

Abstract
Background and Purpose
The succinate receptor (formerly GPR91 or SUCNR1) is described as a metabolic sensor that may be involved in homeostasis. Notwithstanding its implication in important (patho)physiological processes, the function of succinate receptors has remained ill‐defined because no pharmacological tools were available. We report on the discovery of the first family of potent synthetic agonists.
Experimental Approach
We screened a library of succinate analogues and analysed their activity on succinate receptors. Also, we modelled a pharmacophore and a binding site for this receptor. New agonists were identified based on the information provided by these two approaches. Their activity was studied in various bioassays, including measurement of cAMP levels, [Ca2+]i mobilization, TGF‐α shedding and recruitment of arrestin 3. The in vivo effects of activating succinate receptors with these new agonists was evaluated on rat BP.
Key Results
We identified cis‐epoxysuccinic acid and cis‐1,2‐cyclopropanedicarboxylic acid as agonists with an efficacy similar to that of succinic acid. Interestingly, cis‐epoxysuccinic acid was 10‐ to 20‐fold more potent than succinic acid on succinate receptors. For example, cis‐epoxysuccinic acid reduced cAMP levels with a pEC50 = 5.57 ± 0.02 (EC50 = 2.7 μM), compared with succinate pEC50 = 4.54 ± 0.08 (EC50 = 29 μM). The rank order of potency of the three agonists was the same in all in vitro assays. Both cis‐epoxysuccinic and cis‐1,2‐cyclopropanedicarboxylic acid were as potent as succinate in increasing rat BP.
Conclusions and Implications
We describe new agonists at succinate receptors that should facilitate further research on this understudied receptor.
Abbreviations
- BrSA
bromosuccinic acid
- cCBDA
cis‐1,2‐cyclobutanedicarboxylic acid
- cCPDA
cis‐1,2‐cyclopropanedicarboxylic acid
- cESA
cis‐epoxysuccinic acid
- ClSA
chlorosuccinic acid
- EMA
ethylmalonic acid
- GRK
GPCR kinase
- MA
malonic acid
- mDMSA
meso‐dimethylsuccinic acid
- MMA
methylmalonic acid
- MSA
methylsuccinic acid
- OAA
oxaloacetic acid
- p‐NPP
p‐nitrophenylphosphate
- SDH
succinate dehydrogenase
- tCPDA
trans‐1,2‐cyclopropanedicarboxylic acid
- WT
wild type
Tables of Links
| TARGETS |
|---|
| GPCRs |
| Succinate receptor, GPR91 |
| LIGANDS |
|---|
| Succinic acid |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
The GPCRs constitute the most broadly targeted proteins by drugs in human medicine (Overington et al., 2006). These receptors are characterized by seven transmembrane domains and are implicated in nearly all physiological processes. GPCRs signal through various intracellular partners that affect cell function. Four main families of G proteins have been described, namely, Gi, Gs, Gq/11 and G12/13 (Wettschureck and Offermanns, 2005). Gi and Gs subunits are respectively able to negatively and positively regulate the activity of AC and thus decrease and increase the cAMP levels in cells. Following activation, the receptor is usually desensitized by phosphorylation of intracellular sites by specific GPCR kinases called GRK (Lefkowitz and Shenoy, 2005). Particular scaffold proteins named arrestins (for non‐visual GPCR, β‐arrestins 1 and 2 or arrestins 2 and 3) strengthen the desensitization and generally induce receptor internalization (Lefkowitz and Shenoy, 2005). It was recently proposed that arrestins adopt receptor‐dependent conformation and activate specific signalling pathways in a manner similar to G proteins (Lee et al., 2016; Nuber et al., 2016).
The succinate receptor (formerly GPR91, also termed SUCNR1) is a member of the rhodopsin‐like GPCR family and was initially identified as an orphan receptor (Wittenberger et al., 2001). In a landmark study, He et al., (2004) paired it with its natural ligand succinate (succinic acid). Succinate receptors display some homology with the purinergic receptor family, although they do not bind nucleotide ligands (He et al., 2004). Few studies have addressed the signalling pathways of the succinate receptor. This receptor is coupled to Gi, and its activation negatively modulates cAMP levels (He et al., 2004; Gilissen et al., 2015). In addition, activation of succinate receptors promotes transient [Ca2+]i mobilization (He et al., 2004; Gilissen et al., 2015). Although it has been suggested that Gq was mediating this effect (He et al., 2004; Robben et al., 2009), more recent investigations in native and heterologous systems could not detect Gq coupling and proposed the Gβγ dimer as the protein responsible for PLC‐β activation (Hakak et al., 2009; Hogberg et al., 2011; Sundström et al., 2013; Gilissen et al., 2015). Notwithstanding, there is also the possibility that coupling to Gq is tissue dependent. Upon activation, the succinate receptor induces Pertussis toxin‐sensitive ERK phosphorylation and is rapidly desensitized and/or internalized (He et al., 2004; Hakak et al., 2009; Robben et al., 2009; Gilissen et al., 2015). It is currently not clear if arrestins or phosphorylation of the receptor takes an active part in the process of desensitization/internalization.
Succinate (as succinic acid) is an intermediate of the citric acid (or Krebs) cycle that takes place in the mitochondria. In conditions of oxygen deprivation, succinate may accumulate and be released in the extracellular space. Succinate and its receptor have been linked to several (patho)physiological processes such as hypertension (He et al., 2004; Toma et al., 2008), diabetes and obesity (Sadagopan et al., 2007; Toma et al., 2008; McCreath et al., 2015), activation of the immune system (Rubic et al., 2008), platelet aggregation (Hogberg et al., 2011; Spath et al., 2012) and retinal angiogenesis (Sapieha et al., 2008).
Very few active ligands for succinate receptors have been described. Maleate is a confirmed agonist with lower potency (He et al., 2004; Gilissen et al., 2015). In 2011, Bhuniya et al. (2011) published the discovery of potent antagonists from a high‐throughput screening campaign. There is currently no full pharmacological characterization of the compounds, although radiotracers based on these scaffolds have been described (Klenc et al., 2015). The paucity of pharmacological tools for the receptor restricts current research and precludes a more thorough understanding of succinate receptor function.
The present study reports the discovery and characterization of the first potent and highly efficacious succinate receptor agonists, namely, cis‐epoxysuccinic acid (cESA) and cis‐1,2‐cyclopropanedicarboxylic acid (cCPDA). In addition, the identification of several other succinate receptor ligands led to the definition of a precise pharmacophore for agonistic activity on these receptors. We refined the succinate binding pocket by implementing a homology model and validated it by site‐directed mutagenesis. cESA showed higher potency than succinate in succinate receptor‐mediated [Ca2+]i mobilization, arrestin binding, TGF‐α shedding and depletion of basal cAMP levels. In addition, cESA and cCPDA have no activity on the mitochondrial SDH and can be utilized to specifically assess the impact of succinate receptor activation without interfering with the citric acid cycle. Furthermore, cESA and cCPDA demonstrated activity in vivo on rat BP. Both agonists are commercially available, which may open new possibilities for the characterization of succinate receptors and their validation as a drug target.
Methods
Cell culture
Cells were cultured at 5% CO2 and 37°C in DMEM adjusted to contain 10% FBS (Biochrom AG, Berlin, Germany), 1% penicillin and streptomycin (Lonza, Verviers, Belgium) and 1% l‐glutamine (Lonza).
Site‐directed mutagenesis
WT human succinate receptors with a FLAG epitope at the N‐terminal end has been cloned into pcDNA3.1 vector bearing a neomycin resistance cassette. All mutagenesis was carried out using the Q5 Site‐Directed Mutagenesis Kit (New England Biolabs, Massachusetts, USA), according to the manufacturer's instructions. Stable cell lines have been established for each clone after selection with G418 (600 mg·L−1), and the expression at the membrane has been verified by FACS measurements (see Supporting Information Figure S2 and the Supporting Information).
