

Abstract
Glycogen synthase kinase (GSK)-3β has been known as a pro-inflammatory molecule in neuroinflammation. The involvement of GSK-3β remains unsolved in acute monophasic rat experimental autoimmune encephalomyelitis (EAE). The aim of this study was to evaluate a potential role of GSK-3β in central nervous system (CNS) autoimmunity through its inhibition by lithium. Lithium treatment significantly delayed the onset of EAE paralysis and ameliorated its severity. Lithium treatment reduced the serum level of pro-inflammatory tumor necrosis factor a but not that of interleukin 10. Western blot analysis showed that the phosphorylation of GSK-3β (p-GSK-3β) and its upstream factor Akt was significantly increased in the lithium-treated group. Immunohistochemical examination revealed that lithium treatment also suppressed the activation of ionized calcium binding protein-1-positive microglial cells and vascular cell adhesion molecule-1 expression in the spinal cords of lithium-treated EAE rats. These results demonstrate that lithium ameliorates clinical symptom of acute monophasic rat EAE, and GSK-3 is a target for the suppression of acute neuroinflammation as far as rat model of human CNS disease is involved.
Keywords: Experimental autoimmune encephalomyelitis, GSK-3 signaling, Lithium, Rat model
Introduction
Experimental autoimmune encephalomyelitis (EAE) is a T-cell-mediated acute monophasic autoimmune central nervous system (CNS) disease that is used as a model of human multiple sclerosis [1]. EAE is characterized by infiltration of autoimmune T cells and reactive gliosis [1,2]. There is general agreement that encephalitogenic T cells play a key role in the induction of EAE [3,4] and that regulatory T cells and alternatively activated M2 macrophages are involved in the remission of paralysis [1,5]. Several transcription factors, including nuclear factor κB (NF-κB) [6], cyclic AMP response element-binding protein [7], peroxisome proliferator-activated receptor [8], and other signaling and transcription molecules [9], are involved in the progression of EAE via the activation of inflammatory cells and glial cells or in EAE remission via counteracting the effects of pro-inflammatory mediators.
Glycogen synthase kinase (GSK)-3 is an important proinflammatory molecule in CNS autoimmune diseases [10]. GSK-3 is involved in the differentiation of Th17 cells, which is important for the induction of EAE in mice [11,12,13] and also to play an important role in neurodegeneration [10]. Thus, the blockage of GSK-3 signaling may be a therapeutic strategy for neuroprotection in CNS disease [14]. Furthermore, GSK-3 signaling is involved in the activation of adhesion molecules, including vascular cell adhesion molecule 1 (VCAM-1), in cultured endothelial cells [15,16], which are important for T-cell migration in EAE [2,17].
An inhibitor of GSK-3, lithium, has been used as a mood stabilizer to treat human bipolar disorders, possibly through neuroprotection [14,18]. Furthermore, lithium has been used to ameliorate myelin oligodendrocyte glycoprotein (MOG)-immunized chronic EAE in a mouse model [13]. Regarding acute monophasic EAE, lithium-induced amelioration of EAE was regarded as a toxic effect in rat EAE [19]; thus, the mechanism of action was unclear. Moreover, the tissue localization of GSK-3 and the quantitative changes in its upstream and downstream signaling cascades in rat spinal cords with acute monophasic EAE are not fully understood.
In the present study, we first examined the immunohistochemical localization of phosphorylated GSK-3β (Ser9), the inactive form of GSK-3β, in the EAE-affected spinal cords of Lewis rats. We also investigated the role of lithium, as an inhibitor of GSK-3β, in the amelioration of rat acute, monophasic EAE by investigating the associated signaling cascades.
Materials and Methods
Animals
Lewis rats were obtained from OrientBio Inc. (Seongnam, Korea) and bred in our animal facility. Rats of both sexes (7–8 weeks old, 160–200 g) were used. All animal experiments were conducted in accordance with the Jeju National University Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committees of Jeju National University. The protocols for the care and handling of animals conformed to current international laws and policies (NIH Guide for the Care and Use of Laboratory Animals, NIH Publication No. 85-23, 1985, revised 1996). Every effort was made to minimize the number of animals and their suffering.
Induction of EAE and behavioral test
EAE induction and all experiments were performed as described in our previous papers [5,20,21]. Briefly, the footpads of both hind feet of rats in the EAE group were injected with 100 µl of an emulsion containing an equal volume of guinea pig myelin basic protein (1 mg/ml) and complete Freund's adjuvant (CFA) supplemented with 3 mg/ml Mycobacterium tuberculosis H37Ra (Difco, Detroit, MI, USA).
Control rats were immunized with CFA only. After immunization, the rats were observed daily for clinical signs of EAE. The progression of EAE was divided into the following eight clinical stages: grade 0 (G.0), no signs; G.0.5, mild floppy tail; G.1, complete floppy tail; G.2, mild paraparesis; G.3, severe paraparesis; G.4, tetraparesis; G.5, moribund condition or death; and R.0, recovery.
Tissue sampling
At each sampling time point, including the paralytic peak stage (G.3, days 12–14 postimmunization [PI]) and the EAE paralysis recovery stage (R.0, day 21 PI) (n=5 per time point) of rat EAE, five rats per group were euthanized under deep ether anesthesia. To assay serum cytokine levels, blood samples were collected from the heart of EAE rats with and without lithium treatment. After clotting, serum was isolated after centrifugation, followed by freezing until use for cytokine assays.
Spinal cords were sampled at the peak and recovery stages of EAE paralysis. Normal and CFA-immunized rats were used as controls (n=5 per group). Pieces of the cervical, thoracic, and lumbar spinal cords ~0.5 cm in length were collected.
For histological analysis, spinal cords were embedded in paraffin wax after fixation in 4% paraformaldehyde in phosphate-buffered saline (PBS, pH 7.4). Paraffin wax-embedded tissues were cut at a thickness of 5 µm using a rotary microtome (Leica, Nussloch, Germany). Tissue sections were routinely stained with hematoxylin and eosin to evaluate inflammation.
