

Abstract
Background and Purpose
A body of evidence suggests activation of metabotropic glutamate 2/3 (mGlu2/3) receptors would be an effective analgesic in chronic pain conditions. Thus, the analgesic properties of a novel mGlu2/3 receptor agonist prodrug were investigated.
Experimental Approach
After oral absorption, the prodrug LY2969822 rapidly converts to the brain penetrant, potent and subtype‐selective mGlu2/3 receptor agonist LY2934747. Behavioural assessments of allodynia, hyperalgesia and nocifensive behaviours were determined in preclinical pain models after administration of LY2969822 0.3–10 mg·kg−1. In addition, the ability of i.v. LY2934747 to modulate dorsal horn spinal cord wide dynamic range (WDR) neurons in spinal nerve ligated (SNL) rats was assessed.
Key Results
Following treatment with LY2934747, the spontaneous activity and electrically‐evoked wind‐up of WDR neurons in rats that had undergone spinal nerve ligation and developed mechanical allodynia were suppressed. In a model of sensitization, orally administered LY2969822 prevented the nociceptive behaviours induced by an intraplantar injection of formalin. The on‐target nature of this effect was confirmed by blockade with an mGlu2/3 receptor antagonist. LY2969822 prevented capsaicin‐induced tactile hypersensitivity, reversed the SNL‐induced tactile hypersensitivity and reversed complete Freund's adjuvant – induced mechanical hyperalgesia. The mGlu2/3 receptor agonist prodrug demonstrated efficacy in visceral pain models, including a colorectal distension model and partially prevented the nocifensive behaviours in the mouse acetic acid writhing model.
Conclusions and implications
Following oral administration of the prodrug LY2969822, the mGlu2/3 receptor agonist LY2934747 was formed and this attenuated pain behaviours across a broad range of preclinical pain models.
Abbreviations
- CFA
complete Freund's adjuvant
- CRD
colorectal distension
- DRG
dorsal root ganglia
- Glu
glutamate
- HEC
hydroxyethylcellulose
- mGlu
metabotropic Glu
- SNL
spinal nerve ligation
- TRPV1
transient receptor potential cation channel subfamily V member 1
- WDR
wide dynamic range
Tables of Links
| TARGETS |
|---|
| mGlu2 receptor |
| mGlu3 receptor |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
The naturally occurring amino acid glutamate is the preeminent excitatory neurotransmitter in the mammalian CNS, mediating fast synaptic responses through activation of ligand‐gated ion channels, ionotropic glutamate receptors (Traynelis et al., 2010), and influencing the production of second messengers through the activation of GPCRs, that is, metabotropic glutamate (mGlu) receptors. The mGlu receptors have been historically segregated into three groups based on sequence homology, agonist pharmacology and signal transduction mechanisms (Conn and Pin, 1997; Schoepp et al., 1999; Pin and Archer, 2002). Group I mGlu receptors consist of two members, mGlu1 and mGlu5. These show predominant dendritic localization, are coupled to Gαq and function to enhance in enhanced post‐synaptic signalling and plasticity. In contrast, group II (mGlu2 and mGlu3) and group III (mGlu4, mGlu6, mGlu7 and mGlu8) receptors are primarily localized on axon terminals and are coupled to Gαi. Activation of group II or III mGlu receptors leads to Gα subunit‐mediated inhibition of adenylate cyclase and diminished synthesis of cAMP, suppressing cAMP‐dependent kinase activity, and Gβ/γ subunit‐mediated inhibition of voltage‐dependent Ca2+ channels and vesicular release machinery.
MGlu2/3 receptors are prominently distributed in cortical and limbic brain structures and are associated with pre‐synaptic, post‐synaptic and glial elements (Neki et al., 1996, Petralia et al., 1996, Richards et al., 2005, Gu et al., 2008, Wright et al., 2013). Agonist activation of the mGlu2/3 receptors in animal models of anxiety and psychosis results in suppression of excessive glutamate transmission and behavioural efficacy (Monn et al., 1997; Helton et al., 1998; Moghaddam and Adams, 1998; Cartmell et al., 1999; Kłodzińska et al., 1999; Nakazato et al., 2000; Cartmell et al., 2000a; Schoepp et al., 2003; Takamori et al., 2003; Swanson et al., 2005; Rorick‐Kehn et al., 2007; Fabricius et al., 2011; Jones et al., 2011; Ago et al., 2012). In addition, abundant evidence (Nicoletti et al., 2011; Chiechio and Nicoletti, 2012) implicates mGlu2/3 receptor involvement in the modulation of glutamate transmission in both the ascending pain pathway (from peripheral nociceptors through the spinal cord ascending through midbrain nuclei to the cortex) and descending pain modulatory pathway (from the cortex to the primary nociceptor synapse in the dorsal horn of the spinal cord). For instance, mGlu2 and/or mGlu3 receptors are localized in brain regions involved in the central response to pain, including the rostal ventral medulla, periaquiductal grey, thalamus and cortex (Varney and Gereau, 2002), as well in structures involved in emotional processing such as the amygdala (Gu et al., 2008). The mGlu2 receptors are also expressed by the dorsal root ganglia (DRG) peripheral nociceptive nerves and co‐localize with transient receptor potential cation channel subfamily V member 1 (TRPV1), residing on axonal projections into the dorsal horn of the spinal cord at the primary pain transmission synapse (Carlton et al., 2009). Thus, activation of mGlu2 receptors would be expected to limit pain sensation by decreasing action potential‐driven release of glutamate and neuropeptides. This has been confirmed as mGlu2/3 receptor agonists infused onto the spinal cord of primates block TRPV1 agonist (capsaicin)‐induced increase in firing of mechano‐sensitive wide dynamic range (WDR) neurons (Neugebauer et al., 2000). Most recently, expression of mGlu2 was confirmed in neonatal and adult human DRGs, and mGlu2/3 receptor agonist‐induced reversal of membrane hyperexcitability in mouse and human cultured DRG sensory neurons exposed to the inflammatory mediator PGE2 was demonstrated (Davidson et al., 2016). In addition, mGlu3 receptors are highly expressed on glial cells in the human the dorsal horn (Ohishi et al., 1993). These spinal mGlu3 receptors have increased expression with the induction of chronic pain, such as chronic constriction injury of the sciatic nerve, inflammation with ultraviolet radiation of the skin and with induction on bone cancer pain. This response is blocked by agents that inhibit glial activation (Boxall et al., 1998; Osikowicz et al., 2009) or with repeated intrathecal administration of an mGlu2/3 receptor agonist (Ren et al., 2012), suggesting mGlu3 may be a compensatory mechanism in chronic pain conditions and plays a critical role in limiting glial activation.