GloSensor cAMP assay
The assay has been conducted with a protocol previously described (Gilissen et al., 2015). Briefly, HEK293 cells stably expressing cAMP Glosensor with or without stable expression of human succinate receptors were starved for 5 h with 1% FBS, detached and incubated 1 h in the dark at room temperature in assay buffer HBSS (120 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 10 mM HEPES; pH 7.4, 10 mM glucose) containing IBMX (300 μM) and Luciferin (GloSensor reagent; Promega, Madison, Wisconsin, USA). Then, cells were distributed into 96‐well plates (150 000 cells per well, white lumitrac®; Greiner Bio‐One, Kremsmünster, Austria) containing the tested compound at different concentrations. After 1 min agitation at 1200 rpm and 9 min additional incubation, basal luminescence level was recorded by using a microplate luminometer (Fluoroskan Ascent FL equipped with two dispensers, Ascent software version 2.6; Thermo Fisher Scientific).
Intracellular calcium mobilization assay
The assay has been conducted according to previous description (Gilissen et al., 2015). Briefly, cells from a confluent T175 flask were detached and incubated in assay buffer (HBSS: 120 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 10 mM HEPES; pH 7.4, 10 mM glucose) containing 5 mM coelenterazine h (Regis Technologies, Morton Grove, Illinois, USA) and 1.8 mM CaCl2 for 1 h in the dark at 37°C. Luminescence was followed for 20 s (40 measures; 500 ms integration) immediately upon ligand addition. Measurements were acquired with a microplate luminometer (Fluoroskan Ascent FL equipped with two dispensers, Ascent software version 2.6; Thermo Fisher Scientific).
Arrestin complementation assay
Stable cell lines for the measurement of arrestin 3 recruitment by complementation of firefly luciferase have been described previously (Gilissen et al., 2015). Cells in suspension in the buffer (HBSS with 20 mM HEPES, pH 7.4, 10 mM glucose) were incubated into 96‐well plates (105 cells per well) containing the ligands at different concentrations for 10 min at room temperature. Following injection of 50 μM luciferin (Synchem, Felsberg, Germany), luminescence was recorded for 30 min using a high‐sensitivity luminometer (Centro XS3 LB 960, MicroWin 2000 software, equipped with two dispensers; Berthold Technologies, Bad Wilbad, Germany).
TGF‐α shedding assay
The procedure and plasmids have been described previously (Inoue et al., 2012). Briefly, expression vectors (a mixture of 2.5 μg AP‐TGF‐α, 1 μg receptor and 0.5 μg promiscuous Gα protein per 100 mm dish; 24 h before the assay) were transfected in HEK293 cells using 12 μL per 100 mm dish of x‐tremegene 9 (Sigma‐Aldrich). Transfected cells were pelleted by centrifugation and resuspended in PBS, followed by incubation for 10 min at room temperature. After centrifugation, cells were suspended in HBSS containing 5 mM HEPES (pH 7.4) and plated in 90 μL per well (40 000 cells per well) in a 96‐well plate and placed in a 37°C incubator with 5% CO2. Thirty minutes later, 10 μL per well of 10× concentration of compounds was added and incubated for 1 h at 37°C under 5% CO2. Plates were centrifuged, and conditioned medium (80 μL per well) was transferred into another 96‐well plate. A solution of p‐NPP (10 mM p‐NPP, 40 mM Tris–HCl (pH 9.5), 40 mM NaCl, 10 mM MgCl2) was added at 80 μL per well into both a conditioned medium and a cell plate. Absorbance at 405 nm of both plates was read before and after 1 h incubation for 37°C using a microplate reader (Infinite m200, TECAN, Zurich, Switzerland). We calculated relative percentage of alkaline phosphatase activity in conditioned medium = ∆OD405 CM/(∆OD405 CM + ∆OD405 Cell), where ∆OD405 CM and ∆OD405 Cell denote changes in OD405 in the conditioned medium and on the cell surface, respectively, before and after 1 h incubation in the presence of p‐NPP.
Succinate dehydrogenase (SDH) activity
SDH activity was measured with the Succinate Dehydrogenase Activity Colorimetric Assay Kit (BioVision, San Francisco, California, USA) following the manufacturer instructions. HEK293 cells were lysed with SDH assay buffer (100 μL for 106 cells) to isolate mitochondrial SDH, and 20 μL of lysate and 1 μL of disodium salts of the compounds, oxaloacetic acid (OAA), cESA and cCPDA, dissolved in PBS were added to a 96‐well plate 5 min before adding the probe. The absorbance was measured using a microplate reader (Infinite m200, TECAN, Zurich, Switzerland).
Animals
All animal care and experimental procedures were compliant with Belgian and European regulations on protection of animals used for scientific purposes (Arrêté Royal du 29 mai 2013 and EU Directive 2010/63/UE) and were approved by the University of Liège Animal Ethics committee (including experimental design and statistical determination of group size; approval number #1651). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
In total, 32 male Wistar rats (250‐350 g, 3 months of age) originating from the University of Liège animal facility (Federal authorization for breeding animals LA1610002) were kept in a pathogen‐free (SPF) facility, at least two per ventilated cages in a controlled temperature and regular light/dark cycle. The non‐invasive tail–cuff method used to measure BP has been extensively described as an adequate method for the estimation of BP and routinely used in our lab (Bialy et al., 2015; Dogne et al., 2016).
Rat BP measurements
One week before the experiment, rats were acclimated to the experimental procedure and restraint devices to prevent stress. They were randomized according to weight in four groups of eight animals to receive vehicle (NaCl 0.9% m/v) or saline solutions of disodium salts of the compounds (succinic acid, cESA, cCPDA and tCPDA). BP was measured by the tail–cuff method using CODA system (Kent Scientific Corporation; NIBP‐CODA8‐PACK). The animals were injected via the tail vein. Immediately after injection, systolic, diastolic and mean BPs were recorded 10 times every 20 s after an acclimation of five runs (CODA software). The method being non‐invasive, no surgical procedures, anaesthesia or analgesia were needed. No animals were killed. General welfare assessment was conducted prior to and following the experiments.
Randomization and blinding
The whole cohort of rats was constituted of animals of similar age and weight. They were attributed to a group randomly (number generated by computer programme). The homogeneity of weight in each group was not significantly different. The scientist in charge of preparing the solutions was different from the one operating the BP‐measuring device. The solutions and their vessels did not differ in volume, colour or shape. The operator of the BP device was not aware of the nature of the injected solution. The results were analysed by a third person who did not know which groups of animals received the solutions containing active ligands.
Pharmacophore model
The pharmacophore model was built using the programme phase 3.3. implemented in the Maestro 9.2 software package (Schrödinger; LLC, New York, New York, USA, 2011) and based on the results obtained from the primary structure–activity relationship study. The 3D structures were initially built using the programme Ligprep 2.5 of Maestro 9.2 (Schrödinger; LLC). Structural conformers were generated with the thorough sampling option. The top‐ranked hypothetical pharmacophore including two hydrogen bond donors and two negative charges (N) was selected for the subsequent virtual screening. Excluded volumes were also added according the superimposition of inactive ligands.