For western blot analysis, spinal cords were removed and frozen at −80℃ until use.
Antibodies
For the immunohistochemical analysis of rat spinal cords, we used mouse monoclonal anti–glial fibrillary acidic protein (Sigma-Aldrich, St. Louis, MO, USA) for astrocytes and rabbit anti–ionized calcium-binding protein-1 (Iba-1) (Wako Pure Chemical Industries, Ltd., Osaka, Japan) for ramified microglia and macrophages.
To detect GSK-3β, monoclonal rabbit anti-phospho-GSK-3β (Ser9) (p-GSK-3β) and monoclonal rabbit anti-GSK-3β antibodies were used (Cell Signaling Technology, Beverly, MA, USA). Monoclonal rabbit anti-β-catenin (Ser675) and PhosphoPlus Akt (Ser473) antibody kits were purchased from Cell Signaling Technology. Rabbit polyclonal anti–vascular cell adhesion molecule-1 (VCAM-1) antibody (H-276, Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used to assess the severity of inflammation. A mouse monoclonal anti-β-actin (Sigma-Aldrich) antibody was also used.
An inhibitor of GSK-3β, lithium, treatment
To assess the effect of lithium in rat EAE, rats were divided into the following three groups (10 animals per each group): normal control group, vehicle-treated group, and lithium-treated group. To rapidly increase the lithium level, lithium chloride (100 mg/kg/day, Sigma) was intraperitoneally administered to the lithium-treated group three times beginning 1 day prior to immunization. Lithium carbonate (40 mg/kg/day, Sigma-Aldrich) was then orally administered from day 3 PI to day 14 PI. This lithium administration in rats is nontoxic and is commonly used to achieve serum levels equivalent to those in human patients [22]. The serum lithium concentration in the rats was measured using a lithium assay kit LS (Catalog number: LI01ME, MG Metallogenics, Chiba, Japan). Throughout the experiment, the body weight and behavioral changes of all rats were checked daily.
Cytokine assays
Serum levels of cytokines, such as tumor necrosis factor α (TNF-α) and interleukin-10 (IL-10), were determined using commercially available immunoassay kits in accordance with the manufacturer's instructions (Biosource, Camarillo, CA, USA). The absorbance at 450 nm was read using a Thermomax microplate reader (Molecular Devices, Sunnyvale, CA, USA). Cytokine levels were calculated with standard curves using recombinant rat cytokines.
Western blotting
Western blotting was performed as described previously [23]. Briefly, spinal cord tissue was homogenized in a modified radioimmunoprecipitation assay buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1% Triton-X 100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1% NP-40, 10 mM NaF, 1 mM ethylenediaminetetraacetic acid, 1 mM ethylene glycol tetraacetic acid, 1 mM Na3VO4, 1 mM phenylmethanesulfonylfluoride, 10 µg/ml aprotinin, and 10 µg/ml leupeptin) by means of 20 strokes of a homogenizer.
The homogenate was centrifuged at 10,900 ×g for 20 minutes, and the supernatant was harvested. For western blotting, supernatants containing 40 µg protein were loaded into individual lanes of 10% sodium dodecyl (or lauryl) sulfate-polyacrylamide gels, electrophoresed, and transferred to nitrocellulose membranes (Schleicher and Schuell, Keene, NH, USA). Any residual binding sites on the membranes were blocked by incubation with 5% skim milk in Tris-buffered saline (TBS; 10 mM Tris-HCl [pH 7.4], and 150 mM NaCl) for 1 hour. The membranes were then incubated with primary antibodies against phospho-Akt (p-Akt) (1:1,000), anti-Akt (1:1,000), anti-p-GSK-3β (1:1,000), anti-GSK-3β (1:1,000), and anti-β-catenin (1:1,000) for 2 hours. The membranes were washed three times in TBS containing 0.1% Tween 20 and incubated with the matching secondary antibodies, horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (Vector Laboratories, Burlingame, CA, USA), for 1 hour. Bound antibodies were detected using a chemiluminescent substrate (WEST-one Kit, iNtRON Biotech, Seongnam, Korea) according to the manufacturer's instructions. After imaging, the membranes were stripped and reprobed using an anti-β-actin antibody (1:10,000). The optical density (OD; per mm2) of each band was measured using a scanning laser densitometer (GS-700, Bio-Rad, Hercules, CA, USA), and the ratio of the density of each band relative to that of β-actin was compared using ImageJ software (NIH, Bethesda, MD, USA).
Immunohistochemistry
Paraffin-embedded tissues were cut at a thickness of 5 µm using a rotary microtome (Leica). Sections were deparaffinized using routine protocols, exposed to citrate buffer (0.01 M, pH 6.0), and heated in a microwave for 3 minutes. All subsequent steps were performed at room temperature. Sections were treated with 0.3% hydrogen peroxide in distilled water for 20 minutes to block endogenous peroxidase activity. After three washes in PBS, the sections were blocked with 10% normal goat serum (Vector ABC Elite Kit, Vector Laboratories) for 1 hour and then allowed to react with the following primary antibodies for 1–2 hours: rabbit anti-Iba-1 (1:1,000), rabbit anti-p-GSK-3β (1:200), and rabbit anti-VCAM-1 (1:400). After three washes in PBS, sections were incubated with biotinylated goat anti-mouse IgG (Vector ABC Elite Kit) for 45 minutes and then incubated with avidin-biotin peroxidase complex (Vector ABC Elite Kit) for 45 minutes according to the manufacturer's instructions. The peroxidase reaction was developed using a diaminobenzidine substrate (DAB kit, SK-401, Vector Laboratories). As a control, primary antibodies were omitted from some test sections in each experiment. After color development, the sections were counterstained with Harris' hematoxylin for 5 seconds, washed under running tap water for 20 minutes, dehydrated using a graded ethanol series, cleared with xylene, and mounted using Canada balsam (Junsei Chemical Co., Tokyo, Japan). The sections were observed under an Olympus microscope (BX-51, Olympus, Tokyo, Japan).