In support of the hypothesis that activation of mGlu2/3 receptors will provide analgesic efficacy, epigenetic up‐regulation of mGlu2 receptors in the spinal cord and cerebral cortex of rats after L‐acetylcarnitine administration induced mGlu2/3 receptor antagonist‐sensitive antinociception (Chiechio et al., 2002), as did reversal of the glutamate‐cystine antiporter with N‐acetylcysteine (Bernabucci et al., 2012; Truini et al., 2015). Accordingly, direct activation of mGlu2/3 receptors with synthetic agonists has been shown to be efficacious in rodent models of inflammatory and neuropathic pain (Neugebauer et al., 2000; Simmons et al., 2002; Varney and Gereau, 2002; Jones et al., 2005; Du et al., 2008; Kumar et al., 2010). We have recently reported the synthesis, in vitro pharmacology and pharmacokinetics of a novel mGlu2/3 receptor agonist, (1S,2S,5R,6S)‐2‐aminospiro[bicyclo[3.1.0]hexane‐4,1″‐cyclopropane]‐2,6‐dicarboxylic acid (LY2934747) (Monn et al., 2015). In that account, we provided preliminary evidence that when administered i.p., LY2934747 exhibited efficacy in rodent models of psychosis and formalin‐induced nocifensive behaviours. Herein, we provide evidence for the broad‐spectrum efficacy of both LY2934747 and its oral prodrug, (1R,2S,4R,5R,6R)‐4‐((4H‐1,2,4‐triazol‐3‐yl)thio)‐2‐((S)‐2‐aminopropanamido)bicyclo[3.1.0]hexane‐2,6‐dicarboxylic acid (LY2969822), in rodents.
Methods
Animals
Male Sprague Dawley rats from Harlan Industries (Indianapolis, IN), weighing between 200 and 250 g upon arrival, were acclimatized in a controlled temperature and lighting (12 h light cycle; 0600 to 1600 h) environment for at least 7 days prior to any procedure or testing. C57BL6/J, male mice from Jackson Laboratory (25 to 30 g), were similarly allowed to acclimatize for at least 7 days before being placed in the study. Animals had free access to food and water at all times except during data collection or during a 12 h period prior to p.o. dosing. All animal studies complied with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications no. 8023, revised 1978) as reviewed and approved by the Eli Lilly Institutional Animal Care and Use Committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Drug administration
LY2969822 dihydrate was administered p.o. in a volume of 10 mL·kg−1 vehicle: 1% hydroxyethylcellulose (HEC), 0.25% Tween 80, 0.05% antifoam in water. Doses listed are given as the active (LY2934747) equivalent dose accounting for molecular weight. Injections, i.p. or i.v., of LY2934747 were administered in 0.9% saline solution at 1 or 2 mg·mL−1 respectively. Tramadol HCl was administered p.o. at 4 mL·kg−1 in 1% HEC, 0.25% Tween 80, 0.05% antifoam in water. LY341495 was administered s.c. in 0.9% saline solution at 1 mL·kg−1. Morphine sulphate was administered s.c. at 1 mL·kg−1 in water. Gabapentin was administered p.o. in 5 mL·kg−1 of water. The TRPV1 antagonist AMG517 was administered p.o. at 5 mL·kg−1 in 1% HEC, 0.25% Tween 80, 0.05% antifoam in water. Naproxen sodium (Sigma) was prepared in water at 5 mL·kg−1 and administered p.o. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Pharmacokinetics of the active LY2934747 following p.o. dosing of the prodrug LY2969822
Following administration of 1, 3 or 10 mg·kg−1 LY2969822 p.o., at 1, 2 or 4 h approximately 30–50 μL CSF and 0.5 mL blood were collected into EDTA anticoagulant tubes. A second group of four animals was dosed with 10 mg·kg−1 LY2969822 p.o. and plasma was collected at 6, 8 and 24 h; the plasma was immediately collected following centrifugation of the blood samples. The CSF was collected via a syringe from the cisterna magna. Plasma and CSF were stored at −70°C until analysed as described in Monn et al. (2015). All values were above the lower detection limit (<5 nM) for the bioanalytical assay utilized.
Rat formalin model
The method used for these studies is a modification of that used by Jett and Michelson (1996). Thirty minutes before the formalin injection, rats were placed into individual test chambers of an automated system for measuring force of movement (SR‐Lab Startle Response System, San Diego Instruments, San Diego, CA). After the 30 min acclimatization period, rats were removed from the test chamber and injected with 50 μL of 5% formalin solution (in 0.9% saline) into the plantar surface of the right hind paw using a 0.5 inch, 27 gauge needle, and then placed back in the test chamber. The test chambers were positioned on a detection system that monitors the force of movement continuously for 50 min in 1 s bins. The dependent factor (number of events) was defined as the number of 1 s bins with a change in dynamic force that exceeded an empirically determined threshold value (a value of arbitrary load units, which corresponds visually with rats quietly breathing or sniffing). The formalin‐induced movements detected by the system included licking and flinching of the affected paw as well as hopping and turning. The number of events was collected and summarized according to the typical biphasic response pattern (Early and Late Phases) in 5 min intervals. Binned data were summed and analysed using one‐way ANOVA for the early phase (first 5 min) and late phase (minute 11 through 40) time points. Post hoc analysis of treatment groups compared with the vehicle group was performed with Dunnett's test using Graphpad Prism(La Jolla, CA) with P < 0.05 considered as significantly different versus vehicle treated rats. Data are expressed as means ± SEM.
Evoked pain responses
For evaluation of mechanical hyperalgesia via the Randall–Selitto paw pressure test, rats were acclimatized in the testing room for at least 1 h. Baseline naïve scores were taken by placing the hind paw in between the pusher and plinth of the apparatus (Ugo Basile Analgesiometer), which, when activated, delivers a ramping weight increase up to a maximum of 250 g over approximately 10 s. The force in g when paw withdrawal occurred was scored with a maximum cut‐off of 250 g. The score for each treatment group at each time point was compared using Graphpad Prism (La Jolla, CA) (two‐way ANOVA followed by post hoc Dunnett's test) with P < 0.05 considered as significantly different versus vehicle treated rats.
