

Abstract
Background and Purpose
Ionotropic GABA receptors are evolutionarily conserved proteins that mediate cellular and network inhibition in both vertebrates and invertebrates. A unique class of excitatory GABA receptors has been identified in several nematode species. Despite well‐characterized functions in Caenorhabditis elegans, little is known about the pharmacology of the excitatory GABA receptors EXP‐1 and LGC‐35. Using a panel of compounds that differentially activate and modulate ionotropic GABA receptors, we investigated the agonist binding site and allosteric modulation of EXP‐1 and LGC‐35.
Experimental Approach
We used two‐electrode voltage clamp recordings to characterize the pharmacological profile of EXP‐1 and LGC‐35 receptors expressed in Xenopus laevis oocytes.
Key Results
The pharmacology of EXP‐1 and LGC‐35 is different from that of GABAA and GABAA‐ρ receptors. Both nematode receptors are resistant to the competitive orthosteric antagonist bicuculline and to classical ionotropic receptor pore blockers. The GABAA‐ρ specific antagonist, TPMPA, was the only compound tested that potently inhibited EXP‐1 and LGC‐35. Neurosteroids have minimal effects on GABA‐induced currents, but ethanol selectively potentiates LGC‐35.
Conclusions and Implications
The pharmacological properties of EXP‐1 and LGC‐35 more closely resemble the ionotropic GABAA‐ρ family. However, EXP‐1 and LGC‐35 exhibit a unique profile that differs from vertebrate GABAA and GABAA‐ρ receptors, insect GABA receptors and nematode GABA receptors. As a pair, EXP‐1 and LGC‐35 may be utilized to further understand the differential molecular mechanisms of agonist, antagonist and allosteric modulation at ionotropic GABA receptors and may aid in the design of new and more specific anthelmintics that target GABA neurotransmission.
Abbreviations
- EXP‐1
expulsion defective
- LGC‐35
ligand‐gated ion channel
Tables of Links
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
GABA is the primary fast inhibitory neurotransmitter in both vertebrate and invertebrate nervous systems (Hosie and Sattelle, 1996). Fast inhibition is produced when GABA binds and activates chloride‐selective ionotropic GABA receptors. These receptors belong to the ‘Cys‐loop’ superfamily of ligand‐gated ion channels, which include the nicotinic acetylcholine (nAChRs), glycine and 5HT3 receptors (Bormann, 2000). Like all members of the superfamily, ionotropic GABA receptors consist of five subunits that pseudo‐symmetrically arrange around a central ion‐conducting pore (Johnston, 2005). Each subunit consists of a large extracellular N‐terminal domain that contains consensus motifs that comprise the orthosteric agonist binding site, four transmembrane‐spanning domains (M1–M4) and a large intracellular loop between M3 and M4, which is thought to mediate receptor trafficking and subcellular localization (Michels and Moss, 2007; Thompson et al., 2010). Within the receptor complex, each subunit is arranged such that the second transmembrane domain (M2) lines the ion channel pore and determines ion selectivity.
In vertebrates, ionotropic GABA receptors are divided into two classes based on their distinct oligomerization and pharmacological profiles: (1) GABAA receptors are heteropentameric (i.e. multiple different subunits) and inhibited by the competitive antagonist bicuculline and (2) GABAA‐ρ receptors are homopentameric (i.e. single subunit) and bicuculline‐insensitive (Bormann, 2000; Chebib and Johnston, 2000). In Caenorhabditis elegans, the ionotropic GABA receptor family consists of chloride‐conducting inhibitory (UNC‐49) and sodium‐conducting excitatory receptors [expulsion defective (EXP‐1) and ligand‐gated ion channel (LGC‐35)] (Bamber et al., 1999; Beg and Jorgensen, 2003; Jobson et al., 2015). Each of these receptors mediate distinct and coordinated neuromuscular contractions. Specifically, the inhibitory UNC‐49 receptor is required to reciprocally inhibit antagonistic body wall muscles to ensure coordinated locomotion (Bamber et al., 1999); whereas, the excitatory EXP‐1 receptor is required for intestinal muscle contraction during defecation (Beg and Jorgensen, 2003). Expanding the unique role of excitatory GABA signalling in the C. elegans nervous system, LGC‐35 was recently shown to act as a spillover receptor that modulates locomotor body bending and movement speed (Jobson et al., 2015).
Although the inhibitory UNC‐49 GABA receptor has been pharmacologically characterized in both free living and parasitic nematodes (Bamber et al., 2003; Kaji et al., 2015), relatively little is known about the pharmacological profile of excitatory GABA receptors in C. elegans. Moreover, excitatory GABA receptor signalling not only regulates a wider variety of neuromuscular contractions that underlie specific behaviours in the nematode but also utilizes a larger repertoire of characterized receptors (EXP‐1 and LGC‐35). Despite these observations, canonical inhibitory GABA signalling and pharmacological profiling of UNC‐49 have been the primary focus of investigation in nematode species.
Here, we have characterized the pharmacological profiles of EXP‐1 and LGC‐35 utilizing a broad panel of agonists, antagonists and allosteric modulators that differentially regulate ionotropic GABAA and GABAA‐ρ receptors. We conclude that EXP‐1 and LGC‐35 more closely resemble the GABAA‐ρ receptor subfamily; however, they both exhibit unique pharmacological profiles despite their highly conserved orthosteric GABA binding sites. In addition, several characteristics distinguish these receptors from vertebrate GABAA and GABAA‐ρ receptors, insect GABA receptors and nematode GABA receptors. Taken together, our data reveal that inhibitory and excitatory GABA receptor signalling can be pharmacologically segregated, which may aid in the differential and specific modulation of nematode GABA receptor neurotransmission. Given the high amino acid sequence identity between EXP‐1 and LGC‐35, these receptors, as a pair, provide a useful framework to better understand the differential mechanisms of agonist, antagonist and allosteric modulation at ionotropic GABA receptors and may aid in the design of new and more specific anthelmintic GABA mimetics.
Methods
Animals
All animal care and experimental procedures followed the guidelines from the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). X. laevis ovaries were obtained from Xenopus1 (Ann Arbor, MI, USA). Briefly, female X. laevis frogs were housed in a climate‐controlled, light‐regulated room. Frogs were anaesthetized by immersion in 0.2% tricaine until non‐responsive to toe pinch. Subsequently, frogs were decapitated, and ovarian lobes were harvested and defolliculated by incubation in 2 mg·mL−1 collagenase (Type I, Worthington, Lakewood, NJ, USA).
Preparation of receptors
The C. elegans EXP‐1 and LGC‐35 cDNAs were isolated and cloned into the X. laevis expression vector pSGEM (courtesy of M. Hollman) as previously described (Beg and Jorgensen, 2003; Jobson et al., 2015). Briefly, plasmids were linearized with the restriction enzyme MscI, and capped RNA (cRNA) was prepared using the mMessage mMachine kit (Life Technologies) following the manufacturer's guidelines. Defolliculated stages V–VI oocytes were sorted and injected with EXP‐1 (50 ng) or LGC‐35 (12.5 ng) cRNA. Injected oocytes were incubated for 2–5 days at 18°C in Barth's solution: 88 mM NaCl, 1 mM KCl, 0.33 mM Ca(NO3)2, 0.41 mM CaCl2, 0.82 MgSO4, 2.4 mM NaHCO3, 10 mM HEPES, supplemented with 1 mM Na+‐pyruvate, and 50 mg·L−1 gentamicin (pH = 7.4, NaOH).
Electrophysiology
Electrophysiological recordings were performed 2–5 days after injection and were carried out at room temperature in Ringer's solution (115 mM NaCl, 2 mM KCl, 1.8 mM CaCl2 and 10 mM HEPES, pH = 7.4 with NaOH). Two electrode voltage clamp recordings were obtained using an OC‐725C oocyte clamp amplifier (Warner Instruments, Hamden, CT, USA) using 3 M KCl‐filled microelectrodes (1–5 MΩ). All recordings were performed at a holding potential of −60 mV. Oocytes were exposed to varying concentrations of compounds using a custom‐built gravity controlled perfusion system with a flow rate of ~8 mL·min−1. Dose–response curves were generated on single oocytes by applying compounds at increasing concentrations (low to high). Oocytes were continually perfused with control Ringer's for 2 min between each dose.
Sequence alignments
Protein sequences were aligned using the Clustal W algorithm in the MacVector software suite. The GenBank identifier numbers for the protein sequences used for comparative sequence analysis are as follows: EXP‐1 (550540762), LGC‐35 (71998246), GABRA1 (27808653), GABRB2 (292495010), GABRG2 (189083762), GABRR1 (223590210) and RDL (635377460).