Semi-quantitative analysis of immunohistochemistry
Iba-1 and VCAM-1 immunoreactivity in spinal cords were quantified using ImageJ software. Three different regions of the spinal cords (cervical, thoracic, and lumbar) of each of three animals per group were photographed at 4× magnification. We measured the area of immunopositivity per total spinal area [(positive area/total area)×100 (%)]. The results are shown as means±standard error of the mean (SEM).
Statistical analysis
All measurements were from three independent experiments, and all values are presented as means±SEM. The results were analyzed using one-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls post hoc test for multiple comparisons. In all cases, P<0.05 was considered to indicate statistical significance.
Results
GSK-3β phosphorylation was inversely related to EAE paralysis
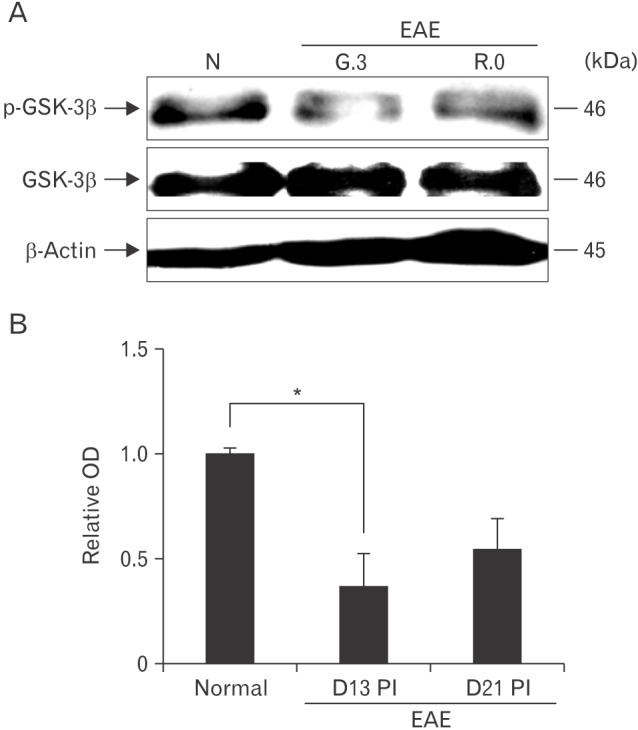
Total GSK-3β was constitutively expressed in the spinal cords of normal rats and EAE-immunized rats (Fig. 1, lower image). p-GSK-3β expression in the spinal cord throughout the course of EAE was semi-quantitatively evaluated by western blotting (Fig. 1, upper image). p-GSK-3β expression was detected in the spinal cords of normal control rats (relative OD value, 1.0±0.02), whereas p-GSK-3β expression was transiently reduced on day 13 PI (1.37±0.15-fold increase, n=5; P<0.05 vs. control groups) and recovered on day 21 PI (0.54±0.14-fold increase, n=5; P<0.05 vs. day 13 PI groups). These findings suggest that p-GSK-3β was constitutively expressed in normal tissues, temporarily decreased in spinal cords at the peak stage of rat EAE, and restored after recovery from EAE paralysis.
Fig. 1. Western blot analysis of phosphorylated glycogen synthase kinase-3β (p-GSK-3β) and total GSK-3β expression in the spinal cords of rats with experimental autoimmune encephalomyelitis (EAE). (A) Representative Western blot of p-GSK-3β, total GSK-3β, and β-actin expression. (B) Semi-quantitative analysis of p-GSK-3β expression in the spinal cord. p-GSK-3β expression was significantly decreased on day 13 postimmunization (PI) compared with normal controls. Values are presented as means±SE. *P<0.05 vs. controls.
Immunohistochemical localization of p-GSK-3β in rat spinal cords with EAE
In normal spinal cords (Fig. 2A), p-GSK-3β was strongly expressed in the astrocytes of white matter (arrows), neurons, and vascular endothelial cells (data not shown). p-GSK-3β expression in CFA-immunized spinal cords did not differ from that in normal spinal cords (data not shown). On day 13 PI, p-GSK-3β immunoreactivity was gradually decreased in the astrocytes (Fig. 2B, arrows) of the white matter; in contrast, it was expressed in some inflammatory cell types, including macrophages (Fig. 2B, arrowheads), in the parenchyma. In addition, p-GSK-3β was also detected in neurons (Fig. 2C, arrows), vascular endothelial cells, and ependymal cells (Fig. 2D). On day 21 PI, p-GSK-3β immunoreactivity remained, albeit weakly, in the astrocytes (Fig. 2E, arrows) and vascular endothelial cells. A small number of inflammatory cells showed p-GSK-3β immunoreactivity on day 21 PI (Fig. 2E, arrowhead) compared with on day 13 PI. Primary antibody was omitted (Fig. 2F). Table 1 summarizes the results of the p-GSK-3β immunohistochemical analysis in normal, CFA-immunized control, and EAE-affected rat spinal cords.
Fig. 2. Immunohistochemical staining of phosphorylated glycogen synthase kinase-3β (p-GSK-3β) in the spinal cords of normal (A) and experimental autoimmune encephalomyelitis (EAE) rats on day 13 postimmunization (PI) (G.3) (B–D) and day 21 PI (R.0) (E). p-GSK-3β in astrocytes (arrows) in the spinal cords of normal controls. On day 13 PI, p-GSK-3β was expressed by infiltrating inflammatory cells (B and E, arrowheads), astrocytes (B and E, arrows), neurons (C, arrows), vascular endothelial cells (D, “V”) and ependymal cells (D) in the spinal cord. Primary antibody was omitted (F). Counterstained with hematoxylin. Scale bars=25 µm (A–F).