Binding site and docking
The succinate receptor model was built by means of the SYBYL 8.0 molecular modelling package (SYBYL, version 8.0; Tripos Inc., St. Louis, Missouri, USA, 2008). First, the human sequence of the succinate receptor obtained from the Universal Protein Resource (code entry: Q9BXA5) were aligned with the sequence of another GPCR, the human β2 adrenoceptor (code entry P07550) by using the FUGUE module (Shi et al., 2001). The next step was the copy of a set of constraints derived from the crystal structure of the human β2 adrenoceptor to the corresponding residues of the sequences to be modelled using the ORCHESTRAR protein structure modelling module (Dilly and Liegeois, 2011). As succinate is an agonist at succinate receptors, the receptor model was built in its active form from a crystal structure of the activated turkey β2 adrenoceptor (PDB code 3P0G) (Rasmussen et al., 2011).
The binding mode of succinate to the succinate receptor was then investigated by molecular docking using the GOLD 5.2 program (Jones et al., 1997). The binding pocket was defined from the one proposed by He et al. (2004). The structure of the resulting succinate–receptor complex was finally refined by a 2 ns molecular dynamics simulation using the MMFF94 force field implemented in SYBYL 8.0 (Halgren, 1996), a temperature of 310 K and a time step of 1 fs.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Statistical analysis and plotting of concentration–response curves were performed with GraphPad Prism version 5.0. The EC50 were calculated by the software following nonlinear regression (curve fit) with four parameters. In the screening experiments and in vitro determination, all the compounds have been tested at least in three independent experiments (n ≥ 3). Determinations of assay performance have previously demonstrated the robustness and variability of the procedure, which is sufficient for this number of independent experiments. For the in vitro cAMP determinations, the results have been normalized to the activity of succinate to be able to compare the activity of compounds from different experiments, the absolute (but not the relative) response being influenced by the number of receptors expressed by the cells, with slight variations from one day to another. No statistical analysis has been performed in datasets of less than five independent experiments. For the evaluation of significant differences in the in vivo determination of BP, a one‐way ANOVA followed by Bonferroni's multiple comparison test was performed. The post hoc tests were run only when F achieved P < 0.05. There was no significant variance in homogeneity. P< 0.05 between means were considered as statistically significant.
Materials
All chemicals used were from Sigma‐Aldrich (St Louis, Missouri, USA) unless otherwise stated. cESA, citraconic acid, acetylenedicarboxylic acid, (R)‐methylsuccinic acid (MSA), (S)‐MSA, (S)‐malic acid, (R)‐malic acid, (S)‐aspartic acid and (R)‐aspartic acid were from Tokyo Chemical Industry (Tokyo, Japan). (S)‐Bromosuccinic acid (BrSA) and adipic acid (AA) were from Santa Cruz Biotechnology (Dallas, Texas, USA). cCPDA was from Diverchim (Roissy, France). trans‐CPDA (tCPDA) was from Enamine (Kiev, Ukraine). trans‐1,2‐Cyclobutanedicarboxylic acid was from abcr (Karlsruhe, Germany). Antibiotics for cell culture were from InvivoGen (San Diego, California, USA). Except for (S)‐BrSA (Sigma‐Aldrich), Halogeno‐succinic acids were synthesized by us according to published procedures (see Supporting Information Figure S1, the Supporting Information and Zurwerra et al., 2012). The pGloSensor™‐22F cAMP plasmid was obtained from Promega Corporation (Fitchburg, Wisconsin, USA). Stable cell lines expressing wild‐type (WT) human succinate receptors and the GloSensor system have been described previously (Gilissen et al., 2015). pcDNA3.1 was from Thermo Fisher Scientific (Waltham, Massachusetts, USA). HEK293 cells were from ATCC (Manassas, Virginia, USA).
Results
Screening of a library of succinic acid analogues
In the search for novel succinate receptor agonists, we tested a small library of 32 analogues of succinic acid (Figure 1A). The library was built using three parameters: (i) the substituent borne by the backbone; (ii) the size of the carbon backbone; and (iii) the influence of negative or positive charges. For ease of the presentation, we refer to the acidic form of the tested analogues even if the carboxylic acid moieties of the interacting molecules are deprotonated at physiological pH. The chemical properties and formulas of all tested compounds in this report are summarized in (Supporting Information Table S1). Each compound was tested at a single concentration of 500 μM on an HEK293 cell line stably expressing succinate receptors together with the cAMP GloSensor (Gilissen et al., 2015). We evaluated their ability to inhibit the basal cAMP levels according to a procedure established in the laboratory (Gilissen et al., 2015). None of the evaluated analogues were more active than succinic acid at 500 μM (Figure 1A). Compounds characterized by a backbone of more than four as well as less than three carbon atoms were completely inactive (Figure 1A and Supporting Information Table S1). Compounds with no negative charges were also unresponsive at 500 μM. Succinamic acid and monomethyl succinate, both having a single negative charge at physiological pH (Supporting Information Table S1), showed partial response (Figure 1A). Besides, we evaluated all the compounds as antagonists to make sure that inactive analogues were not binding to the receptor without activating it. We did not detect antagonistic activity of inactive compounds toward succinate receptors (Figure 1B). We performed complete concentration–response curves on compounds that displayed at 500 μM an activity higher than 10% (normalized to succinic acid activity set at 100%, Figure 1A). We chose 10% because we determined it was a significant level compared with the background of the assay (Gilissen et al., 2015). None of the tested compounds showed significant activity on cells not transfected with succinate receptors.
Figure 1.
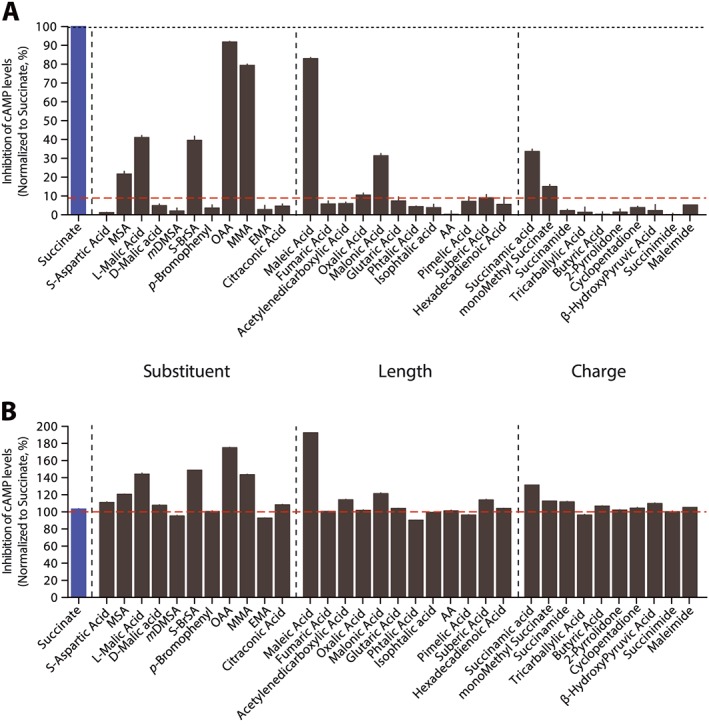
Screening of library of succinic acid analogues. We selected diverse compounds that share some characteristics with succinic acid. The compounds were grouped into three criteria: (i) nature of substituent; (ii) length of carbon backbone; and (iii) charge of the molecule. See (Supporting Information Table S1) for a complete list of molecular structures. (A) The different compounds were tested at 500 μM on succinate receptors. The agonist activity was evaluated by measuring the levels of cAMP in the presence of compounds compared with vehicle control and the response to succinic acid. No compounds showed activity on cell lines lacking succinate receptors. (B) 100 μM of compounds were added prior to the addition of 500 μM succinic acid to evaluate their antagonistic activity. Results are given as percentage of succinic acid activity for ease of comparison and as mean ± SEM of three independent experiments.