Homology modelling
The homology models of EXP‐1 and LGC‐35 were constructed based on the crystal structure of the homopentameric human β3 GABA receptor (PDB ID: 4COF) (Miller and Aricescu, 2014). The homology models were built using Modeller 9.17 software (Marti‐Renom et al., 2000). An initial 20 homology models were generated for EXP‐1 and LGC‐35, which were then further evaluated with Procheck software (Laskowski et al., 1996). The model with the lowest DOPE value and highest LG score was selected for molecular docking. To compare the differences in ligand binding, GABA was docked to the crystal structure of the dimeric human GABA receptor (chain A and B) and the homology models of EXP‐1 and LGC‐35 using Autodock Vina software (Trott and Olson, 2010). The ligand box was centred in the ligand binding pocket of human GABA receptor with a dimension of 20 × 24 × 28 Å. The docked results were viewed and graphed using pymol software (http://www.pymol.org). Residues were numbered beginning at the start methionine for each receptor.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are reported as mean ± SEM, with n = the number of oocytes recorded and N = the number of animal replicates. The response of each compound dose was normalized to the maximal GABA response (3 mM) of each individual recorded oocyte. To determine the overall efficacy of agonists, the maximal response of each drug tested was normalized relative to the maximal GABA response (3 mM). Antagonists and allosteric modulator dose–response recordings were performed as follows. Individual oocytes were exposed to GABA alone (10 μM), followed by GABA plus increasing concentrations of drug (10 μM GABA + [Drug]), and control responses were re‐tested at the end of each recording with GABA alone (10 μM) to confirm complete drug washout and cell health. Dose–response curves from individual oocytes were normalized to the maximum values and averaged using Prism 7.0 software (GraphPad, San Diego, CA, USA). Normalized data were fit to the four parameter Hill equation: Y = Min + (Max − Min)/(1 + 10H(LogEC50 − X)),
where Max is the maximal response, Min is the response at lowest drug concentration, X is the logarithm of agonist concentrations, EC50 is the half‐maximal response and H is the Hill coefficient. Statistical analyses were performed using Prism 7 (GraphPad, San Diego, CA). One‐way ANOVA with Holm–Sidak post hoc test was used for comparisons involving more than two groups. Statistical tests with P < 0.05 were considered significant.
Materials
The following compounds were obtained from Tocris Bioscience (Minneapolis, MN, USA): trans‐4‐aminocrotonic acid (TACA); (Z)‐3‐[(aminoiminomethyl)thio]prop‐2‐enoic acid sulfate (ZAPA); 4,5,6,7‐tetrahydroisoxazolo[5,4‐c]pyridin‐3‐ol hydrochloride (THIP); (1,2,5,6‐tetrahydropyridin‐4‐yl)methylphosphinic acid (TPMPA); (3α,5α)‐3‐hydroxypregnane‐11,20‐dione (alphaxalone); and (3α,5β)‐3‐hydroxy‐pregnan‐20‐one (pregnanalone). The following compounds were obtained from Sigma‐Aldrich: GABA; β‐alanine; muscimol), glycine, imidazole‐4‐acetic acid (IMA); piperdine‐4‐sulphonic acid (P4S); 4‐piperidinecarboxylic acid (isonipecotic acid); 5‐aminovaleric acid (DAVA); isoguvacine; bicuculline methchloride, mecamylamine hydrochloride and dieldrin. All compound stocks (≥10 mM) were dissolved in Ringer's solution and stored at −20°C. The water insoluble compounds alphaxalone and pregnanalone were dissolved in absolute ethanol. Due to their hydrophobicity, the maximum soluble concentration of these compounds in Ringer's solution was 30 μM (pregnanalone) and 100 μM (alphaxalone).
Results
The C. elegans excitatory GABA receptors EXP‐1 and LGC‐35 are homopentameric ‘Cys‐loop’ ligand gated ion channel proteins that share 53% identity and 68% similarity across their entire protein coding sequences. Sequence alignments against human ionotropic GABAA and GABAA‐ρ1 receptor subunits reveal that EXP‐1 and LGC‐35 have highly homologous residues that comprise the orthosteric GABA binding site (Figure 1A). The neurotransmitters acetylcholine, glycine, serotonin and glutamate exhibit no activity at EXP‐1 or LGC‐35 when expressed in X. laevis oocytes (Beg and Jorgensen, 2003; Jobson et al., 2015). Given the specific GABA‐gating, we characterized a broad range of agonists, antagonists and modulators to determine the pharmacological profiles of EXP‐1 and LGC‐35 and to functionally classify the receptor family as GABAA or GABAA‐ρ like. We expressed each receptor in X. laevis oocytes and used two‐electrode voltage clamp to test the concentration–response of a variety of compounds known to differentially activate, inhibit or modulate GABAA and GABAA‐ρ receptors.
Figure 1.
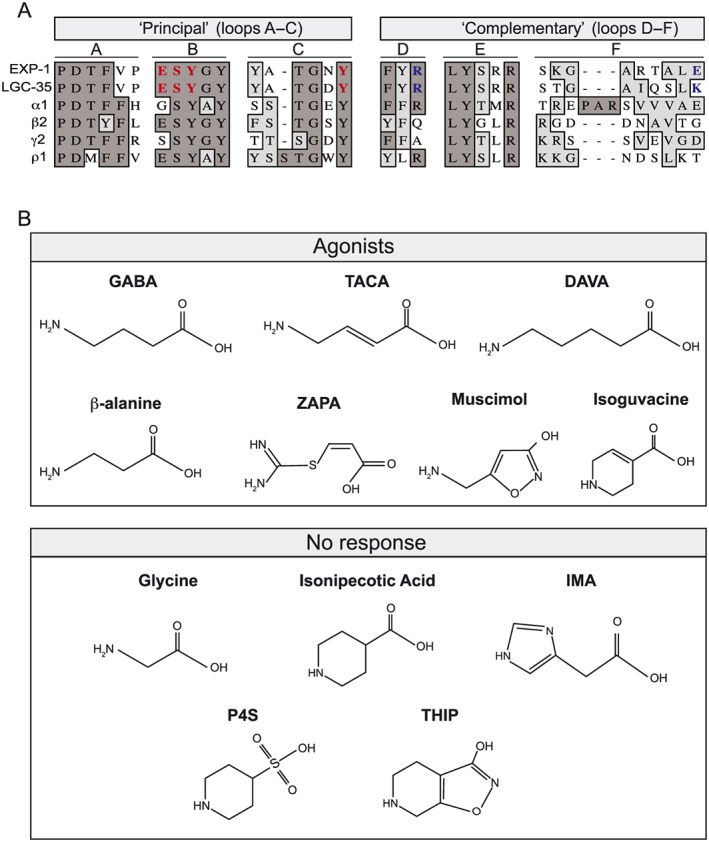
The orthosteric binding sites of EXP‐1 and LGC‐35 are highly conserved. (A) Sequence alignment of amino acid motifs (loops A–F) that comprise the orthosteric GABA binding site. GABA binding occurs at the subunit interface of two adjacent subunits termed the ‘principal’ and ‘complementary’ subunit. The orthosteric binding site is formed from three loops on the ‘principal’ subunit (loops A–C) and three β‐sheets on the ‘complementary’ subunit (loops D–F). Residue identity is indicated in dark grey and similarity in light grey highlights. Residues involved in ligand binding in this study are bold‐highlighted red (principal), and blue (complementary). Aligned protein sequences, C. elegans EXP‐1 and LGC‐35, human GABAA receptor subunits (α1, β2 and γ2) and human GABAA‐ρ subunit (ρ1). (B) Structures of known agonists at GABAA receptors that were assayed for agonist activity at EXP‐1 and LGC‐35. Compounds that directly activated are categorized as agonists, and those that elicited no activity are categorized as no response.
Agonist profiling of EXP‐1 and LGC‐35
Application of the endogenous neurotransmitter GABA produced robust and concentration‐dependent current responses with a EC50 value comparable with previously published data (Figures 2A, 3A and Table 1). Muscimol, a potent orthosteric agonist that differentially activates ionotropic GABA receptors, is a full agonist at vertebrate GABAA receptors but only a partial agonist at vertebrate GABAA‐ρ receptors and nematode UNC‐49 receptors (Siddiqui et al., 2010; Johnston, 2014). Dose–response curves revealed that muscimol exhibited partial agonist activity at both EXP‐1 and LGC‐35 with reduced efficacy and potency compared with GABA, comparable with that observed in nematode UNC‐49 GABA receptors (Siddiqui et al., 2010) (Figures 2, 3 and Table 1).
Figure 2.