Table 1. p-GSK-3β immunoreactivity in the spinal cords of control and EAE-affected ratsa).
| Cell type | Control | EAE | |||
|---|---|---|---|---|---|
| Normal | CFA control | Paralysis stage | Recovery stage | ||
| G.1 | G.3 | R.0 | |||
| Inflammatory cellsb) | ND | ND | + | + | + |
| Astrocytes | +++ | +++ | +++ | + | +++ |
| Neurons | + | + | + | + | + |
| Ependymal cells | + | + | + | + | + |
| Vascular endothelial cells | +++ | +++ | +++ | +++ | +++ |
p-GSK-3β, phosphorylated glycogen synthase kinase-3β; EAE, experimental autoimmune encephalomyelitis; CFA, complete Freund's adjuvant; ND, inflammatory cells were not detected in the spinal cord; +, weak; ++, moderate; +++, strong. a)Three blind observers examined three sections from each of five animals. b)Inflammatory cells included p-GSK-3β-positive T cells and macrophages.
An inhibitor of GSK-3β, lithium, ameliorates rat EAE paralysis
To assess the role of GSK-3β in rat EAE, myelin basic protein (MBP)-immunized Lewis rats were treated with lithium. Lithium treatment significantly reduced the incidence of paralysis in MBP-immunized rats (40% suppression, 6/10 rats) compared with vehicle-treated EAE rats (100% incidence, 10/10 rats). The onset of EAE paralysis in lithium-treated rats with EAE (day 12.4±0.79 PI) was significantly delayed compared with that in vehicle-treated controls (day 10.3±0.15 PI) (P<0.05). Furthermore, lithium treatment at days 12–13 PI significantly ameliorated the clinical severity of EAE compared with vehicle treatment (P<0.01) (Fig. 3A).
Fig. 3. (A) Clinical signs during lithium treatment of experimental autoimmune encephalomyelitis (EAE). Animals (n=5) were euthanized at the time of peak paralysis (day 13 postimmunization [PI], arrow). (B–D) Histopathological examination of the spinal cords of rats with EAE on day 13 PI. The spinal cords of the control group showed a normal architecture (B). However, the spinal cords of vehicle-treated rats contained many inflammatory cells in the parenchyma (C), whereas there were fewer inflammatory cells in the spinal cords of lithium-treated rats (D). (B–D) Hematoxylin and eosin staining. Values are presented as mean±SE. **P< 0.01 vs. vehicle treatment. Scale bars=50 µm (B–D).

Effects of lithium on histological parameters
Histopathological examination showed no inflammatory cells in the spinal parenchyma of normal controls (Fig. 3B). In vehicle-treated EAE rats, a large number of inflammatory cells were seen in the spinal cord parenchyma (Fig. 3C), whereas inflammatory lesions were reduced in the spinal cords of lithium-treated rats (Fig. 3D). These findings matched the clinical signs (Fig. 3A).
To assess the effect of lithium treatment on rat EAE, we examined the expression of Iba-1, which is a marker of microglia and macrophages, by immunohistochemistry (Fig. 4A). In brief, Iba-1-positive ramified microglial cells were detected diffusely in the normal spinal cord. In vehicle-treated, EAE-affected spinal cords, the number of Iba-1-positive microglia and macrophages increased. In contrast, the intensity of Iba-1 immunolabeling was decreased in the lithium-treated group. A semiquantitative analysis of Iba-1 immunoreactivity in the spinal cord using ImageJ (Fig. 4B) revealed significantly reduced microglial positivity in the spinal cords of rats with EAE treated with lithium (16.23±0.83%) compared with those given vehicle (30.79±3.67%, P<0.01). These results suggest that lithium-induced amelioration of rat EAE is associated with the suppression of microglia and macrophage activation.
Fig. 4. Immunohistochemical localization of ionized calcium-binding protein-1 (Iba-1) in the spinal cords of normal control, vehicle-treated, and lithium-treated rats (A). Semi-quantitative analysis of Iba-1 immunoreactivity in normal control and vehicle- and lithium-treated rats with experimental autoimmune encephalomyelitis (EAE) on day 13 postimmunization (PI) (B). Counterstained with hematoxylin. Values are presented as mean±SE. **P<0.01 vs. normal controls (n=5 per group), ##P<0.01 vs. vehicle treatment. Scale bars=200 µm (A).

An inhibitor of GSK-3β treatment reduces serum TNF-α levels in EAE rats
We determined whether lithium treatment suppressed the expression of proinflammatory cytokines, such as TNF-α, and anti-inflammatory cytokines, such as IL-10 in the serum levels (Fig. 5). TNF-α expression was significantly decreased in lithium-treated rats (relative OD value, 0.15±0.12-fold increase; P<0.05) compared with vehicle-treated EAE rats (relative OD values, 0.35±0.2-fold increase; P<0.05) (Fig. 5A). In contrast, expression of IL-10 did not significantly differ in the vehicle and lithium-treated groups (Fig. 5B).
Fig. 5. Serum tumor necrosis factor α (TNF-α) and interleukin 10 (IL-10) levels. Sera were collected on day 13 postimmunization (PI) and TNF-α (A) and IL-10 (B) levels were measured by enzyme-linked immunosorbent assay. EAE, experimental autoimmune encephalomyelitis. Values are presented as mean±SE. *P<0.05 vs. normal controls (n=5 per group), #P<0.05 vs. vehicle treatment.

Change of Akt/GSK-3β/β-catenin signaling in spinal cords of lithium-treated EAE rats
To determine whether lithium treatment inhibits GSK-3 activity in the spinal cords of EAE rats, we examined GSK-3β phosphorylation and upstream and downstream signaling, such as phosphorylation of Akt and total β-catenin expression, by western blotting (n = 5 rats per group).
The p-Akt level in spinal cords was significantly increased in the lithium-treated group (relative OD value, 1.570±0.015-fold increase; P<0.05) compared with the vehicle-treated group at 13 days PI (0.990±0.003-fold increase) (Fig. 6A).