Tactile hypersensitivity behaviour was evaluated by measuring the sensitivity of the injured paw to von Frey filaments with incremental bending forces (0.2 to 15 g) as described by Chaplan et al. (1994). The scoring pattern was calculated using the Dixon method (Dixon, 1980). The data for each treatment group at each time point were compared using Graphpad Prism (La Jolla, CA) (two‐way ANOVA followed by post hoc Dunnett's test) with P < 0.05 considered as significantly different versus vehicle treated rats.
The degree of thermal hyperalgesia was assessed by use of a focused infrared beam (Hargreaves et al. 1988). Data are expressed as mean 50% paw withdrawal threshold in g (tactile endpoint) or mean paw withdrawal latency in s (thermal endpoint). The data for each treatment group at each time point was compared using Graphpad Prism (La Jolla, CA) (two‐way ANOVA followed by post hoc Dunnett's test) with P < 0.05 considered as significantly different versus vehicle treated rats.
Spinal nerve ligation methods
Male Sprague–Dawley rats underwent unilateral tight ligation of the L5 and L6 spinal nerves as previously described by Kim and Chung (1992). Nerve injury was produced by tightly ligating the left L5 and L6 spinal nerves under gaseous anaesthesia with a mixture of isoflurane (3% for induction and 2% for maintenance) and oxygen. After surgery, rats were returned to their home cages and monitored post‐operatively. The development of tactile hypersensitivity occurred over the subsequent 2 weeks, aided by repetitive mechanical stimulation of the affected hind paw. Baseline values were required to be ≤2.0 g force for the animal to be included in the study.
Capsaicin sensitization
For assessment of either tactile hypersensitivity or thermal hyperalgesia, groups of rats were tested in the naïve state for baseline threshold responses prior to compound dosing. Rats were then pretreated with vehicle or drug as indicated prior to dosing with capsaicin. Capsaicin (1.2%, 18 μg in 15 μL olive oil) was injected unilaterally into the hind paw (s.c. into the mid‐plantar region from the toe towards the heel) using a Hamilton gas‐tight glass syringe and 27 gauge (0.5 in) needle. The degree of tactile hypersensitivity was assessed at 15, 30 and 60 min post‐capsaicin. Similarly, the degree of thermal hyperalgesia was assessed at 0.5, 1 and 2 h post‐capsaicin in a separate experiment.
Complete Freund's adjuvant (CFA) sensitization
A modified version of the CFA model described by Stein et al. (1988) was utilized. Following baseline mechanical hypersensitivity measures, CFA (50 μL neat, Sigma F5881‐10ML) was injected via the intraplantar route into the right hind paw under light isoflurane anaesthesia. The next day (24 h post‐CFA injection), rats were acclimatized in their home cage placed inside the test room for approximately 1 h prior to testing. The post‐CFA baseline hyperalgesia was scored followed by animal randomization and dosing with either vehicle or drug. Paw pressure was scored at 1 and 2 h following compound dosing.
Plantar incision post‐surgical sensitization
The methods of Brennan (Brennan et al., 1996) with modifications described by Whiteside et al. (2004) were utilized. Rats were anaesthetised, and an incision was made on the left hind paw pad 1 cm in length and penetrating the skin, fascia and muscle layers that was then briefly stretched/retracted with forceps. The resulting wound was then closed with two 5.0 vicryl sutures, and the animals allowed to recover for 24 h. Tactile hypersensitivity was assessed, animals randomized and dosed with vehicle or drug.
Colorectal distension (CRD) model
The methods of Ness and Gebhart (1988) were utilized with modifications. Specifically, rats were anaesthetised and surgically implanted with a pair of EMG electrodes in the external oblique abdominal muscle and were exteriorized at the nape of the neck to allow connection to the barostat inflation and EMG contraction recording unit. Following surgery, rats were allowed to recover for 1 week prior to testing. After the recovery period, a 6.0‐cm‐long balloon constructed from a latex glove finger was inserted transanally into an awake rat followed by baseline measurement using a 60 mmHg pressure to confirm that the electrodes/EMG recording was functioning properly. Following this step, rats were randomized to treatment groups. Rats were then dosed with vehicle or drug and monitored for visceromotor/EMG responses to CRD at increasing graded pressures of 20, 40 and 60 mmHg starting at 90 min following dosing. At each pressure, rats were tested three times for a duration of 20 s per inflation followed by a 3 min recovery period after each inflation.
For the acetic acid sensitized version of the CRD model, groups of rats, following EMG electrode implantation surgery and recovery period, were tested for the EMG responses at 60 mmHg pressure to establish baseline responses. Following this pretest, rats received an intracolonic instillation of 1% acetic acid (2 mL) to sensitize the colon. Approximately 24 h later, baseline measurements at 60 mmHg were conducted to ensure sensitization, followed by randomization to treatment groups. Vehicle or drug was administered and monitored for visceromotor/EMG responses to CRD at increasing graded pressures of 20, 40 and 60 mmHg starting at 90 min following dosing. Data were recorded as mean AUC for the visceromotor response (mV ∙ s) ± SEM. Statistical analysis was performed using JMP Statistical Software (one‐way ANOVA followed by post hoc Dunnett's test) with P < 0.05 considered as significantly different versus vehicle treated rats.
Acetic acid writhing
Four hours prior to injection with acetic acid, mice were administered vehicle or drug. Thirty minutes before acetic acid injection, mice were placed into Plexiglas observation chambers to acclimatize. Following the acclimatization period, the mice received an i.p. injection of 0.5% acetic acid (10 mL·kg−1), after which the mice were observed for writhing/stretching behaviour. A writhe is defined as a stretching of the hind limbs and lifting of the tail followed by the front limbs and head arching upwards and then a return to normal posture. The number of times this behaviour occurred over a period of 20 min was counted and recorded by a blinded observer. Data were recorded as mean number of writhes ± SEM. Statistical analysis was performed using JMP software (one‐way ANOVA followed by post hoc Dunnett's test) with P < 0.05 considered as significantly different versus vehicle treated mice.