Effects of the nature and stereochemistry of substituents
We analysed the effects of the nature of substituents and their stereochemistry on potency and maximal efficacy. In the screening, MSA showed some activity at 500 μM (Figure 1A). The methyl substituent introduces a chiral centre, and two stereoisomers exist. The compound evaluated in the screening was a racemic mixture of two stereoisomers. Thus, we performed concentration–response curves on both R and S enantiomers (Figure 2A). Interestingly, these two analogues were characterized by an important difference in their activity, (S)‐MSA being inactive in the range of tested concentrations. We followed a similar strategy for the evaluation of BrSA. In this case, we observed an opposite response with regard to stereochemistry compared to MSA. The (S)‐BrSA enantiomer could not reach its maximal efficacy in the range of tested concentrations, whereas the R enantiomer was inactive at the same concentrations (Figure 2B). We reasoned that the size of the bromine atom could preclude an efficient interaction in the binding pocket of the receptor. Therefore, we synthesized (R)‐ and (S)‐chlorosuccinic acid (ClSA), the chlorine atom being smaller than the bromine atom (for the synthesis route, see Supporting Information Figure S1). The S derivative was also the most potent, being an agonist with potency and efficacy close to that of succinic acid in our assay (succinic acid pEC50 = 4.54 ± 0.08, EC50 = 29 μM; (S)‐ClSA pEC50 = 4.14 ± 0.04, EC50 = 72 μM, Figure 2B). We investigated other types of substituents such as the hydroxy group (malic acid, Figure 2C), which is able to establish hydrogen bond (as a donor). As observed with BrSA, the (S)‐malic (or (L)‐malic) acid was the only one showing an agonist behaviour although weaker than succinic acid. We also addressed the effect of positively charged substituents such as amines by evaluating aspartic acid (Figure 2D). Both S and R isomers were inactive (Figure 2D). OAA is characterized by a carbonyl group on the succinic acid backbone (see Supporting Information Table S1) that is able to form hydrogen bonds as an acceptor. This compound behaved as an agonist with similar efficacy but lower potency than succinic acid (pEC50 = 4.85 ± 0.08, EC50 = 14 μM; OAA pEC50 = 4.15 ± 0.04, EC50 = 70 μM, Figure 2D).
Figure 2.
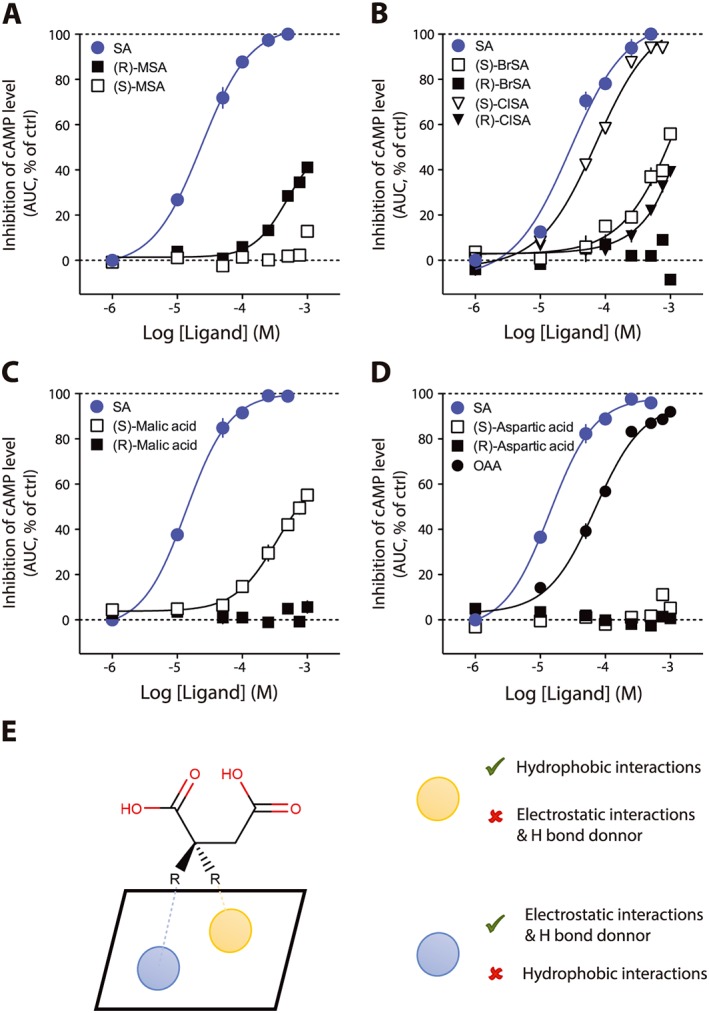
Effects of the nature and stereochemistry of substituents. Some of the compounds showing activity in screening at one concentration were evaluated with full concentration–response curves. Concentration–response curve for the effect on cAMP of (A) (R)‐ and (S)‐MSA; (B) BrSA and ClSA; (C) malic acid; (D) aspartic and OAA. In all experiments, succinic acid (SA) has been used as a reference compound and the data are normalized accordingly. Data are expressed as mean ± SEM of independent three experiments. (E) Model for the interaction of substituents with the binding pocket. AUC, area under curve; ctrl, control.
cis Conformation of the negative charges is an essential feature for agonist activity at succinate receptors
In the screening results (Figure 1A), we noticed that although maleic acid showed activity as expected, fumaric acid, a very close derivative, was inactive at this concentration. We confirmed with full concentration–response curves that fumaric acid was lacking activity on the receptor even at the highest concentration tested, whereas maleic acid was an agonist with lower potency but similar efficacy to that of succinic acid (maleic acid pEC50 = 4.24 ± 0.07, EC50 = 57 μM, Figure 3A). Surprisingly, the only difference between the two derivatives is the cis (maleate) or trans (fumarate) configuration of the carboxylic acids. We reasoned that the orientation of the negative charges had to be in close vicinity to interact with two adjacent positively charged amino acids in the binding pocket. We tested this hypothesis with meso‐dimethylsuccinic acid (mDMSA) that has two bulky substituents (Supporting Information Table S1) that orient the carboxylic acids in a pseudo trans configuration. mDMSA was inactive in the range of tested concentrations (Figure 3B). We evaluated malonic acid (MA) and its substituted analogues methylmalonic (MMA) and ethylmalonic (EMA) acids (see Supporting Information Table S1 for chemical structures). Consistent with the idea that promoting a conformation where the negative charges are closer will produce compounds with increased potency, MMA (pEC50 = 3.77 ± 0.05, EC50 = 169 μM, Figure 3B) was more potent than MA (pEC50 > 3.15, EC50 > 700 μM, Figure 3B). EMA was inactive in the range of tested concentrations (Figure 3B). With all these results, we developed an in silico model of the succinate receptor. We achieved a docking of succinic acid into the binding pocket proposed by He et al., (2004) (Figure 3C). The binding results predicted that the negatively charged oxygen atoms in succinic acid (the carboxylate functions) would establish ionic interactions with positively charged nitrogen atoms (guanidinium functions) of arginines 252 (R2526.55, superscript indicates the residue topology according to the Ballesteros–Weinstein numbering system, Ballesteros and Weinstein, 1995) and 281 (R2817.39) (Figure 3C). In our model, arginine 99 (R993.29) and histidine 103 (H1033.33), although involved in the interaction with succinic acid, were not critical. To confirm our model, we generated mutants for the four AA presumably involved in succinic acid binding. We generated stable cell lines expressing the mutants and evaluated the ability of the different receptors to be activated by succinic acid (Figure 3D). R252A and R281A mutants were unresponsive to succinic acid at concentrations up to 100 mM, whereas R99A and H103A could be activated by succinic acid although at very high concentrations (succinic acid, pEC50 on R99A < 1.50, EC50 on R99A > 30 mM; succinic acid pEC50 on H103A < 0.85, EC50 on H103A > 140 mM, Figure 3D).