Fig. 6. Western blot analysis of the effect of lithium treatment on glycogen synthase kinase-3 (GSK-3) activity in the spinal cords of experimental autoimmune encephalomyelitis (EAE) rats. (A) Representative immunoblots of p-Akt (Ser473), total Akt (~51 kDa), p-GSK-3β (Ser9), total GSK-3β (~46 kDa), β-catenin (~92 kDa), and β-actin (~45 kDa). (B, C) Bar graphs show significant decreases in p-Akt, p-GSK-3β, and β-catenin expression in the spinal cords of vehicle-treated rats. Lithium treatment markedly increased the expression levels of these factors. To quantify phosphorylation of Akt and GSK-3β, levels of the phosphorylated forms were normalized to total Akt or GSK-3β. For normalization of β-catenin expression, the membranes were reprobed using an anti–β-actin antibody. Values are mean±SE. n = 6 per group. *P<0.05 vs. controls, #P<0.05 vs. lithium treatment. N, normal controls; V, vehicle-treated EAE; Li, lithium-treated EAE.

The vehicle-treated EAE rats (0.680±0.001-fold increase) showed significantly decreased p-GSK-3β levels compared with normal control rats (1.00±0.18-fold increase, P<0.05). However, lithium treatment (1.120±0.017-fold increase, P<0.05) resulted in increased p-GSK-3β expression compared with vehicle treatment in EAE rats (Fig. 6B).
In addition, β-catenin levels were decreased in vehicle-treated EAE rats (0.370±0.197-fold increase, P<0.05) compared with normal control rats, whereas lithium treatment resulted in increased total β-catenin levels in EAE rats (1.110±0.064-fold increase) (Fig. 6C).
Immunohistochemical evaluation of VCAM-1 in the spinal cords of lithium-treated EAE rats
In normal control rats, VCAM-1 expression was moderate in the vascular endothelial cells and astrocytes (Fig. 7A). In contrast, VCAM-1 was strongly expressed in the vascular endothelial cells, astrocytes, and infiltrated inflammatory cells of lithium-treated (Fig. 7B) and vehicle-treated (Fig. 7C) rats. However, the intensity of VCAM-1 immunolabeling was weaker in lithium-treated rats than in vehicle-treated rats. A semiquantitative analysis of VCAM-1 immunoreactivity in the spinal cord using ImageJ was significantly increased in vehicle-treated rats (20.7±0.3%, P<0.05) compared with normal control rats (11.1±0.6%). However, we observed a significant decrease in the VCAM-1 immunoreactivity in lithium-treated (13.1±0.5%, P<0.05) compared with vehicle-treated rats (Fig. 7D).
Fig. 7. Immunohistochemical detection of vascular cell adhesion molecule 1 (VCAM-1) in the spinal cords of normal control (A), vehicle-treated (B), and lithium-treated (C) rats. VCAM-1 in vascular endothelial cells (asterisk) and astrocytes (arrow) of the spinal cords of normal control rats (A). VCAM-1 immunoreactivity was detected mainly in the vascular endothelial cells (asterisk), infiltrated inflammatory cells, and astrocytes (arrows) in vehicle-treated rats (B), but it was rarely detected in the spinal cords of lithium-treated rats (C). Semi-quantitative analysis of VCAM-1 immunoreactivity in normal control and vehicle- and lithium-treated rats with experimental autoimmune encephalomyelitis (EAE) on day 13 postimmunization (PI) (D). Counterstained with hematoxylin. Values are presented as mean±SE. *P<0.05 vs. normal controls (n=5 each group), #P<0.05 vs. vehicle treatment. Scale bars=25 µm (A–C).

Discussion
In the present study, we first examined the quantitative changes and immunohistochemical localization of the phosphorylated form of GSK-3β in EAE-affected rat spinal cords and also determined the effect of lithium, an inhibitor of GSK-3β, on the neuropathogenesis of acute monophasic rat EAE.
Regarding the cellular localization of GSK-3β in rat CNS tissue, there is general agreement that GSK-3β, which is involved in cell signaling, is constitutively expressed in a variety of cell types, including neurons and glial cells, under normal conditions [24]. Inactivation of GSK-3β by phosphorylation of Ser-9 in the N-terminal domain of GSK-3β is involved in the interruption of cell signaling pathways, including the Akt and β-catenin pathways in acute monophasic rat EAE.
In terms of rat EAE, GSK-3β was constitutively expressed in spinal cord tissues with or without EAE, and GSK-3β phosphorylation was transiently suppressed at the peak stage of EAE, suggesting that autoimmune neuroinflammation reduces the phosphorylation of GSK-3β (Ser9), leading to enhancement of inflammation.
Regarding the involvement of astroglial activation in rat EAE, we postulate that astrogliosis in rat EAE is influenced by transient GSK-3β activation, as shown in a mouse EAE model in which the loss of p-GSK-3β in radial glia was involved in astrogliosis [25]. GSK-3β involvement in astrocytes has also been demonstrated in the spinal cords of both sham-operated rats and rats that have undergone partial sciatic nerve ligation [26], as well as in the white matter radial astrocytes and gray matter neurons of CFA-immunized mice [25].
For more than 50 years, lithium been used as a mood stabilizer to treat human bipolar disorders, and it exerts a neuroprotective effect [14,18]. Therefore, lithium is used to treat various CNS diseases. In the present study, we found that lithium treatment significantly delayed the onset of EAE paralysis and reduced the clinical severity of acute monophasic rat EAE, suggesting that lithium exerts an anti-inflammatory effect in the rat EAE model, as reported previously [19]. Lithium-induced suppression of neuroinflammation, and possibly suppression of microglial cells, has also been demonstrated in a variety of CNS injury models, including neonatal rat hypoxhypoxia-ischemia [27], intracerebral hemorrhage of rats [28], and lipopolysaccharide (LPS)-induced microglial activation [29]. These findings suggest that lithium has anti-inflammatory effects.