Spontaneous and evoked wide dynamic range neuron firing in spinal nerve‐ligated rats
Surgery
Adult male Sprague–Dawley rats, weighing 220 to 250 g at the time of surgery, were used. They were housed in groups of four in an air‐conditioned room on a 12 h light/dark cycle. Food and water were available ad libitum. All procedures in this study were undertaken in compliance with the UK Animals (Scientific Procedures) Act 1986. Rats were prepared as described by Kim and Chung (1992). Each rat was anaesthetized, and the L5 spinal nerve was carefully isolated and tightly ligated with 6/0 silk suture. The baseline tactile hypersensitivity was examined for three consecutive days before surgery and reassessed on the 7th and 12th through to the 14th day after surgery. Only those rats with significant allodynia (paw withdrawal threshold ≤4 g) were selected for electrophysiological experiments.
Electrophysiological recording
The isolation and recording of lumbar spinal cord dorsal horn WDR neurons utilized the methods of Arendt‐Nielsen and Petersen‐Felix (1995) with modifications. The spinal nerve‐ligated rats were anaesthetized initially with urethane (1.2 to 1.6 g·kg−1, i.p.) and maintained non‐reflexive throughout the experiment by supplemental doses of urethane (0.4 g·kg−1, i.v.) as required. The arterial blood pressure and heart rate (through an ECG lead II) were routinely monitored, and rectal temperature was continuously monitored and maintained within a normal physiological range using a thermo‐blanket system.
The left external jugular vein was cannulated for drug administration. A laminectomy spanning the L2 to L6 vertebral segments was conducted, and the dura mater over the exposed spinal cord was removed. An oil pool was formed with warm mineral oil to cover the exposed tissue. For electrophysiological recording, carbon‐fibre microelectrodes (impedance, 0.4 to 0.8 MΩ at 1 KHz) were lowered into the dorsal horn of the spinal cord using a manual hydraulic manipulator. The neurons were recorded from lamina IV or V at about 500 to 900 μm from the surface of the L4 and L5 segments of the spinal cord. The proprioceptive neurons innervating muscle spindles, joint receptors were excluded based upon their firing pattern and responses to joint movement. The neural activity was amplified and monitored using standard electrophysiological recording techniques and recorded on to a PC using CED Spike 5 software (Cambridge Electronics Design, CED).
A defined protocol of mechanical stimulation was used to identify WDR neurons after mapping the peripheral receptive field in the hind paw. A single unit was identified as a WDR neuron if (1) the unit responded to gentle brushing of the hind paw for 10 s; (2) a graduated response to von Frey hair stimulation (4.17/1.15 g, 4.56/4 g, 5.18/15.6 g; 1 s on and 1 s off, repeated 10 times with the interval between two von Frey Hair applications of 10 s) of the RF was observed; and (3) a 10 s squeeze‐stimulus using a standard artery clip evoked discharge of the neuron.
After a WDR neuron was identified, a pair of fine‐needle electrodes was inserted into the receptive field to deliver electrical stimulation. Thresholds for evoking C‐fibre‐mediated responses were determined by delivering 1 ms duration single electrical pulses of increasing stimulus strength. Once the threshold was determined, a train of electrical pulses (16 pulses in 5 s, 1 ms duration) at an intensity of two times threshold was delivered once every 10 min. The neural activity (spontaneous firing and evoked responses) was recorded for at least 20 min before vehicle or compound administration, and then for 60 min after thevehicle or compound was injected.
The average spontaneous firing frequency over consecutive 4.5 min periods (expressed as numbers of action potentials min‐1) immediately before electrical stimulation was measured prior to and after vehicle or compound injection.
Wind‐up discharge of spinal dorsal horn neurons was calculated as the total number of evoked action potentials of a neuron in response to all 16 electrical pulses minus 16 times the action potentials induced by the first electrical pulse in that train. The number of action potentials within 300 ms after first electrical stimulation pulse (A) was counted, then the total number of action potentials induced by the whole train of electrical pulses (16 pulses) (B) was counted. The number of wind‐up action potentials was calculated as follows: Wind‐up action potential number = B − (A × 16). The wind‐up action potential number immediately before compound administration (0 min) and every 10 min after compound injection was counted. In some cases, wind‐up discharge was completely inhibited after compound administration; the number of wind‐up action potentials (B) was even lower than control levels (A × 16), leading to a negative reading. In such cases, the wind‐up was set to 0 (completely inhibited) for statistical analysis.
The total number of action potentials recorded within 10 s starting from 300 ms after the last electrical pulse of the train (i.e. the 16th electrical stimulus) was used as an indicator of the extent of after‐discharge for that neuron.
All data are expressed as mean ± SEM. Comparisons within animal between baseline measurements (Time 0 min) and subsequent readings after treatment utilized a repeated measures, one‐way ANOVA followed by a Dunnett's test (Graphpad Prism, La Jolla, Ca) with P < 0.05 considered as significantly different versus baseline response.
Results
Confirmation of prodrug hydrolysis in vivo to form active mGlu2/3 receptor agonist
The resulting plasma and CSF levels of the active mGlu2/3 agonist formed (Figure 1A, LY2934747) following a single dose of 1–10 mg·kg−1 (dose given in active equivalent units) of the prodrug LY2969822 p.o. are presented in Figure 1B. As seen in Figure 1B, doses of 1–10 mg·kg−1 of the prodrug LY2969822 resulted in CSF levels of the active LY2934747 that exceed, for at least 4 h, the potency (EC50 = 14.2 nM) of the mGlu2/3 receptor agonist to stimulate the rat mGlu2/3 receptors in cortical synaptosomes in vitro (Monn et al., 2015).
Figure 1.