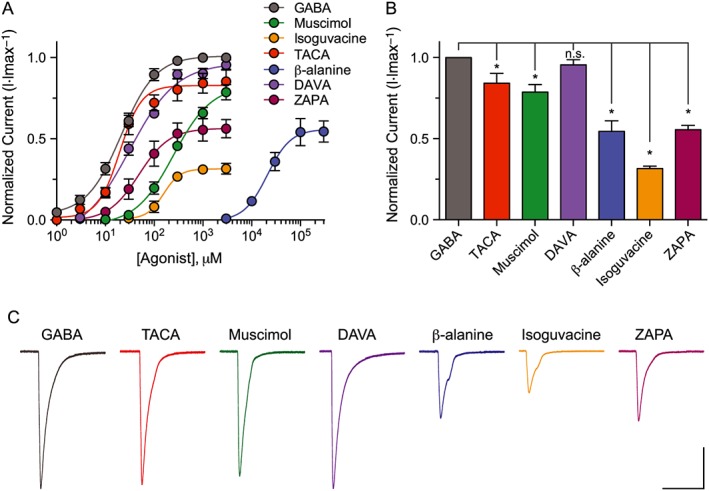
EXP‐1 is differentially activated by GABA agonists. (A) Dose–response curves comparing the responses of each agonist tested normalized to maximal GABA responses (3 mM) for EXP‐1. Oocytes were voltage clamped at −60 mV, and agonists were bath applied in series (1–3000 μM) for 5 s. Due to its low potency, β‐alanine was applied in series at higher concentrations (3–300 mM). Each data point represents the mean ± SEM with n ≥ 10 and N = 4. (B) Bar graph illustrating the efficacy of compounds tested relative to maximal GABA responses (3 mM). Each bar represents the mean ± SEM with n ≥ 10. n.s, not significant (P ≥ 0.05), *P ≤ 0.05; significantly different from GABA; one‐way ANOVA, Holm–Sidak post hoc test. (C) Representative current responses from an EXP‐1 expressing oocyte with each compound tested in series at maximal dose (3 mM; β‐alanine = 300 mM). Scale bar = 1 μA, 10 s.
Figure 3.
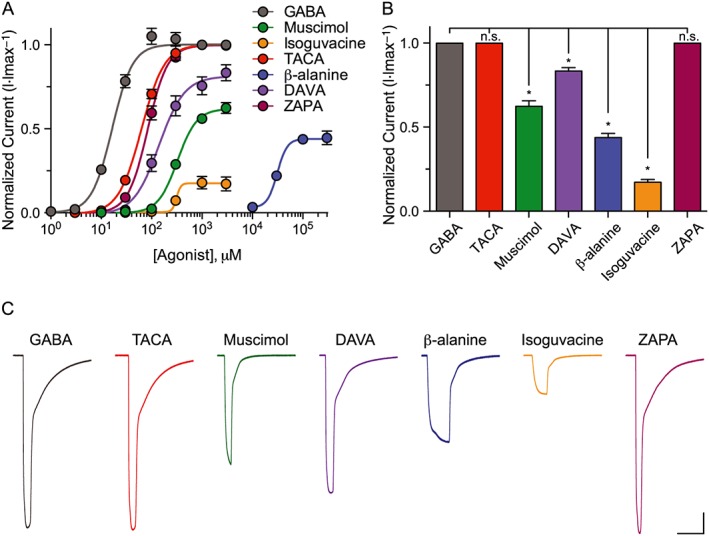
LGC‐35 is differentially activated by GABA agonists. (A) Dose–response curves comparing the responses of each agonist tested relative to maximal GABA responses (3 mM) for LGC‐35. Oocytes were voltage clamped at −60 mV, and agonists were bath applied in series (1–3000 μM) to plateau. Due its low potency, β‐alanine was applied in series (3–300 mM). Each data point represents the mean ± SEM with n ≥ 7 and N = 3. (B) Bar graph illustrating the efficacy of compounds tested relative to maximal GABA responses (3 mM). Each bar represents the mean ± SEM with n ≥ 7. n.s, not significant (P ≥ 0.05), *P ≤ 0.05; significantly different from GABA; one‐way ANOVA, Holm–Sidak post hoc; (C) Representative current responses from an LGC‐35 expressing oocyte with each compound tested in series at maximal dose (3 mM; β‐alanine = 300 mM). Scale bar =1 μA, 30 s.
Table 1.
EC50, Hill slope and maximal current response efficacy for agonists at EXP‐1 and LGC‐35 receptors
| Compound | EXP‐1 | LGC‐35 | ||||||
|---|---|---|---|---|---|---|---|---|
| EC50 (μM) | Efficacy | Hill | n | EC50 (μM) | Efficacy | Hill | n | |
| GABA | 21 ± 0.69 | 100 | 0.91 ± 0.06 | 15 | 17 ± 0.71 | 100 | 2.1 ± 0.10 | 12 |
| Muscimol | 250 ± 22 | 79 | 1.2 ± 0.16 | 11 | 348 ± 17 | 62 | 2.1 ± 0.32 | 8 |
| TACA | 20 ± 2 | 84 | 1.7 ± 0.19 | 11 | 63 ± 3 | 100 | 1.9 ± 0.11 | 8 |
| DAVA | 31 ± 6 | 95 | 0.99 ± 0.12 | 11 | 142 ± 8 | 83 | 1.7 ± 0.03 | 7 |
| Isoguvacine | 155 ± 8 | 32 | 2.5 ± 0.53 | 10 | 316 ± 9 | 17 | 2.9 ± 0.21 | 7 |
| β‐alanine | 26 074 ± 2739 | 55 | 1.4 ± 0.10 | 11 | 37 275 ± 1058 | 44 | 1.7 ± 0.08 | 11 |
| ZAPA | 51 ± 4 | 56 | 1.4 ± 0.07 | 12 | 86 ± 6 | 100 | 2.1 ± 0.12 | 10 |
Agonist activity at EXP‐1 and LGC‐35. EC50 (μM), efficacy (% maximal GABA), Hill slope and replicate values (n) are shown as mean ± SEM.
GABA is a flexible molecule thought to bind and activate GABAA receptors in a partially folded conformation (Krogsgaard‐Larsen et al., 2002). Supporting this model, a number of partially folded conformationally restricted GABA mimetics are potent agonists at GABAA receptors (Kusama et al., 1993; Woodward et al., 1993; Hosie and Sattelle, 1996). In contrast, these partially folded analogues are weak partial agonists or antagonists at GABAA‐ρ receptors; whereas, extended and planar GABA analogues like TACA exhibit high potency and efficacy at GABAA‐ρ receptors (Johnston, 2005). To classify the agonist profile of EXP‐1 and LGC‐35, we determined dose–response relationships using a panel of conformationally restricted analogues of GABA and muscimol that show differential potencies and efficacies at GABAA versus GABAA‐ρ receptors (Figure 1B). Known agonists at GABAA receptors that displayed no current responses at maximal doses (3 mM) in either EXP‐1 or LGC‐35 expressing oocytes were P4S, IMA and THIP. The muscimol analogue isonipecotic acid (3 mM) showed <5% activity at EXP‐1 and LGC‐35 and was not further characterized. Compounds that exhibited initial agonist activity were further characterized by dose–response relationships. The rank order of potency for EXP‐1 was, TACA > GABA > DAVA > ZAPA > isoguvacine > muscimol > β‐alanine (Figure 2A, Table 1). The rank efficacy order for EXP‐1 was GABA > DAVA > TACA > muscimol > ZAPA > β‐alanine > isoguvacine (Figure 2B, Table 1). TACA was as potent as GABA and demonstrated strong partial agonist activity at EXP‐1. DAVA was the only compound that showed full agonist activity at EXP‐1 but was slightly less potent than GABA. All tested agonists produced fast‐desensitizing currents that were similar to those elicited by GABA (Figure 2C). For LGC‐35, the rank order of potency was similar to EXP‐1, GABA > TACA > ZAPA > DAVA > isoguvacine > muscimol > β‐alanine (Figure 3A, Table 1). The rank order of efficacy for LGC‐35 was GABA, TACA, ZAPA > DAVA > muscimol > β‐alanine > isoguvacine (Figure 3B, Table 1). Maximal agonist concentrations produced slow‐desensitizing currents that were similar to GABA responses (Figure 3C). The agonist profile of LGC‐35 differed from that of EXP‐1 in that both TACA and ZAPA showed full agonist activity, but their potencies were threefold and fivefold decreased compared with GABA (Table 1). Taken together, these data demonstrate that the agonist profiles of EXP‐1 and LGC‐35 more closely resemble GABAA‐ρ receptors.
Compared with inhibitory nematode UNC‐49 GABA receptors, EXP‐1 and LGC‐35 exhibit a unique agonist profile. First, IMA (94% efficacy) and isonipecotic acid (81% efficacy) are full and strong partial agonists at UNC‐49, respectively, but fail to activate either EXP‐1 and LGC‐35 (Kaji et al., 2015). Second, isoguvacine is a strong partial agonist at UNC‐49 (86% efficacy) but a very weak partial agonist at EXP‐1 and LGC‐35 (Table 1). Third, DAVA is a very weak partial agonist at UNC‐49 (31% efficacy) but is a full agonist at EXP‐1 and strong partial agonist at LGC‐35 (Table 1). Fourth, ZAPA is an extremely weak partial agonist at UNC‐49 (11% efficacy) (Kaji et al., 2015) but exhibits differential activity as a partial agonist at EXP‐1 (56% efficacy) and full agonist at LGC‐35. Together, these data suggest that excitatory and inhibitory GABA neurotransmission in C. elegans may be selectively activated using distinct GABA mimetics.