The present study demonstrated that lithium treatment reduced the number of Iba-1-positive macrophages/activated microglia in the spinal cords of rats with EAE at the peak stage. Activated microglia are a source of cytotoxic molecules, such as the pro-inflammatory cytokines TNF-α, IL-6 [29], and IL-1β [30]. The suppression of microglial activation is important for the treatment of inflammation. In the present study, we found that lithium treatment resulted in a decrease in the Iba-1-positive area in the spinal cord and a reduction in the serum TNF-α level, suggesting that lithium both suppresses the levels of circulating pro-inflammatory mediators and reduces the number of CNS microglial cells. Recently, it was reported that a peripheral increase in the TNF-α level in EAE is important for the activation of the microglial cells and astrocytes in the EAE cerebral cortex prior to cell migration into the parenchyma [31].
In terms of the mechanism of action of lithium, Dong et al. [29] reported downregulation of pro-inflammatory cytokines (such as TNF-α and IL-6) in LPS-induced primary rat microglial cells treated with lithium due to the inhibition of Toll-like receptor 4 expression and microglial activation through the phosphoinositide 3-kinase (PI3K)/Akt/FoxO1 signaling pathway. In addition, lithium pretreatment reduced in vitro production of interferon γ , IL-6, and IL-17 by splenocytes isolated from MOG35–55-immunized mice on day 10 PI [13]. Lithium treatment also induced the production of anti-inflammatory cytokines, such as IL-10, in Pseudomonas aeruginosa keratitis [32], human monocyte-derived dendritic cells [33], and an animal model of mania induced by dextroamphetamine [34].
We found no significant change in the serum IL-10 level in lithium-treated rats, suggesting that lithium-induced amelioration of rat EAE is not directly linked to increased serum IL-10 levels. A similar result in terms of no change in the IL-10 level was reported in splenocytes isolated from MOG33–55-immunized mice with and without lithium treatment on day 10 PI [13]. Therefore, we postulated that lithium suppresses production of pro-inflammatory cytokines, but not the M2 milieu, as indicated by the upregulation of the anti-inflammatory cytokine IL-10 in the rat model of EAE.
GSK-3, which is a multifunctional serine/threonine kinase, is involved in the development and progression of neuroinflammation [35,36,37] and is regulated by the PI3K/Akt and/or Wnt signaling pathway [14,38,39]. In the PI3K/Akt signaling pathway, PI3K and Akt are involved in the ability of Toll-like receptors to mediate the phosphorylation of GSK-3β (Ser9) and NF-κBp65 by regulating the production of pro- and anti-inflammatory cytokines [40]. In addition, the histone deacetylase inhibitor Scriptaid increased the phosphorylation of GSK-3β in the microglia of traumatic brain injury, thereby promoting PI3K/Akt signaling and depolarizing microglia toward M2 [41]. In human macrophages treated with LPS, lithium pretreatment significantly enhanced NF-κBp65 phosphorylation and markedly inhibited ERK1/2 phosphorylation. However, lithium had no effect on p38 and JNK signaling [42]. Furthermore, it was reported that the accumulation of β-catenin caused by GSK-3 inhibition through the activation of Wnt signaling plays an important role in neuroprotection [37,38,43]. In the present study, which used a rat model of EAE, we found that lithium treatment significantly inactivated GSK-3β by phosphorylation of Ser 9. We also found an accumulation of β-catenin in the spinal cord, suggesting that the Akt/GSK-3β/β-catenin signaling pathway is associated with amelioration of the EAE inflammation caused by lithium treatment. In a previous study, we found that all three MAP kinases—ERK, JNK, and p38—were transiently activated during the inflammatory stage of rat EAE; however, these factors are present in all inflammatory cell types as well as in brain cells [44], suggesting that MAP kinases are less affected in EAE rats.
In CNS autoimmune diseases, the activation of adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1) and VCAM-1, in the vascular endothelial cells and astrocytes initiates inflammation [2,45,46].
In the present study, we found moderate VCAM-1 expression in vascular endothelial cells and some astrocytes in the normal spinal cord, whereas VCAM-1 expression was significantly increased in the spinal cords of EAE rats, particularly in astrocytes and infiltrating inflammatory cells, which is in agreement with our previous report [2]. VCAM-1 expression by astrocytes is vital for T cell entry into spinal cord parenchyma in MOG35–55-immunized mice and is required for CNS inflammation [14,45]. Consistent with the activation of vascular endothelial cells, the close link between GSK-3β and VCAM-1 was confirmed; this is involved in vascular permeability via endothelial adhesion molecules [47]. Taken together, the previous studies and our findings suggest that suppression of ICAM-1 and VCAM-1 in vascular endothelial cells and astrocytes is involved in the amelioration of rat monophasic EAE. This assumption is supported by reports that matrine, a quinolizidine alkaloid, significantly mitigated EAE severity in Wistar rats by suppressing the levels of adhesion molecules, such as ICAM-1 and VCAM-1 [48], and that lithium-induced amelioration by methylglyoxal is associated with the inhibition of endothelial GSK-3 [47]. Therefore, our results suggest that lithium suppressed inflammation in the spinal cord of EAE rats by reducing inflammatory cell infiltration via the inhibition of GSK-3β expression in vascular endothelial cells and astrocytes.
Taken all into considerations, these findings suggest that GSK-3β is a target for acute CNS autoimmune diseases, and that lithium treatment, possibly through GSK-3β inhibition, would be a strategy for the amelioration of acute neuroinflammation as far as rat model of human CNS autoimmune diseases is involved.
Acknowledgements
This research was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), funded by the Ministry of Education (Grant number: NRF-2014R1A1A2055965).