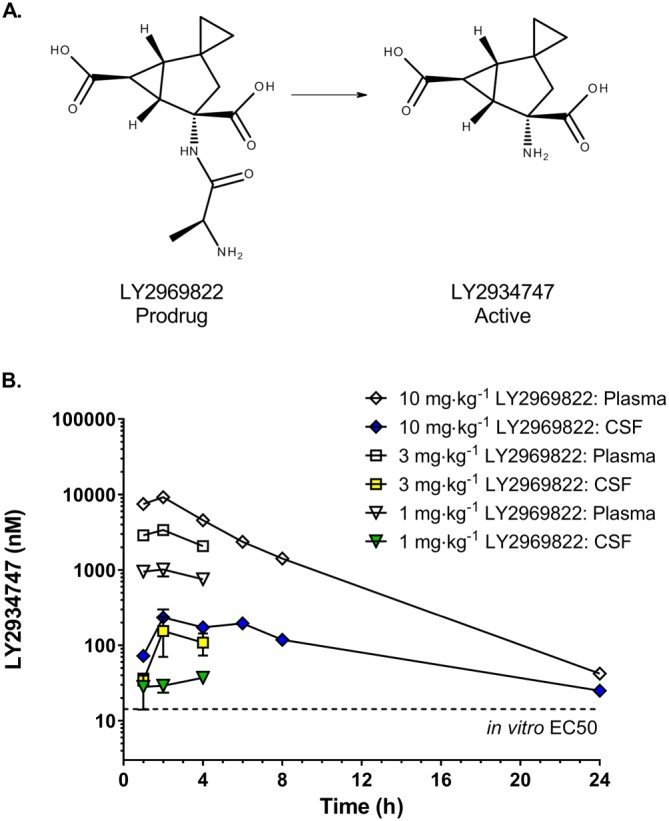
Chemical structures of LY2969822 (prodrug) and LY2934747 (active) and the resulting plasma and CSF levels of the active form after dosing orally with the prodrug in rats. (A) The chemical structure of the prodrug LY2969822 and the resulting structure after absorption and cleavage to the active molecule LY2934747. (B) Resulting plasma and CSF levels of the active LY2934747 follow p.o. dosing with 1, 3 or 10 mg·kg−1 (units in active equivalents) of the prodrug LY2969822. Values are mean ± SEM, n = 4. The dotted line at 14 nM represents the potency of LY2934747 for the rat mGlu2 receptor as reported in Monn et al., 2015.
Inhibition of formalin‐induced nocifensive behaviours in rats
As seen previously after i.p. dosing with the active molecule LY2934747 (Monn et al., 2015), p.o. administration of the prodrug LY2969822, 1 to 10 mg·kg−1 (expressed as LY2934747 equivalent doses), resulted in a significant dose‐dependent decrease in the formalin‐induced nocifensive behaviour as measured in the rat automated formalin test (Figure 2A). The absolute ED50 value for dose–response curve of % inhibition of LY2969822 dihydrate was 3.2 mg·kg−1. To confirm the specificity of the effect, the ability of the mGlu2/3 receptor antagonist to block the response was tested. As seen in Figure 2B, the decrease in the formalin‐induced nocifensive behaviour seen with a 3 mg·kg−1 i.p. dose of LY2934747 was blocked by a 10 min pretreatment of either 1 or 3 mg·kg−1 doses of an mGlu2/3 receptor antagonist, LY341495. No effect was observed with dosing the antagonist LY341495 alone. In order to test whether the effect on formalin induced nocifensive behaviours would persist with repeated administration, a subchronic dosing regimen of once a day p.o. dosing of the prodrug LY2969822 (1–10 mg·kg−1) for 3 days was examined and compared with a single acute dose. Formalin was injected into the hind paw 2 h after the single or last dose as indicated in Figure 2C. The repeated (3 days) p.o. administration of LY2969822 resulted in a significant decrease in the formalin‐induced nocifensive behaviour, which was similar to the effect seen with an acute single dose when measured in the same study in the rat automated formalin test.
Figure 2.
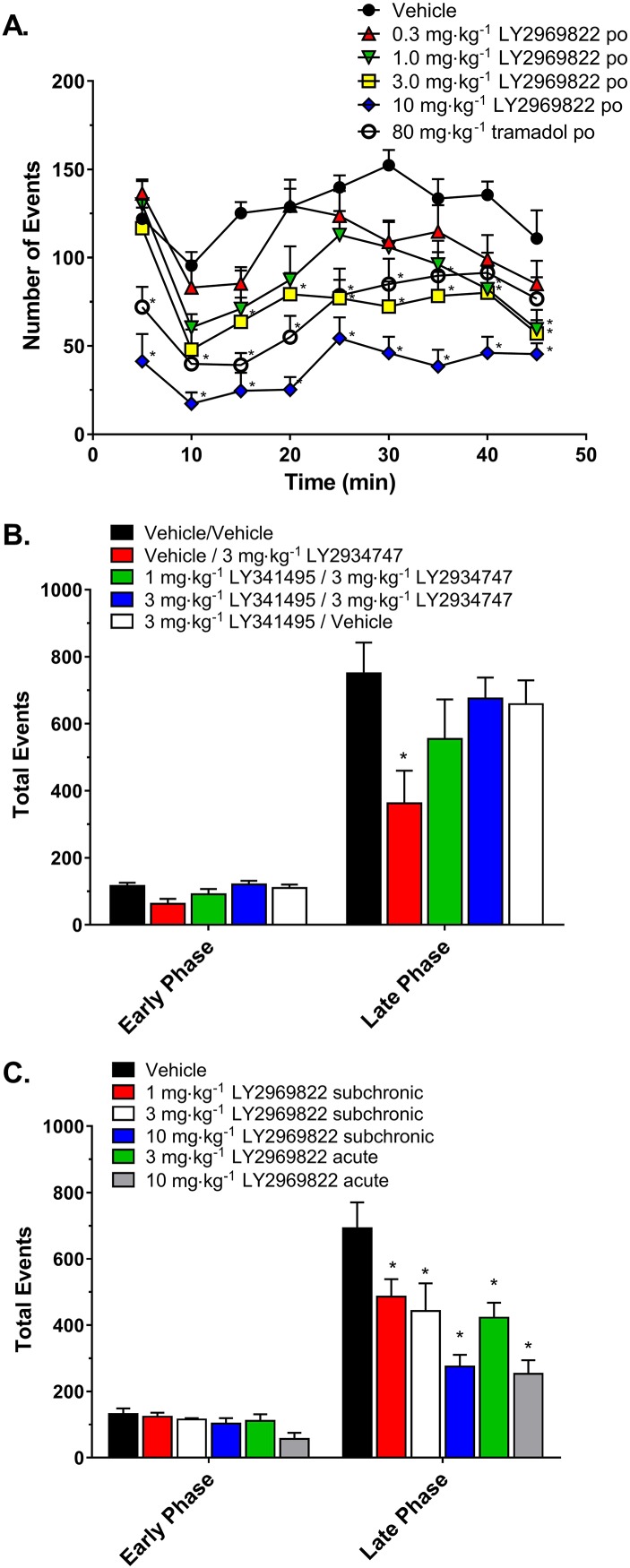
Effects of LY2969822 and LY2934747 in blocking formalin‐induced nocifensive behaviours. (A) Vehicle, tramadol (80 mg·kg−1) or LY2969822 (0.3 to 10 mg·kg−1, active equivalents) was dosed p.o. 2 h prior to formalin injection into the hind paw of the rat. Nocifensive responses were monitored and quantified in 5 min bins for 50 min. Values are mean ± SEM, n = 7–8 per group. *Indicates significantly different versus vehicle response at the corresponding time point (P < 0.05, two‐way ANOVA followed by Dunnett's test). (B) LY341495 or vehicle was administered at 1 or 3 mg·kg−1 via the s.c. route 70 min prior to formalin injection into the hind paw, while LY2934747 or vehicle was administered at 3 mg·kg−1 via the i.p. route 60 min prior to formalin injection. (C) LY2969822 was dosed p.o. at 1 to 10 mg·kg−1 (active equivalent) with a single treatment (acute) or daily treatments for 3 days (subchronic), and 2 h after the last dose formalin was injected into the hind paw of the rat. Binned data were summed for early phase (first 5 min) and late phase (minutes 11 through 40) time points. Values are mean ± SEM, n = 7–8 per group. *Indicates significantly different versus vehicle response (P < 0.05, one‐way ANOVA followed by Dunnett's test).