Antagonist profiling of EXP‐1 and LGC‐35
EXP‐1 is highly resistant to the orthosteric competitive GABAA antagonist bicuculline and the pore blocker picrotoxin (Beg and Jorgensen, 2003). The pore domain of EXP‐1 is functionally analogous to the cation‐conducting members of the ‘Cys‐loop’ family, yet EXP‐1 is also highly resistant to the use‐dependent nicotinic acetylcholine receptor blocker mecamylamine. Co‐application of GABA (10 μM) with increasing concentrations of bicuculline, picrotoxin and mecamylamine corroborated previous data showing that EXP‐1 is strongly resistant to these classical antagonists (Figure 4A–C). As the agonist profile of EXP‐1 and LGC‐35 more closely resembles GABAA‐ρ receptors, we tested the inhibitory effects of the selective and competitive GABAA‐ρ antagonist TPMPA. Current amplitudes of GABA (10 μM) were dose‐dependently inhibited by increasing concentrations of TPMPA (Figure 4, IC50 = 172 ± 7 μM). THIP (gaboxadol) is widely used as a high efficacy partial agonist at GABAA receptors but exhibits antagonist activity at GABAA‐ρ receptors (Krogsgaard‐Larsen et al., 2002). The lack of agonist activity at EXP‐1 and LGC‐35 suggested that THIP may be an antagonist at these receptors. Application of increasing concentrations of THIP dose‐dependently decreased GABA currents, but inhibition was no greater than bicuculline, picrotoxin or mecamylamine, which all reduced GABA (10 μM) currents by ~50% (Figure 4A–C).
Figure 4.
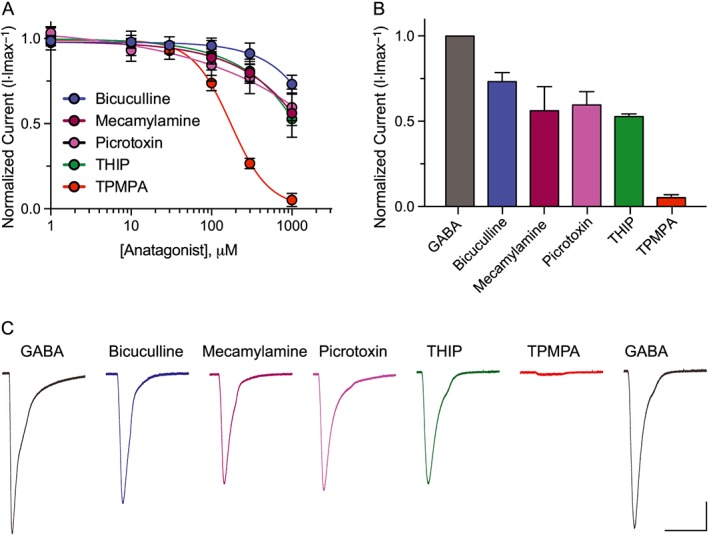
EXP‐1 is resistant to ionotropic GABA receptor antagonists. (A) Inhibitor dose–response curves were generated by applying GABA (10 μM) alone followed by GABA plus increasing concentrations of antagonist (10 μM GABA + 1–1000 μM antagonist). TPMPA was the only compound that potently inhibited EXP‐1 GABA‐induced currents (IC50 = 172 ± 7 μM). Responses were normalized to maximal GABA (10 μM) responses. Each data point represents the mean ± SEM with n ≥ 8 and N = 3. (B) Bar graph illustrating the efficacy of compounds tested (10 μM GABA + 1 mM antagonist) relative to maximal GABA only responses (10 μM). Each bar represents the mean ± SEM with n ≥ 8 and N = 3. (C) Representative current responses from an EXP‐1 expressing oocyte with each compound tested in series at maximal dose (10 μM GABA + 1 mM antagonist). GABA responsiveness fully recovers (2 min wash) after complete TPMPA inhibition. Scale bar = 0.5 μA, 10 s.
LGC‐35 was recently demonstrated to be an excitatory GABA receptor in C. elegans, but the receptor's pharmacological profile remains completely unknown (Jobson et al., 2015). We tested a panel of antagonists against LGC‐35. The antagonist profile of LGC‐35 was similar to EXP‐1 with some notable differences. First, bicuculline, picrotoxin and THIP all showed relatively similar antagonist activity at LGC‐35, with each compound capable of blocking GABA (10 μM) currents by ~75% (Figure 5A, B). Compared with EXP‐1, these antagonists were not only more efficacious at blocking GABA‐induced currents but also exhibited greater potency. Interestingly, mecamylamine showed no difference in its antagonism at LGC‐35, compared with EXP‐1. To further probe the pore‐forming domain, we applied known pore blockers of ionotropic GABA receptors to EXP‐1 and LGC‐35 expressing oocytes. The insecticide dieldrin, β‐lactam antibiotic penicillin G, and widely used pro‐convulsant pentylenetetrazol all failed to block GABA‐induced currents (data not shown). Taken together, these data suggest that the nearly identical M2 pore forming domain (85% identity, 100% similarity) shared between EXP‐1 and LGC‐35 is distantly related to other anion‐ and cation‐conducting pores of the ‘Cys‐loop’ superfamily. Lastly, TPMPA was the only compound tested that was able to fully block LGC‐35 GABA‐induced currents (Figure 5, IC50 = 61 ± 4 μM). From a pharmacological perspective, the antagonist and agonist profiles suggest that EXP‐1 and LGC‐35 more closely resemble the GABAA‐ρ receptor subfamily.
Figure 5.
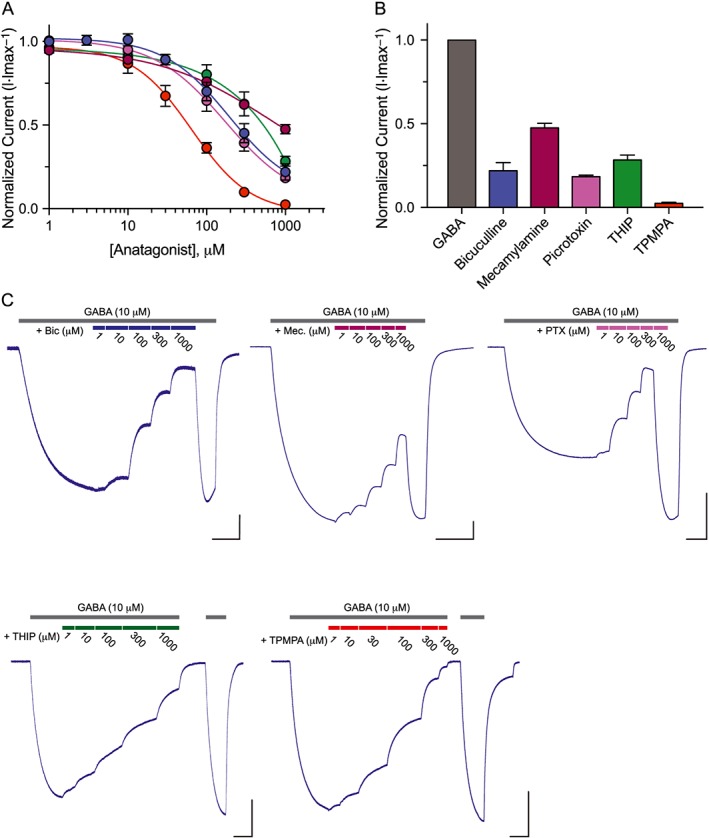
LGC‐35 is sensitive to ionotropic GABA receptor antagonists. (A) Inhibitor dose–response curves were generated by applying GABA (10 μM) alone followed by GABA plus increasing concentrations of antagonist (10 μM GABA + 1–1000 μM antagonist). TPMPA was the most potent inhibitor of LGC‐35 GABA‐induced current (IC50 = 61 ± 4 μM). Responses were normalized to maximal GABA only responses. Each data point represents the mean ± SEM with n ≥ 7 and N = 3. (B) Bar graph illustrating the efficacy of compounds tested (10 μM GABA + 1 mM antagonist) relative to maximal GABA only responses (10 μM). Each bar represents the mean ± SEM with n ≥ 7 and N = 3. (C) Representative current responses of individual LGC‐35 expressing oocytes. GABA (10 μM) was applied until a steady‐state non‐desensitizing plateau was reached. Subsequently, increasing concentrations of antagonist (1–1000 μM antagonist) were applied in series in the presence of constant GABA (10 μM). The grey bars above each trace denote GABA application, and the coloured bars illustrate antagonist concentration co‐applications. GABA responsiveness fully recovers after the highest antagonist concentration block. Scale bar = 1 μA, 30 s.