Figure 3.
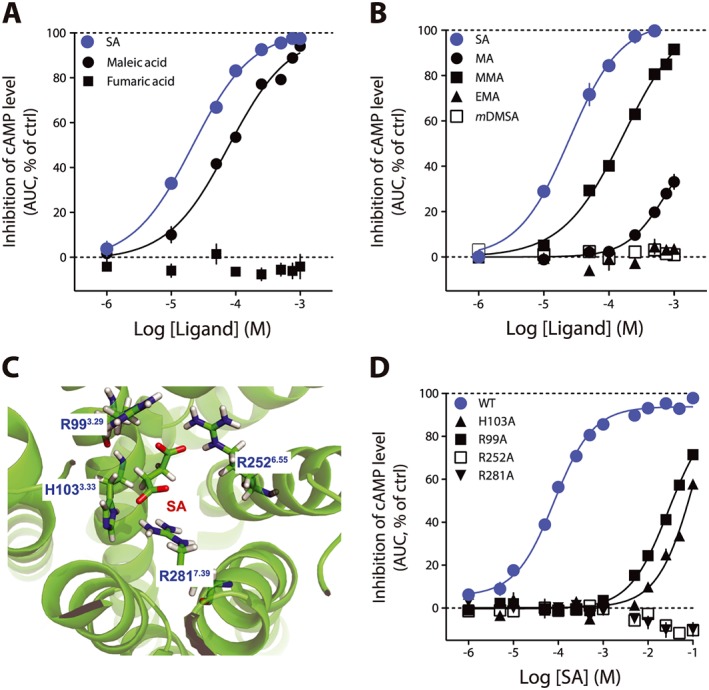
cis Conformation of the negative charges is an essential feature for succinate receptor agonists. (A) Maleic acid is a full agonist for succinate receptors, albeit with a lower potency than succinic acid (SA). Fumaric acid is completely inactive. (B) MA, MMA, EMA and mDMSA concentration–response curves on basal cAMP levels. (C) Homology modelling of the succinic acid binding pocket. succinic acid is positioned in a pseudo cis conformation. (D) Evaluation of the effect of succinic acid on basal cAMP level in several HEK293 cell lines stably transfected by GloSensor system and the succinate receptor mutants (H103A, R99A, R252A, R281A). Data are presented as mean ± SEM of three independent experiments. AUC, area under curve; ctrl, control.
cis‐Cyclic derivatives of succinic acid are succinate receptor agonists
Based on the active and inactive compounds that we identified so far, we developed a pharmacophore for activity at succinate receptors (Figure 4A). The yellow spheres represent exclusion volume. Pink and red spheres indicate the hydrogen bond acceptor and negative sites respectively. We screened the ZINC library and obtained as hits with cyclic compounds such as cCPDA (Figure 4B). We evaluated this compound with our cAMP assay on succinate receptors and found that cCPDA was an agonist of similar potency and efficacy compared with succinic acid (cCPDA pEC50 = 3.31 ± 0.02, EC50 = 49 μM, Figure 4C). Consistent with our binding site model, the trans analogue of 1,2‐cyclopropanedicarboxylic acid was inactive (Figure 4C). We evaluated the cis‐1,2‐cyclobutanedicarboxylic (cCBDA) acid and measured an activity for the cis isomer (pEC50 < 3.60, EC50 > 250 μM, Figure 4C), although it did not reach maximal efficacy in the range on tested concentrations, whereas the trans isomer was inactive (Figure 4C). The cis‐ and trans‐1,2‐cyclopentanedicarboxylic acid were both inactive (data not shown). Given the good potency obtained with OAA (pEC50 = 4.15 ± 0.04, EC50 = 70 μM, Figure 2D), we reasoned that a hydrogen bond acceptor in the cycle could have beneficial effect on the activity of the cyclic dicarboxylic acids. This led to the evaluation of cESA on succinate receptors. This compound displayed a 100% efficacy and was more potent than succinic acid (cESA pEC50 = 5.57 ± 0.02, EC50 = 2.7 μM, Figure 4D).
Figure 4.
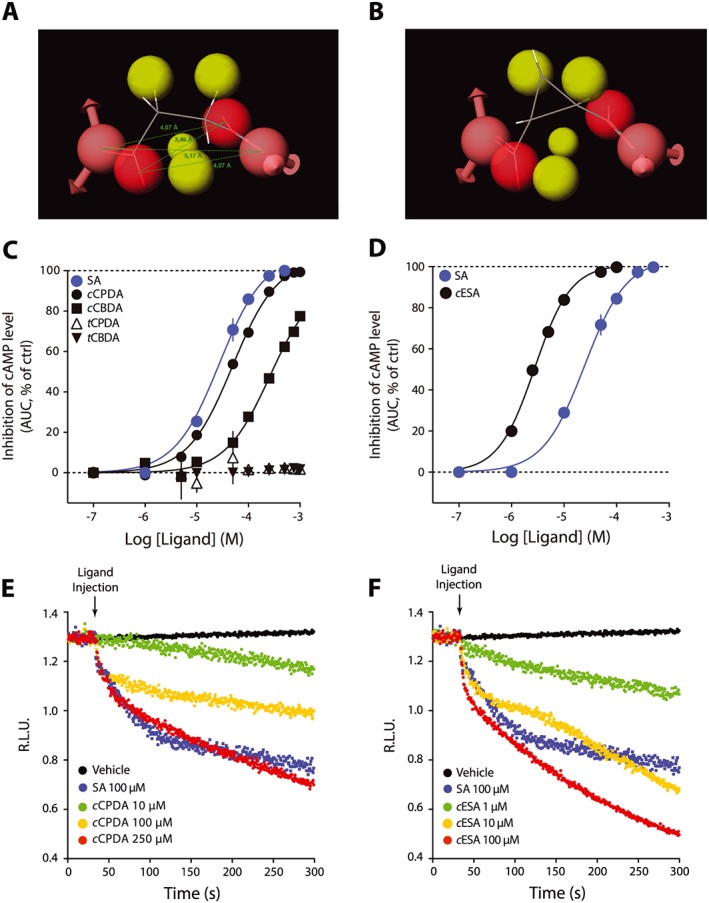
cis‐Cyclic derivatives of succinic acid (SA) are succinate receptor agonists. (A) Model for the succinate receptor agonist pharmacophore. Yellow spheres represent exclusion volume and red spheres negative charges. (B) cCPDA fit with the pharmacophore model. (C) Evaluation of cis‐ and trans‐cyclic compounds on the inhibition of basal cAMP levels. (D) cESA concentration–response curve on the inhibition of basal cAMP level. (E, F) Kinetic measurement of the inhibition of cAMP levels followed in HEK293 cells stably expressing cAMP Glosensor and succinate receptor upon addition of the succinate receptor agonists cCPDA (E) and cESA (F).Succinic acid was used as a positive control (ctrl). Data are expressed as mean ± SEM of three independent experiments. AUC, area under curve.