Inhibition of spontaneous and evoked spinal wide dynamic range neuronal activity in spinal nerve‐ligated neuropathic rats
Following an i.v. dose of vehicle in rats that had undergone spinal nerve ligation (SNL) and developed mechanical allodynia, neither the observed spontaneous activity (Figure 3B) nor the increase neuronal firing with repeated stimulation (wind‐up, Figure 3C) was changed for the duration of monitoring in the animals. In contrast, 3 mg·kg−1 morphine significantly decreased spontaneous activity and wind‐up of WDR neuronal activity. Similarly, an i.v. dose of 1 or 10 mg·kg−1 LY2934747 (active form of the mGlu2/3 receptor agonist prodrug LY2969822) rapidly and significantly decreased spontaneous activity (Figure 3A, B) and wind‐up (Figure 3A, C) in the WDR neurons. The similar response seen with 1 and 10 mg·kg−1 may indicate a maximal efficacy in this assay possibly driven by a higher cmax and exposure reached by dosing i.v. with the active molecule. Note that the magnitude of the effect on spontaneous activity and wind‐up with LY2934747 was similar to that seen after a 3 mg·kg−1 i.v. dose of morphine. Similar significant decreases in after‐discharge measurements were seen with morphine and 10 mg·kg−1 LY2934747 as well (data not shown).
Figure 3.
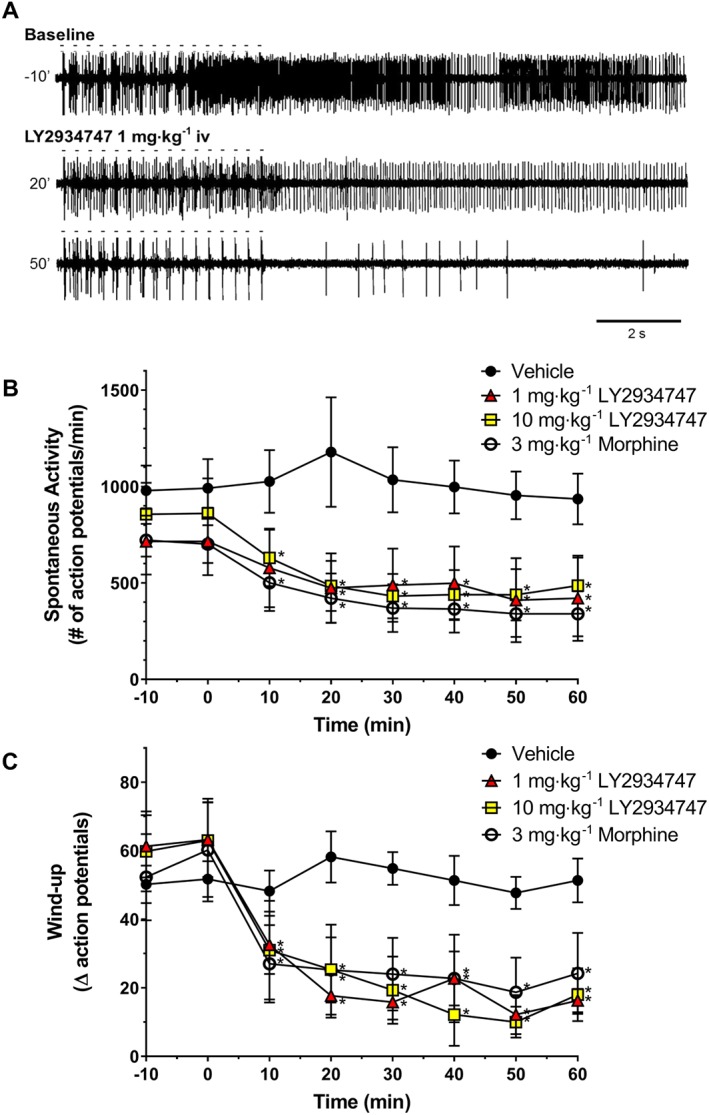
Effects of LY2934747 (active form of the mGlu2/3 receptor prodrug LY2969822) on in vivo spontaneous and evoked electrical activity of WDR neurons in spinal nerve‐ligated rats. WDR neurons in the lumbar region of the dorsal horn of the spinal cord that had receptive fields in the hind paw were identified, and the number of spontaneous and stimulus‐evoked action potential discharges was determined in rats that had undergone SNL with confirmed tactile allodynia. (A) Representative tracing from an animal's baseline measurements of evoked and spontaneous activity at baseline and 20 or 50 min after an i.v. dose of LY2934747. Note that the train of 16 stimulations is indicated by the dashes above the tracing. (B) Values are the average number of spontaneous discharges over a 60 min period measured every 10 min recorded after i.v. injection of vehicle, 1 or 10 mg·kg−1 LY2934747 or 3 mg·kg−1 morphine. (C) Values are the average change in the number of discharges due to repeated stimulation (see methods for calculation detail) measured every 10 and 60 min period recorded after i.v. injection of vehicle, 1 or 10 mg·kg−1 LY2934747 or 3 mg·kg−1 morphine. Values are mean ± SEM, n = 6. *Indicates P < 0.05 versus response at time 0 min (repeated measures one‐way ANOVA followed by Dunnett's test).