Homology modelling of EXP‐1 and LGC‐35
The crystal structure of the human homopentameric β3 GABAA receptor was used as a template for homology modelling of the nematode EXP‐1 and LGC‐35 receptors (Miller and Aricescu, 2014). GABA docked into the highly conserved aromatic‐rich binding pocket that is located between the ‘principal’ and ‘complementary’ subunit interface (Figure 6A, E). In EXP‐1, the amino group of GABA is positioned to hydrogen bond with the side chain of Glu250 (loop B), the backbone carbonyl groups of Ser251 and Tyr252 (both loop B), and have potential cation–π interactions with Tyr252 and Tyr300 (loop C), similar to the binding mode observed in the human GABA receptor (Figures 1A, 6A) (Miller and Aricescu, 2014). GABA also docked in a highly conserved manner in the LGC‐35 model (Figures 1A, 6E). In both models, the agonists TACA, DAVA and β‐alanine docked with their amine nitrogen in a similar pose to that observed for GABA (Figure 6B–D, F–H). The binding energies (ΔG) for GABA, TACA, DAVA and β‐alanine were −4.2, −4.4, −4.4 and −3.5 kcal·mol−1 for EXP‐1 and −4.3, −4.4, −4.8 and −3.6 kcal·mol−1 for LGC‐35 respectively. One of the major differences between the models is that a lysine residue, Lys245 (loop F), is located in the proximity of ligands in LGC‐35, as opposed to a Glu272 (loop F) in the corresponding position in EXP‐1 (Figures 1A, 6E–H). The resulting charge reversal orients the ligand carboxyl group in closer proximity to Lys245, as opposed to Arg153 (loop D) in EXP‐1. Finally, compared with the other agonists, β‐alanine fails to extend across the binding pocket, losing energetically favourable electrostatic interactions with ‘complementary’ subunit residues, which is likely to reduce binding energy and contributes to the weak partial agonist activity.
Figure 6.
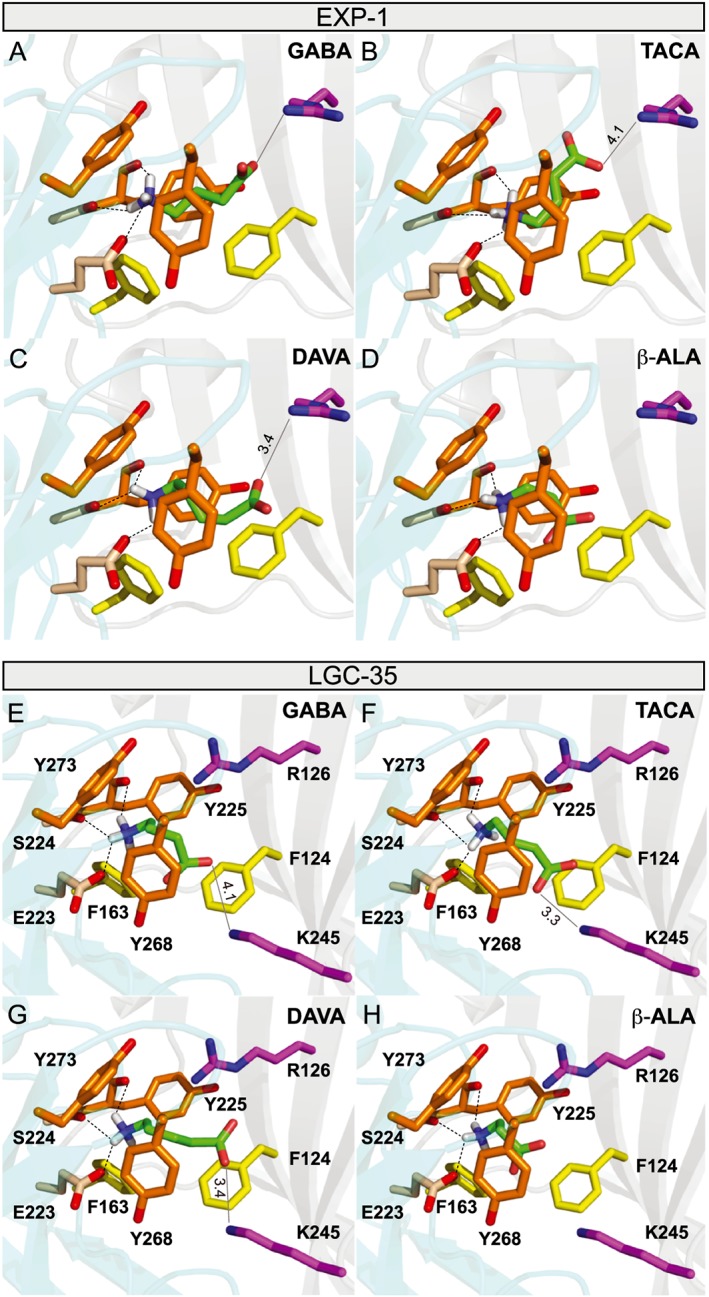
Homology models of EXP‐1 and LGC‐35. (A–D) Molecular modelling of the binding modes of: (A) GABA, (B) TACA, (C) DAVA and (D) β‐alanine in the homology model of EXP‐1. The homology model of EXP‐1 was constructed based on the crystal structure of human GABA receptor, and the ligands were docked to the model with Autodock Vina software as described in the Methods section. Ligands are coloured green with oxygen labelled red and nitrogen blue. Dotted lines represent potential hydrogen bonds between ligand and receptor residues. Solid light grey line represents potential electrostatic interactions of the ligand carboxyl group with Arg126. Labelled distances are in angstroms. Unlabelled potential hydrogen bonds were less than 3.1 Å. Residues were numbered beginning at the start methionine. (E–H) Molecular modelling of the binding modes of: (A) GABA, (B) TACA, (C) DAVA and (D) β‐alanine in the homology model of LGC‐35. The homology model of EXP‐1 was constructed based on the crystal structure of human GABA receptor, and the ligands were docked to the model with Autodock Vina software as described in the Methods section. Ligands are coloured green with oxygen labelled red and nitrogen blue. Dotted lines represent potential hydrogen bonds between ligand and receptor residues. Solid light grey line represents potential electrostatic interactions of the ligand carboxyl group with Lys 245. Labelled distances are in angstroms. Unlabelled potential hydrogen bonds were less than 3.2 Å. Residues were numbered beginning at the start methionine.
Neurosteroids do not modulate EXP‐1 and LGC‐35
Neuroactive steroids, metabolites of the stress hormone corticosterone and progesterone, can potently modulate (low nanomolar concentration) or directly activate (micromolar concentration) vertebrate and invertebrate ionotropic GABA receptors (Lambert et al., 2003). The endogenous neurosteroid pregnanalone and the synthetic neurosteroid alphaxalone potently potentiate mammalian GABAA receptors. In contrast, vertebrate GABAA‐ρ receptors are differentially modulated by these neurosteroids, where alphaxalone potentiates, but pregnanalone inhibits GABA‐induced currents (Morris et al., 1999). We tested if neurosteroids can modulate or directly activate EXP‐1 and LGC‐35. In the absence of GABA, application of high concentrations of pregnanalone (30 μM) or alphaxalone (100 μM) failed to directly activate either receptor, demonstrating that these neurosteroids have no agonist‐like activity at EXP‐1 or LGC‐35. However, co‐application of increasing concentrations of alphaxalone and pregnanalone with GABA weakly inhibited EXP‐1 and LGC‐35 GABA‐induced currents, when compared with GABA alone (Figure 7A, B). Although weak, the neurosteroid inhibition of LGC‐35 was markedly increased compared with EXP‐1 (Figure 7C, D). Like EXP‐1 and LGC‐35, the inhibitory C. elegans GABAA receptor UNC‐49 is only slightly inhibited by alphaxalone but can be completely blocked by pregnanalone (Bamber et al., 2003). These data demonstrate that the site for neuroactive modulation is likely to be unique in that EXP‐1 and LGC‐35 receptors resemble neither the modulatory properties of GABAA, GABAA‐ρ or nematode UNC‐49 receptors.
Figure 7.