cESA and cCPDA are agonists for all known succinate receptor pathways and do not interfere with SDH
We performed a complete validation of cESA and cCPDA pharmacology on succinate receptors. First, we analysed the agonist behaviour of the compounds on a recently described transcatheter arterial chemoembolization‐induced TGF‐α shedding assay (Inoue et al., 2012). We detected activation of the receptor with the transient transfection of the α subunit of the chimeric Gqi1 that couples to Gi1 receptors and induces the Gq pathway. In this assay, cESA was the most potent agonist and cCPDA displayed a similar potency and efficacy to that of succinic acid (cESA pEC50 = 4.38 ± 0.04, EC50 = 42 μM; cCPDA pEC50 = 3.05 ± 0.04, EC50 = 887 ± 80 μM; succinic acid, pEC50 = 3.46 ± 0.03, EC50 = 350 μM, Figure 5A). A similar experiment with the chimeric Gqi3 that couples to Gi3 receptors and induces the Gq pathway gave similar results (data not shown). Next, we used a firefly luciferase complementation assay (Gilissen et al., 2015) to evaluate the arrestin 3 recruitment to the receptor. cESA and succinic acid, but not cCPDA, activated the receptor with similar efficacies at the highest concentrations tested (cESA pEC50 = 4.13 ± 0.04, EC50 = 74 μM; succinic acid pEC50 = 3.06 ± 0.07, EC50 = 865 μM; cCPDA pEC50 = 2.97 ± 0.07, EC50 = 1076 μM, Figure 5B). Succinate receptors are able to elicit [Ca2+]i mobilization, and the three tested compounds elicited an activation of this pathway in the range of tested concentrations (cESA pEC50 = 3.72 ± 0.01, EC50 = 191 μM; cCPDA pEC50 = 2.98 ± 0.01, EC50 = 1040 μM; succinic acid, pEC50 = 3.23 ± 0.01, EC50 = 581 μM, Figure 5C). We tested the activity of the two synthetic agonists on SDH activity. At concentrations up to 500 μM, neither cESA nor cCPDA had an effect on SDH activity (Figure 5D). OAA was used as a positive control for SDH inhibition. None of the agonists displayed activity on cells not expressing succinate receptors.
Figure 5.
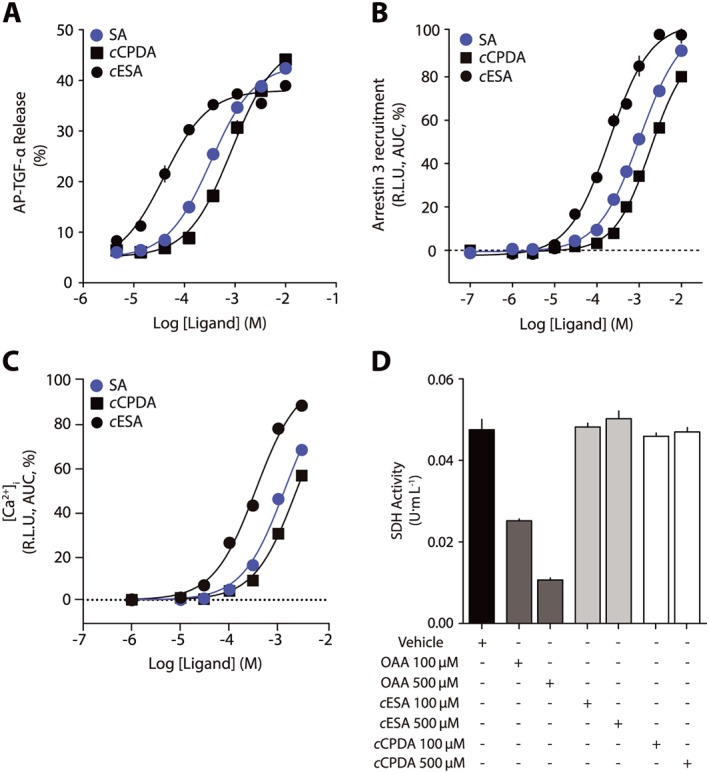
cESA and cCPDA are agonists for all known succinate receptor pathways and do not interfere with SDH. Concentration–response curves for succinic acid, cESA and cCPDA on (A) TGF‐α shedding, (B) arrestin 3 recruitment, (C) [Ca2+]i mobilization and (D) SDH activity. Data are presented as mean ± SEM of six independent experiments. AUC, area under curve; SA, succinic acid.
cCPDA and cESA are active in vivo
We finally tested the agonists of succinate receptors in an in vivo model. We non‐invasively measured BP in rats following intravenous infusion of succinic acid and observed a significant increase in BP, as previously reported (He et al., 2004; Vargas et al., 2009) (Figure 6). We observed a similar increase in response to the injection of cCPDA (10 mg·kg−1) and succinic acid (10 mg·kg−1). At a dose of 1 mg·kg−1, cESA injection was followed by an increase in BP that was not statistically different from that induced by succinic acid or cCPDA (Figure 6).
Figure 6.
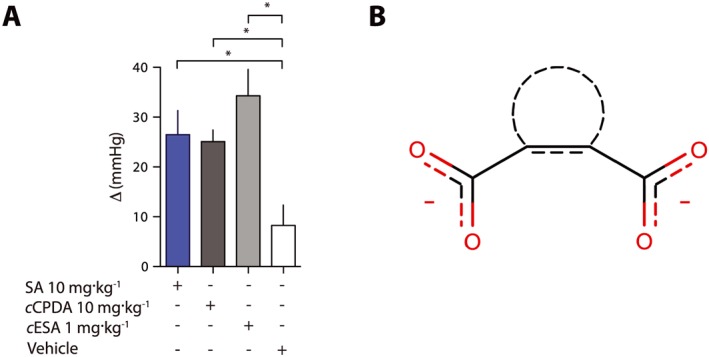
cESA and cCPDA are as active as succinic acid (SA) in vivo. (A) Repeated non‐invasive measures of BP in rats (n = 8 in each group) injected intravenously with a saline solution of the test compounds. These experiments have been performed at least three times for each condition on different animal cohorts. Data are expressed as the difference between mean BP before injection and 15 min post‐injection. Data are presented as mean ± SD. *P <0.05, significantly different as indicated: one‐way ANOVA. (B) Proposed pharmacophore for succinate derivatives as agonists.
Discussion
Although the human genome sequence was published in 2001 (Venter et al., 2001), many proteins continue to be understudied, presumably because of a lack of research tools such as low MW ligands (Edwards et al., 2011). Despite extensive knowledge on the function and pharmacology of some receptors and despite their potential as drug targets, the majority of the GPCR family is actually understudied (Roth and Kroeze, 2015). Thus, an important number of GPCRs are poorly characterized or even devoid of known ligands and labelled as ‘orphans’. Recently, Roth and Kroeze (2015) proposed that it was the availability of good ligands that made some GPCR popular and not the other way around. The receptor for succinic acid, the succinate receptor, belongs to this category of attractive drug target (Gilissen et al., 2016) whose pharmacology is poorly defined. Notwithstanding its demonstrated involvement in immune responses and inflammation (Rubic et al., 2008; Littlewood‐Evans et al., 2016), retinal angiogenesis (Sapieha et al., 2008) and regulation of renin release (Toma et al., 2008; Peti‐Peterdi et al., 2013), surprisingly few research tools are available and only sparse information about its molecular structure has been published.