References
- 1.Shin T, Ahn M, Matsumoto Y. Mechanism of experimental autoimmune encephalomyelitis in Lewis rats: recent insights from macrophages. Anat Cell Biol. 2012;45:141–148. doi: 10.5115/acb.2012.45.3.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shin T, Kojima T, Tanuma N, Ishihara Y, Matsumoto Y. The subarachnoid space as a site for precursor T cell proliferation and effector T cell selection in experimental autoimmune encephalomyelitis. J Neuroimmunol. 1995;56:171–178. doi: 10.1016/0165-5728(94)00144-d. [DOI] [PubMed] [Google Scholar]
- 3.Swanborg RH. Experimental autoimmune encephalomyelitis in the rat: lessons in T-cell immunology and autoreactivity. Immunol Rev. 2001;184:129–135. doi: 10.1034/j.1600-065x.2001.1840112.x. [DOI] [PubMed] [Google Scholar]
- 4.Matsumoto Y. New approach to immunotherapy against organ-specific autoimmune diseases with T cell receptor and chemokine receptor DNA vaccines. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:73–77. doi: 10.2174/1568008053174732. [DOI] [PubMed] [Google Scholar]
- 5.Ahn M, Yang W, Kim H, Jin JK, Moon C, Shin T. Immunohistochemical study of arginase-1 in the spinal cords of Lewis rats with experimental autoimmune encephalomyelitis. Brain Res. 2012;1453:77–86. doi: 10.1016/j.brainres.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 6.Ellrichmann G, Thöne J, Lee DH, Rupec RA, Gold R, Linker RA. Constitutive activity of NF-kappa B in myeloid cells drives pathogenicity of monocytes and macrophages during autoimmune neuroinflammation. J Neuroinflammation. 2012;9:15. doi: 10.1186/1742-2094-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim H, Moon C, Ahn M, Lee Y, Kim S, Matsumoto Y, Koh CS, Kim MD, Shin T. Increased phosphorylation of cyclic AMP response element-binding protein in the spinal cord of Lewis rats with experimental autoimmune encephalomyelitis. Brain Res. 2007;1162:113–120. doi: 10.1016/j.brainres.2007.05.072. [DOI] [PubMed] [Google Scholar]
- 8.Aleshin S, Strokin M, Sergeeva M, Reiser G. Peroxisome proliferator-activated receptor (PPAR)beta/delta, a possible nexus of PPARalpha- and PPARgamma-dependent molecular pathways in neurodegenerative diseases: Review and novel hypotheses. Neurochem Int. 2013;63:322–330. doi: 10.1016/j.neuint.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 9.Chen G, Shannon M. Transcription factors and th17 cell development in experimental autoimmune encephalomyelitis. Crit Rev Immunol. 2013;33:165–182. doi: 10.1615/critrevimmunol.2013006959. [DOI] [PubMed] [Google Scholar]
- 10.Beurel E. Regulation by glycogen synthase kinase-3 of inflammation and T cells in CNS diseases. Front Mol Neurosci. 2011;4:18. doi: 10.3389/fnmol.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beurel E, Kaidanovich-Beilin O, Yeh WI, Song L, Palomo V, Michalek SM, Woodgett JR, Harrington LE, Eldar-Finkelman H, Martinez A, Jope RS. Regulation of Th1 cells and experimental autoimmune encephalomyelitis by glycogen synthase kinase-3. J Immunol. 2013;190:5000–5011. doi: 10.4049/jimmunol.1203057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rowse AL, Naves R, Cashman KS, McGuire DJ, Mbana T, Raman C, De Sarno P. Lithium controls central nervous system autoimmunity through modulation of IFN-gamma signaling. PLoS One. 2012;7:e52658. doi: 10.1371/journal.pone.0052658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Sarno P, Axtell RC, Raman C, Roth KA, Alessi DR, Jope RS. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2008;181:338–345. doi: 10.4049/jimmunol.181.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 15.Eto M, Kouroedov A, Cosentino F, Lüscher TF. Glycogen synthase kinase-3 mediates endothelial cell activation by tumor necrosis factor-alpha. Circulation. 2005;112:1316–1322. doi: 10.1161/CIRCULATIONAHA.105.564112. [DOI] [PubMed] [Google Scholar]
- 16.Ramirez SH, Fan S, Zhang M, Papugani A, Reichenbach N, Dykstra H, Mercer AJ, Tuma RF, Persidsky Y. Inhibition of glycogen synthase kinase 3beta (GSK3beta) decreases inflammatory responses in brain endothelial cells. Am J Pathol. 2010;176:881–892. doi: 10.2353/ajpath.2010.090671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee MJ, Jang M, Choi J, Chang BS, Kim DY, Kim SH, Kwak YS, Oh S, Lee JH, Chang BJ, Nah SY, Cho IH. Korean red ginseng and ginsenoside-Rb1/-Rg1 alleviate experimental autoimmune encephalomyelitis by suppressing Th1 and Th17 cells and upregulating regulatory T cells. Mol Neurobiol. 2016;53:1977–2002. doi: 10.1007/s12035-015-9131-4. [DOI] [PubMed] [Google Scholar]
- 18.Jope RS. Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci. 2003;24:441–443. doi: 10.1016/S0165-6147(03)00206-2. [DOI] [PubMed] [Google Scholar]
- 19.Levine S, Saltzman A. Inhibition of experimental allergic encephalomyelitis by lithium chloride: specific effect or nonspecific stress? Immunopharmacology. 1991;22:207–213. doi: 10.1016/0162-3109(91)90045-z. [DOI] [PubMed] [Google Scholar]
- 20.Kim S, Moon C, Wie MB, Kim H, Tanuma N, Matsumoto Y, Shin T. Enhanced expression of constitutive and inducible forms of nitric oxide synthase in autoimmune encephalomyelitis. J Vet Sci. 2000;1:11–17. [PubMed] [Google Scholar]
- 21.Kim MD, Cho HJ, Shin T. Expression of osteopontin and its ligand, CD44, in the spinal cords of Lewis rats with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;151:78–84. doi: 10.1016/j.jneuroim.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 22.De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 23.Kim H, Moon C, Ahn M, Byun J, Lee Y, Kim MD, Matsumoto Y, Koh CS, Shin T. Heat shock protein 27 upregulation and phosphorylation in rat experimental autoimmune encephalomyelitis. Brain Res. 2009;1304:155–163. doi: 10.1016/j.brainres.2009.09.060. [DOI] [PubMed] [Google Scholar]
