregulation of various receptor tyrosine kinases and reduced growth factor secretion.1g Apratoxin A (1, Figure 1) possesses broad-spectrum differential in vitro activity;2 however, it also showed irreversible toxicity in vivo and was not well tolerated.3a We suspected earlier that the Michael acceptor in the modified cysteine (moCys) unit could be a liability in vivo. Apratoxin E (2, 6, Figure 1) has been the only natural apratoxin to date without the Michael acceptor in this position; however, the C34–C35 double bond in this compound reduced the antiproliferative activity against different colon cancer cells by 15- to 31-fold.1c,3a In order to overcome the drawbacks of apratoxin A and develop lead compounds with better activity, we previously designed and synthesized apratoxin S4, a hybrid of apratoxins A and E, containing the hydroxylated polyketide unit (from A) and saturated moCys unit (from E), as well as a series of analogues, including published apratoxins S5–S9.3 Apratoxin S4 (3, Figure 1) showed greater potency and efficacy in a colorectal tumor xenograft model than the parent molecule (apratoxin A), and was much better tolerated in vivo.3a Apratoxin S9 (5, Figure 1), the C30 epimer of apratoxin S4, exhibited even greater in vitro potency with subnanomolar IC50 values. The C30 configuration is central to the present report.
Figure 1.

The structures of apratoxins A and E and related synthetic hybrid compounds.
In the synthesis of apratoxins S4–S7 and S9, we noticed that the hydroxy group at C35 tended to dehydrate to form a conjugated system with the thiazoline.3 Specifically, this tendency is higher for apratoxin S7 (4, Figure 1) than other analogues. Initially, we expected that the dehydration product in the synthesis of apratoxin S7 should be apratoxin E (Supporting Information, Figure S1).1c Contrary to our expectation, the NMR spectra of the dehydration product did not match with those of the natural product apratoxin E (SI, Figure S2). In order to resolve the discrepancy, we aimed to synthesize apratoxin E using a novel strategy (Scheme 1). We found that the product obtained via the new total synthesis was the same as the dehydration product from the initial synthesis of apratoxin S7 and also did not match the published apratoxin E (SI, Figure S12–S14).
Scheme 1.

Attempt to the synthesis of apratoxin E with preassembly carbon-carbon double bond
We suspected that apratoxin E must have 30R configuration for the thiazoline because C30 was the only stereocenter that was not experimentally proven but only assumed to be 30S, as the same with other natural apratoxins from the same location.1c We embarked on the synthesis of naturally occurring apratoxin E. Since apratoxin A/E hybrids are now in preclinical development, it was imperative to unambiguously determine the absolute configuration. The final results confirmed our speculation. In this paper, we report the corrected structure of apratoxin E, in which a new synthetic strategy was utilized, which could be used for general route for the synthesis of other apratoxins.
As shown in Scheme 1, treatment of the commercially available and inexpensive (−)-citronellal with tert-butyl lithium gave the alcohol 7 in 95% yield. Oxidation of the alcohol and simultaneous bromination of the olefin with 1,3-dibromo-5,5-dimethylhydantoin (DDH)4 afforded ketone 8 in good yield. Reduction of this ketone by using (R)-CBS5 in the presence of BH3-Me2S followed by treatment with KOH gave alcohol 10 in high yield. The configuration of the newly generated stereocenter and the diastereoselectivity (dr = 11:1) in the reduction step were determined by conversion of 10 to the corresponding (R)- and (S)-Mosher’s esters6 (see SI). Installation of proline residue, ozonolysis, and then exposure of the resulting aldehyde to allyl 2-(diethoxyphosphoryl)acetate under basic conditions furnished ester 12. Cleavage of the allyl ester, followed by coupling with the free amine of modified cysteine 133 gave 14 in a yield of 56% over three steps. Next, the thiazoline unit was assembled using a tandem deprotection/cyclodehydration strategy developed by Kelly.7 Removal of the allyl ester protection of 15 and coupling with the free amine from 16 gave the linear peptide 17 in 73% yield. Cleavage of allyl ester by Pd(PPh3)4/N-methylaniline (NMA) and the removal of Fmoc by Et2NH/CH3CN from 17 smoothly gave unmasked cyclization precursor. However, only trace amount of the expected product 2, (30S)-apratoxin E, was obtained under various coupling conditions. It’s possible that the preformed double bond prevented the formation of macrocyclization. Similarly, in our previous work with the dehydrated intermediate 15, only 5% cyclized final product was obtained (SI, Figure S1, Route A).