In the initial report describing its pairing with succinic acid, He et al. evaluated 200 carboxylic acids, of which succinic acid was the more potent (He et al., 2004). They disclosed the activity for two other ligands, the less potent agonists maleate and methylmalonate. They identified one important feature for agonist activity, the mandatory dicarboxylic nature of active molecules (He et al., 2004). Here, we aimed to go several steps further and propose a full structure–activity relationship analysis. Compared with previous work, our investigation adds novel essential features required for the activation of succinate receptors In summary, succinate derivatives with an agonist profile must have (i) two negative charges at physiological pH; (ii) a distance from three to five carbon atoms between these two negatively charged atoms; and (iii) the possibility for the molecule to adopt a cis conformation of the two charges. The pharmacophore for agonist ligands is schematically summarized in Figure 6B. This refined pharmacophore led to the establishment of an improved model for the succinate binding pocket. It was initially proposed that four aminoacid residues were critical for succinate binding to the receptor (He et al., 2004). However, the published data showed only the results at one concentration of succinic acid (200 μM) on the mutant (He et al., 2004). Having defined the three criteria for agonists of the succinate receptor, we speculated that the most important interactions should be between two positively charged amino acids located in close proximity and on the same side inside the binding pocket. We performed docking simulation in an improved binding pocket model (Figure 3B) and identified R2526.55 and R2817.39 as the probable candidates. Mutagenesis experiments confirmed that these two residues are indeed critical for the interaction, whereas R993.29 and H1033.33 were important but not crucial. Interestingly, residues 3.33, 6.55 and 7.39 are topologically involved in agonist binding in several other class A GPCRs (Venkatakrishnan et al., 2013). A better delineation of the succinate binding mode is important for future modelling of the receptor to identify new ligands. Subsequently, we identified cis‐cyclic dicarboxylic acids as potent agonists, in particular cESA, which is characterized by a 10‐ to 20‐fold improved potency compared with succinic acid in a range of assays. The presence of an oxygen atom in this cyclic structure, together with the fact that OAA is an agonist with good potency, suggests that a putative aminoacid able to establish a hydrogen bond is ideally positioned inside the binding pocket.
In all bioassays that were used, the ligands showed the same rank order of potencies. This observation suggests that the ligands are not biased for the measured response, although more thorough experiments would be required to analyse biased signalling (or its absence). Interestingly, the potency of the ligands was lower compared with cAMP‐related pathways (Figure 5B). This is consistent with previous reports by us (Gilissen et al., 2015) and others (Southern et al., 2013). The luciferase complementation assay utilized to monitor arrestin recruitment is very different from the physiological environment of the receptor, which may affect the observed potency. The discrepancy between the assays could also be the consequence of a receptor reserve for the Gi‐mediated inhibition of AC. This raises the interesting possibility that in native tissues, in a given (patho)physiological context, the activation of some pathways might occur only in certain conditions of very high succinate concentrations, whereas the other pathways (such as Gi‐related ones) are activated at lower concentrations, due to receptor reserve and amplification.
The other synthetic ligands that have been described as antagonists are reported as being able to inhibit succinate‐mediated [Ca2+]i mobilization in CHO‐K1 cells overexpressing human succinate receptors in the nM range for the best compounds (2c and 4c) (Bhuniya et al., 2011; Gilissen et al., 2016). Their selectivity has been evaluated, and they represent valuable tools to characterize the receptor (Klenc et al., 2015). Interestingly and in stark contrast with the compounds described here, these ligands have no clear structural relationship to succinate. In addition, they display no negative charges at physiological pH. It would be interesting to test whether these antagonists can block the action of cESA and cCPDA with the same potency. Although the compounds reported by Bhuniya et al. are not available commercially, the novel agonists that we have identified will allow a more precise investigation of the mechanism of action and the binding mode (competitive vs. irreversible orthosteric or negative allosteric modulators ...) of the succinate receptor antagonists.
We also addressed two important characteristics in the perspective of the use of our compounds as pharmacological tools: specificity and in vivo efficacy. Historically, several succinic acid analogues such as OAA, malonate or l‐malate have been described as mitochondrial complex II (or SDH) inhibitors. Although they show agonist activity at succinate receptors, they would not be useful as tools because they also disrupt cellular respiration and could lead to artefacts. Similarly, there are some limitations in using succinic acid itself to assess the roles of succinate receptors, the endogenous ligand being also an important cellular metabolite. Our results clearly show that cESA and cCPDA are devoid of activity on SDH activity and suggest that they can be used to specifically address the consequences of the activation of the succinate receptors in cells, native tissues and organs, although we cannot exclude other off‐target effects. Another important aspect for chemical tools is their usefulness in vivo. More specifically, cESA has an ‘epoxy’ (see Supporting Information Table S1) function that may be metabolically unstable. Succinic acid is known to increase BP through succinate receptors and the release of renin in mice (He et al., 2004; Vargas et al., 2009). Therefore, we assessed in rats the ability of our compounds to reproduce the effects of succinic acid in vivo after intravenous injection. Both new agonist compounds were able to significantly increase BP with similar amplitude to that of succinic acid. Although it is tempting to speculate that it is the consequence of an activation of succinate receptors, additional experiments are needed (with antagonist or knockout animals) to substantiate the direct link between cESA‐ and cCPDA‐mediated increase in BP and succinate receptors. The oxoglutarate receptor (OXGR1) and the purinergic P2Y1 receptor are two GPCRs, most closely related to the succinate receptor (Gilissen et al., 2016). Although succinate has been shown previously to be unable to activate these two other GPCRs in various assays (He et al., 2004), further investigation is needed to address the activity of succinic acid and other succinate receptor ligands with regard to these receptors.
In conclusion, this is the first study, to the best of our knowledge, reporting on a synthetic agonist for the succinate receptors (GPR91, also termed SUCNR1) that is more potent than the endogenous ligand, succinic acid. The clinical relevance of these results is the possibility that the succinate receptors might represent an innovative drug target, for instance, in hypertension‐related diseases. However, it needs thorough preclinical validation first. We expect that the emergence of a properly characterized tool, which is readily commercially available, will spur new research on the understudied succinate receptors that should advance its validation as a drug target.
Author contributions
J.H. designed and supervised the study. P.G., J.G., N.D., C.L. and D.A. performed the in vitro experiments and acquired the data. S.D. modelled the pharmacophore and receptor, identified privileged structures and docked succinate. L.P. and F.J. designed and performed the in vivo experiments. P.G., F.J., B.P. and J.H. analysed the data, interpreted the results, created the figures and wrote the paper. A.I. designed and set up the shedding assay and provided research tools.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Synthesis of halogeno succinic acid.
Figure S2 FACS Analysis of HEK293 stably transfected with several succinate receptor mutants. A. WT; B. R252A; C. R281A; D. R99A; E. H103A.
Table S1 Chemical structure of tested compounds. Activity at 500 µM is reported and correspond to the data of Figure 1.
Acknowledgements
This work was supported by the Fonds pour la Recherche Scientifique (F.R.S.‐FNRS) Incentive grant for scientific research (F.4510.14), University of Liège (Crédit de Démarrage‐Fonds Spéciaux) and Léon Fredericq Foundation. J.H. and C.L. are an F.R.S.‐FNRS research associate and a PhD fellow respectively. N.D. is an FRIA PhD fellow. A.I. was funded by JST, PRESTO. The authors wish to thank Céline Piron for excellent technical assistance and Vincent Seutin for critical reading of the manuscript. We thank the GIGA Imaging Platform for technical support in FACS analysis.