Inhibition of nociceptive sensitization induced with capsaicin in rats
In order to determine if pretreatment with the mGlu2/3 receptor prodrug LY2969822 would alter the capsaicin‐induced sensitization, LY2969822 (1–10 mg·kg−1) was p.o. dosed 2 h prior to a unilateral capsaicin injection into the hind paw. Tactile allodynia was measured from 15 to 60 min later, or thermal hyperalgesia assessed at 0.5 to 2 h. LY2969822 dose‐dependently decreased tactile allodynia at all time points assessed (Figure 4A). The degree of efficacy was similar to that seen with a 100 mg·kg−1 dose of gabapentin. In contrast, the ability of LY2969822 to block capsaicin‐induced thermal hyperalgesia was less clear. A significant increase in latency was seen with a dose of 3 mg·kg−1 LY2969822 dihydrate at the 1 h post‐capsaicin, but significant effects were not evident at any time point assessed with the higher dose of LY2969822 dihydrate (10 mg·kg−1 active equivalent). It is worth noting that the experimenter observed that animals receiving the highest dose of LY2969822 dihydrate displayed an increased startle response to sound that is often observed in rodents following treatment with mGlu2/3 receptor agonist compounds. This increased startle activity may have interfered with making accurate measurements of thermal hyperalgesia in these animals. Thus, the results are inconclusive regarding the ability of LY2969822 dihydrate to block capsaicin‐induced thermal hyperalgesia as assessed in this study.
Figure 4.
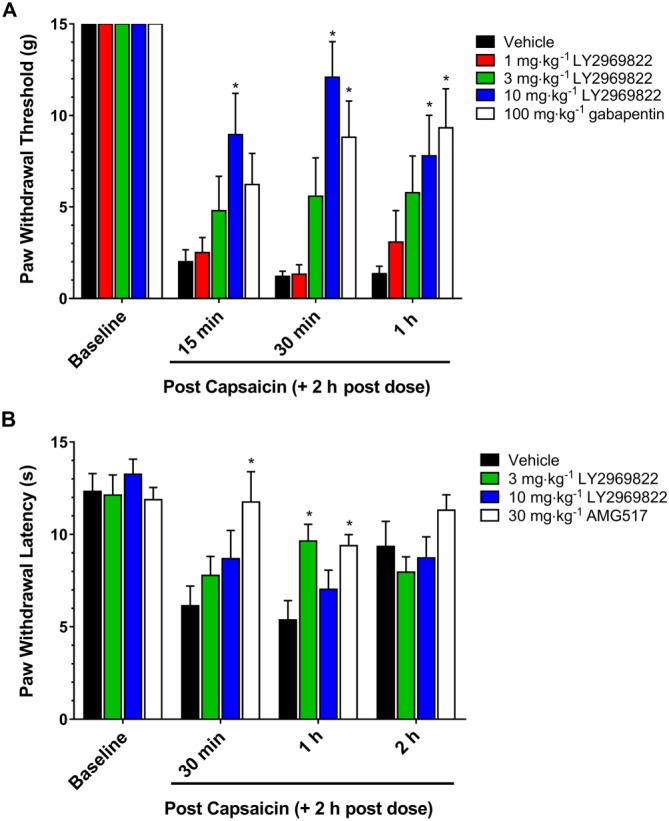
Effect of LY2969822 on the rat capsaicin model of tactile allodynia and thermal hyperalgesia. LY2969822 was dosed at 1, 3 and 10 mg·kg−1 and gabapentin at 100 mg·kg−1 or AMG517 at 30 mg·kg−1. Capsaicin 1.2%, 18 μg in 15 μL olive oil, was injected intraplantar 2 h post‐compounds. (A) Tactile allodynia score was measured at 15, 30 and 60 min post‐capsaicin. (B) Thermal hyperalgesia scores were measured at 0.5, 1 and 2 h post‐capsaicin. Values are mean ± SEM, n = 8–10 per group. *Indicates significantly different versus vehicle response at the corresponding time point (P < 0.05, two‐way ANOVA followed by post hoc Dunnett's test).
Reversal of mechanical hyperalgesia induced with complete Freund's adjuvant (CFA) in rats
Rats were injected in a hind paw with an intraplantar injection of CFA and the resulting mechanical hyperalgesia assessed by measuring withdrawal to a pressure gradient the following day. Rats confirmed to have hyperalgesia were randomized to group and vehicle or 1–10 mg·kg−1 LY2969822 dihydrate dosed p.o. As seen in Figure 5, a statistically significant reversal of mechanical hyperalgesia was seen with LY2969822 dihydrate in a dose‐ and time‐dependent manner, with reversal seen at 1 and 2 h post‐treatment with all doses tested (1, 3 and 10 mg·kg−1 active equivalent). The magnitude of the response was comparable with that seen with an NSAID, 30 mg·kg−1 naproxen.
Figure 5.