- 24.Dill J, Wang H, Zhou F, Li S. Inactivation of glycogen synthase kinase 3 promotes axonal growth and recovery in the CNS. J Neurosci. 2008;28:8914–8928. doi: 10.1523/JNEUROSCI.1178-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tafreshi AP, Payne N, Sun G, Sylvain A, Schulze K, Bernard C. Inactive GSK3beta is disturbed in the spinal cord during experimental autoimmune encephalomyelitis, but rescued by stem cell therapy. Neuroscience. 2014;277:498–505. doi: 10.1016/j.neuroscience.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 26.Weng HR, Gao M, Maixner DW. Glycogen synthase kinase 3 beta regulates glial glutamate transporter protein expression in the spinal dorsal horn in rats with neuropathic pain. Exp Neurol. 2014;252:18–27. doi: 10.1016/j.expneurol.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Li Q, Du X, Sun Y, Wang X, Kroemer G, Blomgren K, Zhu C. Lithium-mediated long-term neuroprotection in neonatal rat hypoxia-ischemia is associated with antiinflammatory effects and enhanced proliferation and survival of neural stem/progenitor cells. J Cereb Blood Flow Metab. 2011;31:2106–2115. doi: 10.1038/jcbfm.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang K, Kim YJ, Kim YH, Roh JN, Nam JM, Kim PY, Ryu WS, Lee SH, Yoon BW. Lithium pretreatment reduces brain injury after intracerebral hemorrhage in rats. Neurol Res. 2012;34:447–454. doi: 10.1179/1743132812Y.0000000015. [DOI] [PubMed] [Google Scholar]
- 29.Dong H, Zhang X, Dai X, Lu S, Gui B, Jin W, Zhang S, Zhang S, Qian Y. Lithium ameliorates lipopolysaccharide-induced microglial activation via inhibition of toll-like receptor 4 expression by activating the PI3K/Akt/FoxO1 pathway. J Neuroinflammation. 2014;11:140. doi: 10.1186/s12974-014-0140-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doverhag C, Hedtjarn M, Poirier F, Mallard C, Hagberg H, Karlsson A, Sävman K. Galectin-3 contributes to neonatal hypoxic-ischemic brain injury. Neurobiol Dis. 2010;38:36–46. doi: 10.1016/j.nbd.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 31.Chanaday NL, Roth GA. Microglia and astrocyte activation in the frontal cortex of rats with experimental autoimmune encephalomyelitis. Neuroscience. 2016;314:160–169. doi: 10.1016/j.neuroscience.2015.11.060. [DOI] [PubMed] [Google Scholar]
- 32.Chen K, Wu Y, Zhu M, Deng Q, Nie X, Li M, Wu M, Huang X. Lithium chloride promotes host resistance against Pseudomonas aeruginosa keratitis. Mol Vis. 2013;19:1502–1514. [PMC free article] [PubMed] [Google Scholar]
- 33.Liu KJ, Lee YL, Yang YY, Shih NY, Ho CC, Wu YC, Huang TS, Huang MC, Liu HC, Shen WW, Leu SJ. Modulation of the development of human monocyte-derived dendritic cells by lithium chloride. J Cell Physiol. 2011;226:424–433. doi: 10.1002/jcp.22348. [DOI] [PubMed] [Google Scholar]
- 34.Valvassori SS, Tonin PT, Varela RB, Carvalho AF, Mariot E, Amboni RT, Bianchini G, Andersen ML, Quevedo J. Lithium modulates the production of peripheral and cerebral cytokines in an animal model of mania induced by dextroamphetamine. Bipolar Disord. 2015;17:507–517. doi: 10.1111/bdi.12299. [DOI] [PubMed] [Google Scholar]
- 35.Maixner DW, Weng HR. The role of glycogen synthase kinase 3 beta in neuroinflammation and pain. J Pharm Pharmacol (Los Angel) 2013;1:001. doi: 10.13188/2327-204X.1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32:577–595. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beurel E, Michalek SM, Jope RS. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3) Trends Immunol. 2010;31:24–31. doi: 10.1016/j.it.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu B, Crampton SP, Hughes CC. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity. 2007;26:227–239. doi: 10.1016/j.immuni.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multitasking kinase. J Cell Sci. 2003;116(Pt 7):1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H, Brown J, Martin M. Glycogen synthase kinase 3: a point of convergence for the host inflammatory response. Cytokine. 2011;53:130–140. doi: 10.1016/j.cyto.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y, Pu H, Li WW, Tang B, Wang Y, Gao Y, Zheng P, Bennett MV, Chen J. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci U S A. 2015;112:2853–2858. doi: 10.1073/pnas.1501441112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raghavendra PB, Lee E, Parameswaran N. Regulation of macrophage biology by lithium: a new look at an old drug. J Neuroimmune Pharmacol. 2014;9:277–284. doi: 10.1007/s11481-013-9516-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J Cell Biol. 2001;152:87–96. doi: 10.1083/jcb.152.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin T, Ahn M, Jung K, Heo S, Kim D, Jee Y, Lim YK, Yeo EJ. Activation of mitogen-activated protein kinases in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2003;140:118–125. doi: 10.1016/s0165-5728(03)00174-7. [DOI] [PubMed] [Google Scholar]
- 45.Archambault AS, Sim J, McCandless EE, Klein RS, Russell JH. Region-specific regulation of inflammation and pathogenesis in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2006;181:122–132. doi: 10.1016/j.jneuroim.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 46.Gimenez MA, Sim JE, Russell JH. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J Neuroimmunol. 2004;151:116–125. doi: 10.1016/j.jneuroim.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 47.Su Y, Qadri SM, Cayabyab FS, Wu L, Liu L. Regulation of methylglyoxal-elicited leukocyte recruitment by endothelial SGK1/GSK3 signaling. Biochim Biophys Acta. 2014;1843:2481–2491. doi: 10.1016/j.bbamcr.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 48.Kan QC, Zhu L, Liu N, Zhang GX. Matrine suppresses expression of adhesion molecules and chemokines as a mechanism underlying its therapeutic effect in CNS autoimmunity. Immunol Res. 2013;56:189–196. doi: 10.1007/s12026-013-8393-z. [DOI] [PubMed] [Google Scholar]