The polyketide domain of 2 was simpler than that of 1 (and many of its analogues), having a trans carbon-carbon double bond instead of a flexible saturated chain. We aimed to introduce a hydroxy as a latent group for the olefin at this site. As we disclosed previously, the hydroxy at C35 of apratoxin A was prone to elimination to E-dehydroapratoxin A even when exposed to weak acidic CDCl3.1b Additionally, we also observed similar dehydration in the synthesis of apratoxin A/E hybrids.3 On the other hand, when the hydroxy group was masked with an electron-withdrawing group (2,2,2-trichloroethoxycarbonyl, Troc), the elimination was accelerated and occurred as soon as the thiazoline ring formed, according to Doi’s research.8 Taking inspiration from these known side-reactions, we speculated that masking this free hydroxy with an electron-donating group might avoid the dehydration in the preparation of the intermediates. On the other hand, the protecting group should also be easily removed to release the free hydroxy, which could be converted to the desired trans carbon-carbon double bond after the macrocyclization. Forsyth and Chen9 converted the linear peptide bearing the triethylsilyl (TES) ether at the C35 position to the apratoxin A scaffold and then safely removed the TES protecting group in the last step.
With these ideas in mind, we then advanced intermediate 11 to alcohol 18 via Reformatsky reaction10 (Scheme 2). Protecting group manipulations followed by installation of the modified cysteine residue via HATU-mediated condensation gave 19 in 72% yield over three steps. The thiazoline unit was assembled and the TES protecting group was exchanged for Troc immediately to avoid undesired elimination at this stage. Removal of the allyl group of 22 followed by coupled with the free amine from 16 with the assistance of 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT)11 furnished 23 in 66% yield.
Scheme 2.

The successful synthesis of apratoxin E and its epimer with late-stage formation of the carbon-carbon double bond
With the advanced intermediate 23 in hand, we embarked on the final steps of the total synthesis of apratoxin E. Releasing of the carboxylic acid from allyl ester and removal of the Fmoc moiety on N-terminus followed by DEPBT-mediated macrocyclization furnished the apratoxin scaffold. Notably, part of the TES was destroyed during the coupling steps. Treatment of the mixture of 24/25 with HF-pyridine removed the TES group thoroughly and generated a small amount of dehydrated product (2) (monitored by LC-MS). Finally, exposure of the mixture of 25 and 2 to Martin’s reagent12 afforded the originally proposed apratoxin E (30S), with the carbon-carbon double bond generated at the expected position and desired trans configuration. However, the NMR data of 2 did not match that of the natural product, but were identical to the data of dehydrated product of apratoxin S7 which we previously prepared (SI, Figure S2 and S12).
After the successful synthesis of (30S)-apratoxin E (2), we then turned to the preparation of its C30 epimer (6) by following the same procedure but using ent-13 instead of 13 (Scheme 2).
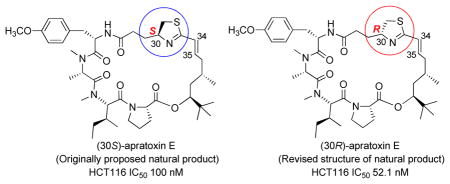
With these two isomers in hand, we were able to compare their spectroscopic data with those of natural apratoxin E and unambiguously confirmed that (30R)-apratoxin E (6) is the natural product (and thus has the same configuration as apratoxin S9) and that (30S)-apratoxin E is actually 30-epi-apratoxin E (SI, Figure S3–S14). NMR data was also obtained in benzene-d6 in order to confirm E configuration of the double bond (SI, Figure S8 and S14).1c
Both isomers were tested for their antiproliferative activities in HCT116 cells. In consistent with the reduced potency of natural apratoxin E compared with apratoxin A in colon cancer cells,1c,3a (30R)-apratoxin E (6) exhibits about 9-fold less activity (IC50 52.1 nM, SI, Figure S15) than apratoxin A (IC50 5.97 nM)3a in HCT116 cells. While (30S)-apratoxin E (2) showed 17-fold less activity with IC50 value of 100 nM (SI, Figure S15), indicating the positive impact of 30R on activity. Importantly, this trend is in concert with the SAR study of apratoxin S4 (3) and S9 (5).3b
In summary, we accomplished the first total synthesis of apratoxin E and its C30 epimer, unambiguously assigning and correcting the originally proposed absolute configuration. This result indicates that apratoxin S9 is the truest apratoxin A/E hybrid with identical configurations of the stereocenters “taken” from the respective portions of the natural products.3b The formal epimerization of C30 for apratoxin A versus apratoxin E also fuels the interest in the biosynthesis of these compounds.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the National Natural Science Foundation of China (81573340), the “Zhuo Xue” Talent Plan of Fudan University and National Institutes of Health grant R01CA172310.