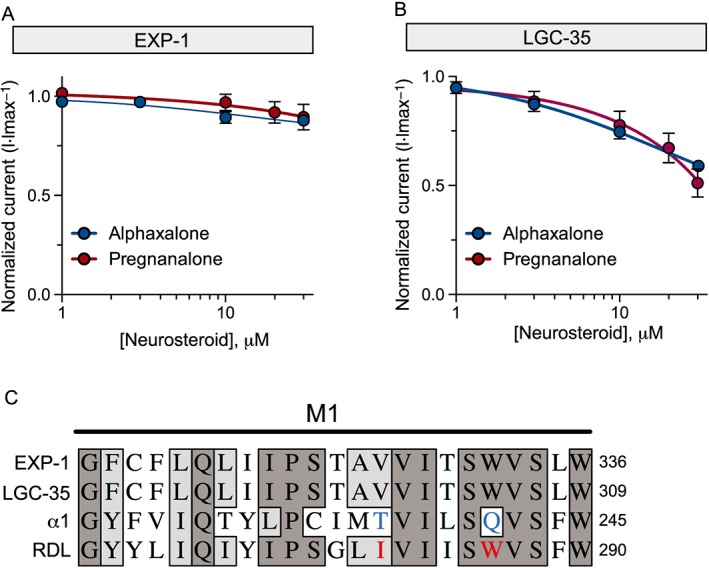
EXP‐1 and LGC‐35 are not modulated by neurosteroids. (A) EXP‐1 neurosteroid dose–response curves were generated by applying GABA (10 μM) alone or GABA plus increasing concentrations of neurosteroids (10 μM GABA + alphaxalone = 1, 3, 10 and 30 μM; pregnanalone = 1, 3, 10, 20 and 30 μM). Responses were normalized to maximal GABA only responses. Each data point represents the mean ± SEM with n ≥ 6 and N = 2. (B) LGC‐35 neurosteroid dose–response curves were generated by applying GABA (10 μM) alone or with increasing concentrations of neurosteroids (alphaxalone = 1, 3, 10, 30 and 100 μM; pregnanalone = 1, 3, 10, 20 and 30 μM). Responses were normalized to maximal GABA only responses. Each data point represents the mean ± SEM with n ≥ 7 and N = 2. (C) Comparative sequence alignment of transmembrane domain (M1), highlighting key residues that confer differential neurosteroid sensitivity (blue) and resistance (red) in the human α1 GABAA subunit and fly RDL subunit respectively. EXP‐1 and LGC‐35 contain a similar valine and an identical asparagine present in the neurosteroid resistant RDL receptor. Dark grey shading marks amino acid identity and light grey shading identifies similarity.
LGC‐35 is selectively potentiated by ethanol
Anaesthetic drugs, including ethanol, can modulate cellular and network signal transduction by altering the function of a wide variety of ion channels, transporters, receptors and enzymes (Lobo and Harris, 2008). However, the contribution of these molecular targets to the intoxicating and behavioural effects of ethanol remain unclear. GABAA receptors are thought to be key targets of ethanol action in the brain, and subunit composition is a critical determinant that mediates the potentiating effect of alcohol in heteropentameric GABAA receptors (Mihic et al., 1997). In contrast, the single subunit homopentameric ρ1 receptor is inhibited by alcohol. At a molecular level, key residues in transmembrane domains 2 and 3 (M2 and M3) have been shown to mediate the potentiating or inhibitory effects of alcohol in GABAA versus GABAA‐ρ receptors respectively. The 15′ serine residue (S270) in M2 and alanine residue (A291) in M3 of the α1 GABAA receptor subunit were shown to critically regulate ethanol potentiation (Figure 8F) (Mihic et al., 1997; Ueno et al., 1999). In the ρ1 receptor, the analogous isoleucine residue in M2 (I307) and asparagine residue in M3 (W328) confer resistance against the potentiating effect of ethanol (Figure 8F) (Mihic et al., 1997; Borghese et al., 2016). We used these distinct structural properties to determine if EXP‐1 and LGC‐35 are modulated by ethanol. Ethanol did not significantly potentiate or inhibit EXP‐1 current responses (expressed as % change of 10 μM GABA responses) at any concentration tested (Figure 8A). Although lower doses of ethanol (10 and 30 mM) did not significantly modulate LGC‐35 current responses, higher doses (100, 200 and 500 mM) did significantly potentiate LGC‐35 current response amplitudes (Figure 8C). Stepwise ramps of increasing concentrations demonstrate the potentiating effect of alcohol in the presence of constant GABA exposure (Figure 8E).
Figure 8.
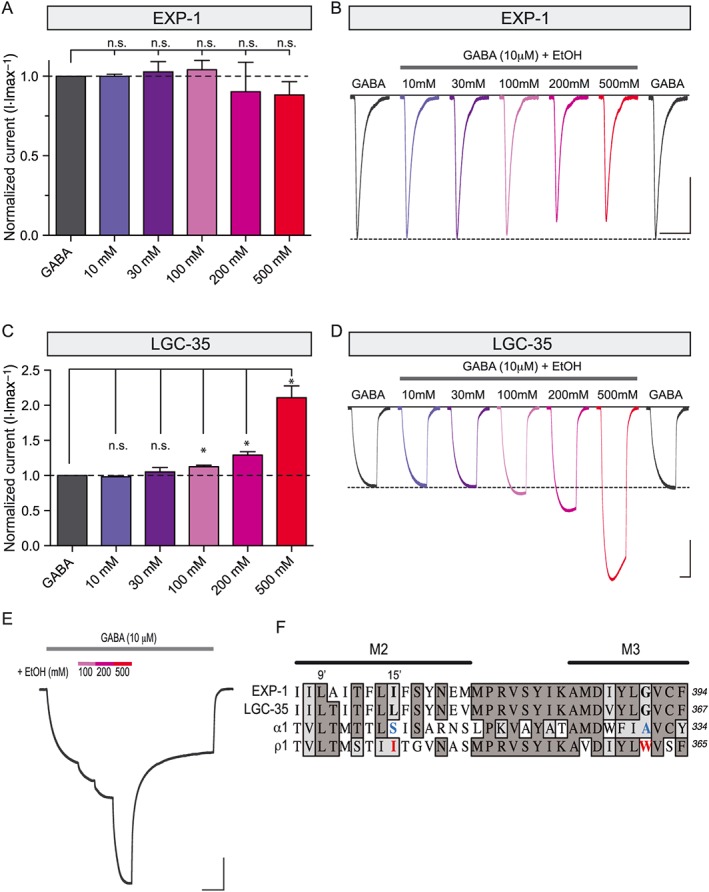
LGC‐35 is selectively potentiated by ethanol. (A) EXP‐1 is neither significantly potentiated nor inhibited by ethanol. Current responses of co‐applied ethanol (10 μM GABA + 10–500 mM ethanol) were normalized relative to maximal GABA only responses (10 μM). For each oocyte, an initial stable GABA response was first achieved. Subsequently, co‐application of 10 μM GABA + increasing concentrations of ethanol were bath applied for 5 s, followed by a 2 min wash. Each bar represents the mean ± SEM with n ≥ 8 and N = 3. (B) Representative current responses from an EXP‐1 expressing oocyte with co‐application of increasing concentrations of ethanol. Scale bar = 0.5 μA, 10 s. (C) LGC‐35 is potentiated by high concentrations of ethanol. Current responses of co‐applied ethanol (10 μM GABA + 10–500 mM ethanol) were normalized relative to maximal GABA only responses (10 μM). For each oocyte, an initial stable GABA response was first achieved. Subsequently, co‐application of 10 μM GABA + increasing concentrations of ethanol were bath applied until a plateau was reached, followed by a 2 min wash. LGC‐35 current responses are significantly potentiated by high concentrations of ethanol (100–500 mM). Each bar represents the mean ± SEM with n ≥ 8 and N = 3. n.s, not significant (P ≥ 0.05), *P < 0.05; One‐way ANOVA with Holm–Sidak post hoc test. (D) Representative current responses from an LGC‐35 expressing oocyte with co‐application of increasing concentrations of ethanol. Note, GABA responsiveness is unchanged compared with initial GABA‐only responses after increasing series of ethanol exposure. Scale bar = 0.5 μA, 30 s. (E) Representative current responses from an LGC‐35 expressing oocyte. GABA (10 μM) was applied until a steady‐state non‐desensitizing plateau was reached. Subsequently, increasing concentrations of ethanol (100–500 mM) were applied in series in the presence of constant GABA (10 μM). The grey bars above each trace denote constant GABA exposure, and the coloured bars illustrate ethanol concentration co‐applications. Scale bar = 1 μA, 60 s. (F) Comparative sequence alignment of transmembrane domains 2 and 3 (M2 and M3), highlighting key residues that confer differential ethanol sensitivity (blue) and resistance (red) in the human α1 and ρ1 receptor subunits respectively. Dark grey shading marks amino acid identity, and light grey shading shows similarity.
Sequence alignments reveal that EXP‐1 contains an identical isoleucine at the 15′ position (I357), which is a critical determinant of alcohol resistance in the ρ1 receptor (Figure 8E). Interestingly, LGC‐35 has a similar leucine at the 15′ position (L330), yet the receptor is significantly potentiated by high concentrations of ethanol (Figure 8E). Both receptors contain a glycine residue in the M3 domain, which is similar to the alanine residue (A291) present in the α1 GABAA receptor subunit that confers alcohol sensitivity but markedly different from the asparagine residue (W328) in the ρ1 subunit that confers alcohol resistance (Figure 8E). Although the potentiating concentrations of ethanol are not likely to be physiologically relevant, these data reveal that the differential effects of ethanol on EXP‐1 and LGC‐35 GABA‐induced current are likely to be distinct from the previously identified residues that confer alcohol sensitivity to ionotropic GABA receptors. Together, these data may provide new insights into molecular determinants that contribute to alcohol modulation in ionotropic GABA receptors.