Geubelle, P. , Gilissen, J. , Dilly, S. , Poma, L. , Dupuis, N. , Laschet, C. , Abboud, D. , Inoue, A. , Jouret, F. , Pirotte, B. , and Hanson, J. (2017) Identification and pharmacological characterization of succinate receptor agonists. British Journal of Pharmacology, 174: 796–808. doi: 10.1111/bph.13738.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J, Weinstein H (1995). Integrated methods for the construction of three‐dimensional models and computational probing of structure–function relations in G protein‐coupled receptors. Meth Neurosci 25: 366–428. [Google Scholar]
- Bhuniya D, Umrani D, Dave B, Salunke D, Kukreja G, Gundu J et al. (2011). Discovery of a potent and selective small molecule hGPR91 antagonist. Bioorg Med Chem Lett 21: 3596–3602. [DOI] [PubMed] [Google Scholar]
- Bialy D, Wawrzynska M, Bil‐Lula I, Krzywonos‐Zawadzka A, Wozniak M, Cadete VJ et al. (2015). Low frequency electromagnetic field conditioning protects against I/R injury and contractile dysfunction in the isolated rat heart. Biomed Res Int 2015: 396593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilly S, Liegeois JF (2011). Interaction of clozapine and its nitrenium ion with rat D2 dopamine receptors: in vitro binding and computational study. J Comput Aided Mol Des 25: 163–169. [DOI] [PubMed] [Google Scholar]
- Dogne S, Rath G, Jouret F, Caron N, Dessy C, Flamion B (2016). Hyaluronidase 1 deficiency preserves endothelial function and glycocalyx integrity in early streptozotocin‐induced diabetes. Diabetes 65: 2742–2753. [DOI] [PubMed] [Google Scholar]
- Edwards AM, Isserlin R, Bader GD, Frye SV, Willson TM, Yu FH (2011). Too many roads not taken. Nature 470: 163–165. [DOI] [PubMed] [Google Scholar]
- Gilissen J, Geubelle P, Dupuis N, Laschet C, Pirotte B, Hanson J (2015). Forskolin‐free cAMP assay for Gi‐coupled receptors. Biochem Pharmacol 98: 381–391. [DOI] [PubMed] [Google Scholar]
- Gilissen J, Jouret F, Pirotte B, Hanson J (2016). Insight into SUCNR1 (GPR91) structure and function. Pharmacol Ther 159: 56–65. [DOI] [PubMed] [Google Scholar]
- Hakak Y, Lehmann‐Bruinsma K, Phillips S, Le T, Liaw C, Connolly DT et al. (2009). The role of the GPR91 ligand succinate in hematopoiesis. J Leukoc Biol 85: 837–843. [DOI] [PubMed] [Google Scholar]
- Halgren TA (1996). Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 17: 490–519. [Google Scholar]
- He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J et al. (2004). Citric acid cycle intermediates as ligands for orphan G‐protein‐coupled receptors. Nature 429: 188–193. [DOI] [PubMed] [Google Scholar]
- Hogberg C, Gidlof O, Tan C, Svensson S, Nilsson‐Ohman J, Erlinge D et al. (2011). Succinate independently stimulates full platelet activation via cAMP and phosphoinositide 3‐kinase‐beta signaling. J Thromb Haemost 9: 361–372. [DOI] [PubMed] [Google Scholar]
- Inoue A, Ishiguro J, Kitamura H, Arima N, Okutani M, Shuto A et al. (2012). TGFalpha shedding assay: an accurate and versatile method for detecting GPCR activation. Nat Methods 9: 1021–1029. [DOI] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R (1997). Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267: 727–748. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenc J, Lipowska M, Taylor AT (2015). Identification of lead compounds for (99m)Tc and (18)F GPR91 radiotracers. Bioorg Med Chem Lett 25: 2335–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Appleton KM, Strungs EG, Kwon JY, Morinelli TA, Peterson YK et al. (2016). The conformational signature of beta‐arrestin2 predicts its trafficking and signalling functions. Nature 531: 665–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK (2005). Transduction of receptor signals by beta‐arrestins. Science 308: 512–517. [DOI] [PubMed] [Google Scholar]
- Littlewood‐Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J et al. (2016). GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med 213: 1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreath KJ, Espada S, Galvez BG, Benito M, de Molina A, Sepulveda P et al. (2015). Targeted disruption of the SUCNR1 metabolic receptor leads to dichotomous effects on obesity. Diabetes 64: 1154–1167. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB et al. (2016). beta‐Arrestin biosensors reveal a rapid, receptor‐dependent activation/deactivation cycle. Nature 531: 661–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington JP, Al‐Lazikani B, Hopkins AL (2006). How many drug targets are there? Nat Rev Drug Discov 5: 993–996. [DOI] [PubMed] [Google Scholar]
- Peti‐Peterdi J, Gevorgyan H, Lam L, Riquier‐Brison A (2013). Metabolic control of renin secretion. Pflugers Arch 465: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS et al. (2011). Structure of a nanobody‐stabilized active state of the beta(2) adrenoceptor. Nature 469: 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robben JH, Fenton RA, Vargas SL, Schweer H, Peti‐Peterdi J, Deen PM et al. (2009). Localization of the succinate receptor in the distal nephron and its signaling in polarized MDCK cells. Kidney Int 76: 1258–1267. [DOI] [PubMed] [Google Scholar]
- Roth BL, Kroeze WK (2015). Integrated approaches for genome‐wide interrogation of the druggable non‐olfactory G protein‐coupled receptor superfamily. J Biol Chem 290: 19471–19477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido‐Perrig N et al. (2008). Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol 9: 1261–1269. [DOI] [PubMed] [Google Scholar]
- Sadagopan N, Li W, Roberds SL, Major T, Preston GM, Yu Y et al. (2007). Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am J Hypertens 20: 1209–1215. [DOI] [PubMed] [Google Scholar]
- Sapieha P, Sirinyan M, Hamel D, Zaniolo K, Joyal JS, Cho JH et al. (2008). The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat Med 14: 1067–1076. [DOI] [PubMed] [Google Scholar]
- Shi J, Blundell TL, Mizuguchi K (2001). FUGUE: sequence‐structure homology recognition using environment‐specific substitution tables and structure‐dependent gap penalties. J Mol Biol 310: 243–257. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern C, Cook JM, Neetoo‐Isseljee Z, Taylor DL, Kettleborough CA, Merritt A et al. (2013). Screening beta‐arrestin recruitment for the identification of natural ligands for orphan G‐protein‐coupled receptors. J Biomol Screen 18: 599–609. [DOI] [PubMed] [Google Scholar]
- Spath B, Hansen A, Bokemeyer C, Langer F (2012). Platelet inhibition by acetylsalicylic acid and P2Y receptor antagonists. Platelets 23: 60–68. [DOI] [PubMed] [Google Scholar]
- Sundström L, Greasley PJ, Engberg S, Wallander M, Ryberg E (2013). Succinate receptor GPR91, a Gαi coupled receptor that increases intracellular calcium concentrations through PLCβ. FEBS Lett 587: 2399–2404. [DOI] [PubMed] [Google Scholar]
- Toma I, Kang JJ, Sipos A, Vargas S, Bansal E, Hanner F et al. (2008). Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Invest 118: 2526–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas SL, Toma I, Kang JJ, Meer EJ, Peti‐Peterdi J (2009). Activation of the succinate receptor GPR91 in macula densa cells causes renin release. J Am Soc Nephrol 20: 1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM (2013). Molecular signatures of G‐protein‐coupled receptors. Nature 494: 185–194. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG et al. (2001). The sequence of the human genome. Science 291: 1304–1351. [DOI] [PubMed] [Google Scholar]
- Wettschureck N, Offermanns S (2005). Mammalian G proteins and their cell type specific functions. Physiol Rev 85: 1159–1204. [DOI] [PubMed] [Google Scholar]
- Wittenberger T, Schaller HC, Hellebrand S (2001). An expressed sequence tag (EST) data mining strategy succeeding in the discovery of new G‐protein coupled receptors. J Mol Biol 307: 799–813. [DOI] [PubMed] [Google Scholar]
- Zurwerra D, Glaus F, Betschart L, Schuster J, Gertsch J, Ganci W et al. (2012). Total synthesis of (−)‐zampanolide and structure–activity relationship studies on (−)‐dactylolide derivatives. Chemistry 18: 16868–16883. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Synthesis of halogeno succinic acid.
Figure S2 FACS Analysis of HEK293 stably transfected with several succinate receptor mutants. A. WT; B. R252A; C. R281A; D. R99A; E. H103A.
Table S1 Chemical structure of tested compounds. Activity at 500 µM is reported and correspond to the data of Figure 1.