Footnotes
H.L. is co-founder of Oceanyx Pharmaceuticals, Inc., which has licensed patents and patent applications related to the subject matter.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, analytical data, and 1H and 13C NMR spectra for all compounds (PDF).
References
- 1.(a) Luesch H, Yoshida WY, Moore RE, Paul VJ, Corbett TH. J Am Chem Soc. 2001;123:5418–5423. doi: 10.1021/ja010453j. [DOI] [PubMed] [Google Scholar]; (b) Luesch H, Yoshida WY, Moore RE, Paul VJ. Bioorg Med Chem. 2002;10:1973–1978. doi: 10.1016/s0968-0896(02)00014-7. [DOI] [PubMed] [Google Scholar]; (c) Matthew S, Schupp PJ, Luesch H. J Nat Prod. 2008;71:1113–1116. doi: 10.1021/np700717s. [DOI] [PubMed] [Google Scholar]; (d) Gutiérrez M, Suyama TL, Engene N, Wingerd JS, Matainaho T, Gerwick WH. J Nat Prod. 2008;71:1099–1103. doi: 10.1021/np800121a. [DOI] [PubMed] [Google Scholar]; (e) Tidgewell K, Engene N, Byrum T, Media J, Doi T, Valeriote FA, Gerwick WH. Chem Bio Chem. 2010;11:1458–1466. doi: 10.1002/cbic.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Thornburg CC, Cowley ES, Sikorska J, Shaala LA, Ishmael JE, Youssef DTA, McPhail KL. J Nat Prod. 2013;76:1781–1788. doi: 10.1021/np4004992. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Liu Y, Law BK, Luesch H. Mol Pharmacol. 2009;76:91–104. doi: 10.1124/mol.109.056085. [DOI] [PubMed] [Google Scholar]
- 2.Luesch H, Chanda SK, Raya RM, De Jesus PD, Orth AP, Walker JR, Belmonte JCI, Schultz PG. Nat Chem Biol. 2006;2:158–167. doi: 10.1038/nchembio769. [DOI] [PubMed] [Google Scholar]
- 3.(a) Chen Q, Liu Y, Luesch H. ACS Med Chem Lett. 2011;2:861–865. doi: 10.1021/ml200176m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen Q, Liu Y, Cai W, Luesch H. J Med Chem. 2014;57:3011–3029. doi: 10.1021/jm4019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Taneja SC, Sethi VK, Andotra SS, Koul S, Qazi GN. Synth Commun. 2005;35:2297–2303. [Google Scholar]; (b) Khazaei A, Abbasi F, Kianiborazjani M, Saednia S. J Braz Chem Soc. 2014;25:361–364. [Google Scholar]
- 5.(a) Corey EJ, Bakshi RK, Shibata S. J Am Chem Soc. 1987;109:5551–5553. [Google Scholar]; (b) Corey EJ, Bakshi RK. Tetrahedron Lett. 1990;31:611–614. [Google Scholar]; (c) Corey EJ, Helal CJ. Angew Chem, Int Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 6.(a) Sullivan GR, Dale JA, Mosher HS. J Org Chem. 1973;38:2143–2147. [Google Scholar]; (b) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 7.You S, Razavi H, Kelly JW. Angew Chem, Int Ed. 2003;42:83–85. doi: 10.1002/anie.200390059. [DOI] [PubMed] [Google Scholar]
- 8.Doi T. Chem Pharm Bull. 2014;62:735–743. doi: 10.1248/cpb.c14-00268. [DOI] [PubMed] [Google Scholar]
- 9.(a) Chen J, Forsyth CJ. J Am Chem Soc. 2003;125:8734–8735. doi: 10.1021/ja036050w. [DOI] [PubMed] [Google Scholar]; (b) Chen J, Forsyth CJ. Proc Natl Acad Sci U S A. 2004;101:12067–12072. doi: 10.1073/pnas.0402752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Reformatsky S. Ber. 1887;20:1210–1211. [Google Scholar]; (b) Fuerstner A. Synthesis. 1989:571–590. [Google Scholar]; (c) Ocampo R, Dolbier WR., Jr Tetrahedron. 2004;60:9325–9374. [Google Scholar]
- 11.(a) Li H, Jiang H, Ye YH, Fan C, Romoff T, Goodman M. Org Lett. 1999;1:91–93. doi: 10.1021/ol990573k. [DOI] [PubMed] [Google Scholar]; (b) Ye YH, Li H, Jiang X. Biopolymers. 2005;80:172–178. doi: 10.1002/bip.20201. [DOI] [PubMed] [Google Scholar]
- 12.Martin JC, Arhart RJ. J Am Chem Soc. 1971;93:4327–4329. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.