Discussion
EXP‐1 and LGC‐35 compose the excitatory GABA receptor family in C. elegans and play important roles in distinct neuromuscular contractions that regulate defecation and locomotion respectively (Beg and Jorgensen, 2003; Jobson et al., 2015). Since their initial cloning and characterization, these receptors have been loosely classified as belonging to the GABAA receptor subtype family. Using a broad panel of pharmacological agonists, antagonists and modulators, we demonstrate this unique receptor family more closely resembles the GABAA‐ρ family. We discuss our findings in regard to the structure, function and pharmacology of ‘Cys‐loop’ ligand‐gated ion channels.
GABA pharmacophores suggest EXP‐1 and LGC‐35 are activated by conformationally extended agonists
Decades of elegant research has demonstrated that vertebrate GABAA and GABAA‐ρ receptors are differentially sensitive to conformationally restricted analogues of GABA (Krogsgaard‐Larsen et al., 2002; Johnston, 2005; Johnston et al., 2010). Specifically, GABAA receptors tend to be preferentially activated by partially folded forms of GABA; whereas, GABAA‐ρ receptors are activated by extended or near planar forms of GABA. Using a collection of conformationally restricted analogues, we conclude that both EXP‐1 and LGC‐35 more closely resemble the GABAA‐ρ family. We draw several conclusions based on our data. First, the homo‐oligomeric assembly of EXP‐1 and LGC‐35 supports their classification in the GABAA‐ρ family. Unlike GABAA‐ρ receptors, the vast majority of GABAA receptors are hetero‐oligomers composed of three distinct subunits, with the (α1)2(β2)2(γ2)1 combination being the most abundant in the human brain (Sigel and Steinmann, 2012). Moreover, EXP‐1 and LGC‐35 are not expressed in the same cells in vivo further supporting that these receptors do not act as hetero‐oligomers (Beg and Jorgensen, 2003; Jobson et al., 2015). Second, dose–response relationships reveal that most of the partially folded GABA analogues tested either showed no agonist activity (nipecotic acid, IMA and P4S) or were weak partial agonists (muscimol and isoguvacine) at both EXP‐1 and LGC‐35. In contrast, the extended and non‐aromatic analogues (TACA and DAVA) showed both higher potency and efficacy at EXP‐1 and LGC‐35. Third, carbon backbone length appears to be a key determinant of agonist potency and efficacy at EXP‐1 and LGC‐35. More specifically, increasing the carbon backbone length by one (DAVA) right shifts the potency in both EXP‐1 and LGC‐35 and decreases efficacy in LGC‐35, but not in EXP‐1. Reduction of the carbon backbone length by one (β‐alanine) severely reduced both potency and efficacy, and reduction by two carbons (glycine) yields no activity at either receptor. Supporting these observations, TACA, which shares the same carbon backbone length as GABA, was the most potent and efficacious compound tested at both receptors. A double bond between carbons 2 and 3 hold TACA in an extended conformation, supporting a model in which EXP‐1 and LGC‐35 are preferentially activated by extended ligands as opposed to folded forms. Fourth, although the pharmacological profile of EXP‐1 and LGC‐35 shares a resemblance to GABAA‐ρ receptors, there are notable exceptions that set these receptors apart as a family. ZAPA is a potent and full agonist at GABAA receptors but is an antagonist at GABAA‐ρ receptors. We demonstrate that ZAPA is an agonist at EXP‐1 and LGC‐35 and not an antagonist as would have been predicted based on our pharmacological profiling. These data demonstrate that EXP‐1 and LGC‐35 exhibit a unique and hybrid pharmacological profile. Comparison of the ligand‐binding loops from EXP‐1 and LGC‐35 with ρ1 subunit demonstrates high conservation in residues implicated in the orthosteric binding site. Furthermore, EXP‐1 and LGC‐35 contain a single non‐conserved amino acid difference in loop C. Whether or not this single change accounts for the differential agonist activity at EXP‐1 and LGC‐35 remains to be determined.
EXP‐1 and LGC‐35 exhibit hybrid antagonist pharmacology
High affinity for GABA, bicuculline resistance and picrotoxin sensitivity are characteristic features of GABAA‐ρ receptors (Johnston et al., 2010). From a pharmacological perspective, the agonist data suggest that EXP‐1 and LGC‐35 more closely resemble GABAA‐ρ receptors. In accordance with the agonist profile, both receptors were highly resistant to bicuculline as expected but were also resistant to picrotoxin block. We previously characterized the molecular mechanisms of ion selectivity in the pore forming domain of both EXP‐1 and LGC‐35 (Beg and Jorgensen, 2003; Jobson et al., 2015). Here, we investigated this unusual pore domain using a panel of well‐characterized channel blockers at ‘Cys‐loop’ ligand‐gated ion channels. We failed to identify a single compound thought to act via pore blockade that could effectively inhibit GABA‐induced currents. These data emphasize that the pore‐forming domain of EXP‐1 and LGC‐35 is highly divergent from any known GABAA or GABAA‐ρ receptors.
Supporting their GABAA‐ρ‐like characteristics, both EXP‐1 and LGC‐35 were dose‐dependently inhibited by the selective GABAA‐ρ antagonist TPMPA. Furthermore, THIP, which differentially acts as a GABAA receptor agonist but a GABAA‐ρ antagonist, exhibited inhibitory activity at EXP‐1 and LGC‐35. These data suggest that EXP‐1 and LGC‐35 more closely resemble GABAA‐ρ receptors, although they exhibit a hybrid pharmacology that is not observed in either GABAA or GABAA‐ρ receptors.
Molecular profiling of neurosteroid resistance
GABAA receptors can be potently modulated by endogenous neurosteroids (Belelli and Lambert, 2005). Low nanomolar concentrations can potentiate GABA‐induced currents (Zhu and Vicini, 1997; Belelli and Herd, 2003; Stell et al., 2003), and micromolar concentrations can directly activate receptors (Majewska et al., 1986). Pregnanalone, an endogenous progesterone metabolite, and the synthetic neurosteroid alphaxalone strongly enhance GABAA‐induced currents. In contrast, GABAA‐ρ receptors are differentially modulated by neurosteroids, where pregnanalone inhibits and alphaxalone potentiates ρ1 receptors (Morris et al., 1999). In comparison to GABAA receptors, which are enhanced in the nanomolar range, neurosteroid modulation of GABAA‐ρ receptors occur in the high micromolar range. We found that pregnanalone and alphaxalone exhibited almost no modulatory activity at EXP‐1 (Figure 7A) but weakly inhibited LGC‐35 only in the high micromolar range (Figure 7B). Previous studies have shown that the inhibitory nematode GABA receptor UNC‐49 is strongly inhibited by pregnanalone but is not modulated by alphaxalone (Bamber et al., 2003); whereas, the fly GABA receptor RDL is weakly potentiated by neurosteroids (Chen et al., 1994). Chimeric studies using fly RDL and mouse α1 subunits identified two discrete residues in transmembrane domain 1 that differentially contribute to direct neurosteroid activation and potentiation of GABA receptors (Hosie et al., 2006). In the α1 subunit, a threonine (T236) in the transmembrane domain 1 was shown to be a major determinant for direct neurosteroid activation (Figure 7C). Substitution with an isoleucine present in RDL (T236I) markedly attenuated direct neurosteroid receptor activation without altering potentiation. Using the same strategy, a glutamine (Q241) in transmembrane 1 was found to be critically important for neurosteroid potentiation (Figure 7C). Substitution with an asparagine present in RDL at this position (Q241W) ablated neurosteroid potentiation. Comparative sequence alignments with RDL reveal that EXP‐1 and LGC‐35 contain a similar valine at that analogous position 236 and an identical asparagine at analogous position 241, which are likely to account for the lack of neurosteroid activation or potentiation (Figure 7C). Taken together, these data demonstrate that EXP‐1 and LGC‐35 exhibit a neurosteroid profile that does not resemble vertebrate GABAA or GABAA‐ρ receptors, or the inhibitory nematode GABA receptor UNC‐49.
Does LGC‐35 play a role in ethanol‐mediated behaviours in C. elegans?
Ethanol produces profound behavioural effects in both humans and C. elegans at equivalent concentrations (McIntire, 2010). However, the relevant molecular targets and biochemical pathways that mediate these effects remain unclear, as ethanol modulates a wide variety of receptors, ion channels and enzymes. The differential functional effect of ethanol on EXP‐1 and LGC‐35 is intriguing given the almost identical sequence conservation shared between the two proteins. Ethanol had no effect on EXP‐1 GABA‐induced currents but significantly potentiated LGC‐35 current responses at higher doses (100–500 mM). From a behavioural perspective, acute exposure to ethanol produces dose‐dependent changes in locomotor behaviours in C. elegans (McIntire, 2010). Increasing concentrations of ethanol flatten the amplitude of body‐bending and decreases speed of movement (Davies et al., 2003). We recently reported that LGC‐35 regulates locomotor body bending and movement speed in C. elegans (Jobson et al., 2015). Specifically, deletion of LGC‐35 results in exaggerated body bending and increases animals' speed of movement. The specific ethanol potentiation of LGC‐35 coupled with our cellular, molecular and behavioural data suggests that LGC‐35 may be a relevant effector of ethanol's action in C. elegans; however, this remains to be tested.
In conclusion, the results from our studies expand the molecular repertoire for studying ionotropic GABA receptor structure–function relationships. The unique pharmacological profiles of EXP‐1 and LGC‐35 provide novel frameworks to differentially probe agonists, antagonists and allosteric modulation at GABAA and GABAA‐ρ receptors. Furthermore, the homo‐oligomeric assembly simplifies identifying critical amino acid residues and motifs, as the binding interfaces and ligand recognition sites are identical in each receptor complex. The highly conserved ligand‐binding loops suggest that the orthosteric GABA binding site is similar to vertebrate GABA receptors, but the hybrid pharmacology and differential sensitivities of EXP‐1 and LGC‐35 may provide a new tool for anthelmintic drug discovery and for the rationale design of GABA mimetics.
Author contributions
G.C.B.N, H.Z. and A.A.B designed the research study and wrote the manuscript. G.C.B.N., A.J., R.W., H.Z and A.A.B performed the experiments and analysed the data. H.Z. performed the homology modelling and docking. A.J. and R.W. prepared constructs, isolated and injected oocytes. G.C.B.N, A.J., R.W., H.Z. and A.A.B. contributed to the discussion and review of the manuscript. All authors read and approved the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
A.A.B is supported by grants from the Alfred P. Sloan Foundation, the Muscular Dystrophy Association (MDA382300), and the National Institute of Neurological Disease and Stroke (NS094678). We kindly thank Xenopus1 (Ann Arbor, MI) for generously providing X. laevis oocytes for this study.
Nicholl, G. C. B. , Jawad, A. K. , Weymouth, R. , Zhang, H. , and Beg, A. A. (2017) Pharmacological characterization of the excitatory ‘Cys‐loop’ GABA receptor family in Caenorhabditis elegans . British Journal of Pharmacology, 174: 781–795. doi: 10.1111/bph.13736.
References
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamber BA, Beg AA, Twyman RE, Jorgensen EM (1999). The Caenorhabditis elegans UNC‐49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J Neurosci 19: 5348–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamber BA, Twyman RE, Jorgensen EM (2003). Pharmacological characterization of the homomeric and heteromeric UNC‐49 GABA receptors in C. elegans . Br J Pharmacol 138: 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Jorgensen EM (2003). EXP‐1 is an excitatory GABA‐gated cation channel. Nat Neurosci 6: 1145–1152. [DOI] [PubMed] [Google Scholar]
- Belelli D, Herd MB (2003). The contraceptive agent Provera enhances GABA(A) receptor‐mediated inhibitory neurotransmission in the rat hippocampus: evidence for endogenous neurosteroids? J Neurosci 23: 10013–10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ (2005). Neurosteroids: endogenous regulators of the GABA(A) receptor. Nat Rev Neurosci 6: 565–575. [DOI] [PubMed] [Google Scholar]
- Borghese CM, Ruiz CI, Lee US, Cullins MA, Bertaccini EJ, Trudell JR et al. (2016). Identification of an inhibitory alcohol binding site in GABAA rho1 receptors. ACS Chem Nerosci 7: 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bormann J (2000). The 'ABC' of GABA receptors. Trends Pharmacol Sci 21: 16–19. [DOI] [PubMed] [Google Scholar]
- Chebib M, Johnston GA (2000). GABA‐activated ligand gated ion channels: medicinal chemistry and molecular biology. J Med Chem 43: 1427–1447. [DOI] [PubMed] [Google Scholar]
- Chen R, Belelli D, Lambert JJ, Peters JA, Reyes A, Lan NC (1994). Cloning and functional expression of a Drosophila gamma‐aminobutyric acid receptor. Proc Natl Acad Sci U S A 91: 6069–6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AG, Pierce‐Shimomura JT, Kim H, VanHoven MK, Thiele TR, Bonci A et al. (2003). A central role of the BK potassium channel in behavioral responses to ethanol in C. elegans . Cell 115: 655–666. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Sattelle DB (1996). Agonist pharmacology of two Drosophila GABA receptor splice variants. Br J Pharmacol 119: 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HM, Smart TG (2006). Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444: 486–489. [DOI] [PubMed] [Google Scholar]
- Jobson MA, Valdez CM, Gardner J, Garcia LR, Jorgensen EM, Beg AA (2015). Spillover transmission is mediated by the excitatory GABA receptor LGC‐35 in C. elegans . J Neurosci 35: 2803–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston GA (2005). GABA(A) receptor channel pharmacology. Curr Pharm Des 11: 1867–1885. [DOI] [PubMed] [Google Scholar]
- Johnston GA (2014). Muscimol as an ionotropic GABA receptor agonist. Neurochem Res 39: 1942–1947. [DOI] [PubMed] [Google Scholar]
- Johnston GA, Chebib M, Hanrahan JR, Mewett KN (2010). Neurochemicals for the investigation of GABA(C) receptors. Neurochem Res 35: 1970–1977. [DOI] [PubMed] [Google Scholar]
- Kaji MD, Kwaka A, Callanan MK, Nusrat H, Desaulniers JP, Forrester SG (2015). A molecular characterization of the agonist binding site of a nematode cys‐loop GABA receptor. Br J Pharmacol 172: 3737–3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogsgaard‐Larsen P, Frolund B, Liljefors T (2002). Specific GABA(A) agonists and partial agonists. Chem Rec 2: 419–430. [DOI] [PubMed] [Google Scholar]
- Kusama T, Spivak CE, Whiting P, Dawson VL, Schaeffer JC, Uhl GR (1993). Pharmacology of GABA rho 1 and GABA alpha/beta receptors expressed in Xenopus oocytes and COS cells. Br J Pharmacol 109: 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JJ, Belelli D, Peden DR, Vardy AW, Peters JA (2003). Neurosteroid modulation of GABAA receptors. Prog Neurobiol 71: 67–80. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM (1996). AQUA and PROCHECK‐NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8: 477–486. [DOI] [PubMed] [Google Scholar]
- Lobo IA, Harris RA (2008). GABA(A) receptors and alcohol. Pharmacol Biochem Behav 90: 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM (1986). Steroid hormone metabolites are barbiturate‐like modulators of the GABA receptor. Science 232: 1004–1007. [DOI] [PubMed] [Google Scholar]
- Marti‐Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A (2000). Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 29: 291–325. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire SL (2010). Ethanol. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.40.1, http://www.wormbook.org. [Google Scholar]
- Michels G, Moss SJ (2007). GABAA receptors: properties and trafficking. Crit Rev Biochem Mol Biol 42: 3–14. [DOI] [PubMed] [Google Scholar]
- Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE et al. (1997). Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature 389: 385–389. [DOI] [PubMed] [Google Scholar]
- Miller PS, Aricescu AR (2014). Crystal structure of a human GABAA receptor. Nature 512: 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KD, Moorefield CN, Amin J (1999). Differential modulation of the gamma‐aminobutyric acid type C receptor by neuroactive steroids. Mol Pharmacol 56: 752–759. [PubMed] [Google Scholar]
- Siddiqui SZ, Brown DD, Rao VT, Forrester SG (2010). An UNC‐49 GABA receptor subunit from the parasitic nematode Haemonchus contortus is associated with enhanced GABA sensitivity in nematode heteromeric channels. J Neurochem 113: 1113–1122. [DOI] [PubMed] [Google Scholar]
- Sigel E, Steinmann ME (2012). Structure, function, and modulation of GABA(A) receptors. J Biol Chem 287: 40224–40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I (2003). Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit‐containing GABAA receptors. Proc Natl Acad Sci U S A 100: 14439–14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Lester HA, Lummis SC (2010). The structural basis of function in Cys‐loop receptors. Q Rev Biophys 43: 449–499. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Wick MJ, Ye Q, Harrison NL, Harris RA (1999). Subunit mutations affect ethanol actions on GABA(A) receptors expressed in Xenopus oocytes. Br J Pharmacol 127: 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward RM, Polenzani L, Miledi R (1993). Characterization of bicuculline/baclofen‐insensitive (rho‐like) gamma‐aminobutyric acid receptors expressed in Xenopus oocytes. II. Pharmacology of gamma‐aminobutyric acidA and gamma‐aminobutyric acidB receptor agonists and antagonists. Mol Pharmacol 43: 609–625. [PubMed] [Google Scholar]
- Zhu WJ, Vicini S (1997). Neurosteroid prolongs GABAA channel deactivation by altering kinetics of desensitized states. J Neurosci 17: 4022–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]