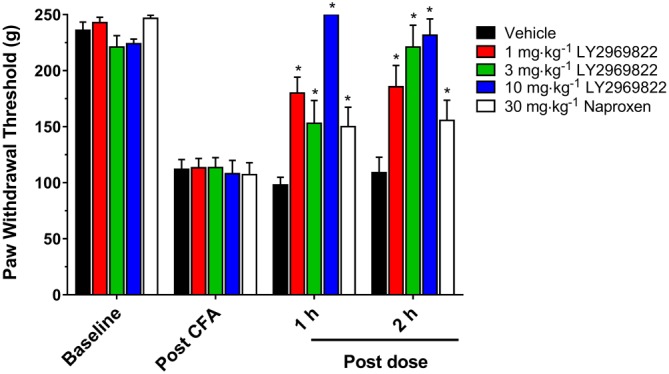
Effect of LY2969822 on the rat CFA model of inflammatory pain. Twenty‐four hours after unilateral injection of CFA into the hind paw mechanical hyperalgesia (baseline) was measured as a change in threshold to pinch (Randall Sellito Test). Vehicle or LY2969822 at 1, 3 and 10 mg·kg−1 was dosed p.o. and mechanical hyperalgesia assessed at 1 and 2 h post‐dosing. Naproxen, 30 mg·kg−1, was similarly dose p.o. and assessed at 1 and 2 h. Values are mean ± SEM, n = 9–10 per group. *Indicates significantly different versus vehicle response at the corresponding time point (P < 0.05, two‐way ANOVA followed by post hoc Dunnett's test).
Reversal of tactile hypersensitivity induced by spinal nerve ligation in rats
At 14 weeks post SNL surgery baseline tactile hypersensitivity was confirmed and rats randomized into treatment groups. The ability of a 10 mg·kg−1 (active drug equivalent) dose of LY2969822 dihydrate to attenuate the surgically induced tactile hypersensitivity was assessed at 1, 2 and 4 h post‐dose. As seen in Figure 6, LY2969822 reversed the tactile hypersensitivity resulting from a chronic SNL. The response persisted for at least 4 h and was similar at its peak to that seen with gabapentin.
Figure 6.
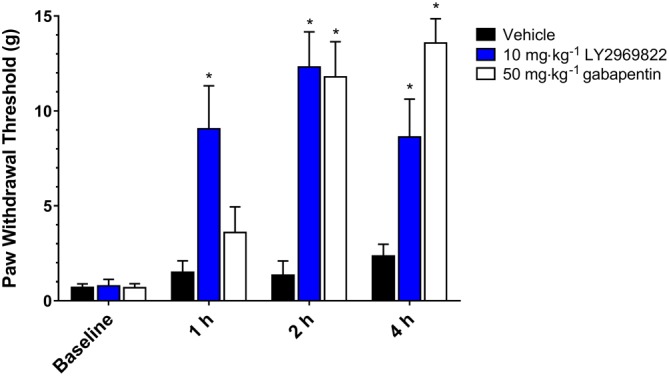
Effect of LY2969822 in the rat SNL model of tactile allodynia. Vehicles or 10 mg·kg−1 LY2969822 were dosed p.o. after the confirmation (baseline) of tactile hypersensitivity in rats that had undergone L5/L6 SNL 14 weeks previously. Tactile hypersensitivity was measured as a change in threshold to increasing force from calibrated von Frey monofilaments, determined at 1, 2 and 4 h post‐dosing. Values are mean ± SEM, n = 7 to 8 per group. *Indicates significantly different versus vehicle response at the corresponding time point (P < 0.05, two‐way ANOVA followed by post hoc Dunnett's test).
Reversal of tactile hypersensitivity induced by plantar incision in rats
Twenty‐four hours after baseline tactile measurements pre surgery and a controlled incision on the left hind paw followed by wound closure, the hypersensitivity of each animal was confirmed and animals randomly assigned to groups. Vehicle, 3–10 mg·kg−1 (active equivalent) of LY2969822 or 3 mg·kg−1 morphine, was administered, and the tactile threshold was determined after 90 min. As seen in Figure 7, a significant reversal of the incision site tactile hypersensitivity was seen after 10 but not 3 mg·kg−1 LY2969822. It should be noted that the magnitude of the response seen after the highest dose of LY2969822 was less than that seen with 3 mg·kg−1 morphine.
Figure 7.
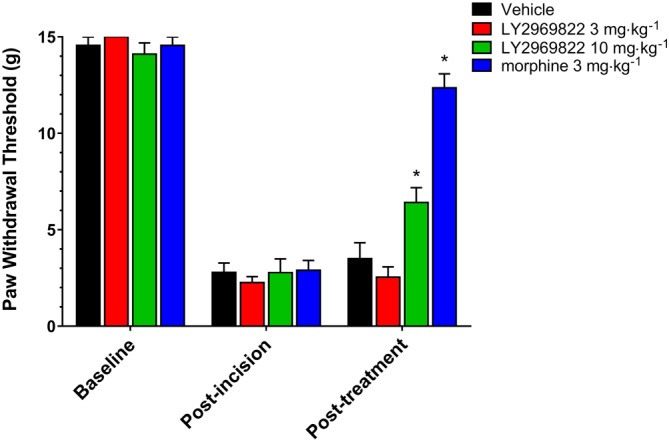
Effect of LY2969822 on the rat paw incision model of tactile allodynia. After measurement of tactile threshold (baseline), a surgical incision was made in the hind paw of rats. After a 24 h recovery period, confirmation of tactile allodynia was established (<4 g) and rats were randomized to treatment groups (post‐incision). At 90 min after administration of vehicle (1% HEC), 3 or 10 mg·kg−1 (active equivalent) of LY2969822 p.o. or 30 min after administration of 3 mg·kg−1 morphine by the s.c. route, the degree of tactile allodynia was measured. Values are mean ± SEM, n = 7 per group. *Indicates significantly different versus vehicle response at the corresponding time point (P < 0.05, two‐way ANOVA followed by post hoc Dunnett's test).
Inhibition of nociceptive response to colorectal distension (CRD) in normal and sensitized rats
In order to confirm the placement of EMG electrodes, the baseline response to a 60 mmHg increase in colon pressure was determined and is shown in Figure 8A in naïve rats and 24 h after sensitization by intracolonic infusion of dilute acetic acid (Figure 8B). Note the expected increased response seen in the rats after sensitization illustrated in Figure 8B. Rats were then treated with vehicle, 1 to 11 mg·kg−1 (active equivalent) of LY2969822 or 3 mg·kg−1 morphine and the response to 20, 40 or 60 mmHg recorded. As seen in Figure 8A/B, naïve or sensitized rats treated with LY2969822 demonstrated decreased nociceptive response (abdominal contraction) in a dose‐ and stimulus‐dependent fashion, with the highest dose tested showing similar effects to morphine. Note that due to an error in formulation, an 11 mg·kg−1 dose rather than a 10 mg·kg−1 dose was administered to the naïve animals.
Figure 8.
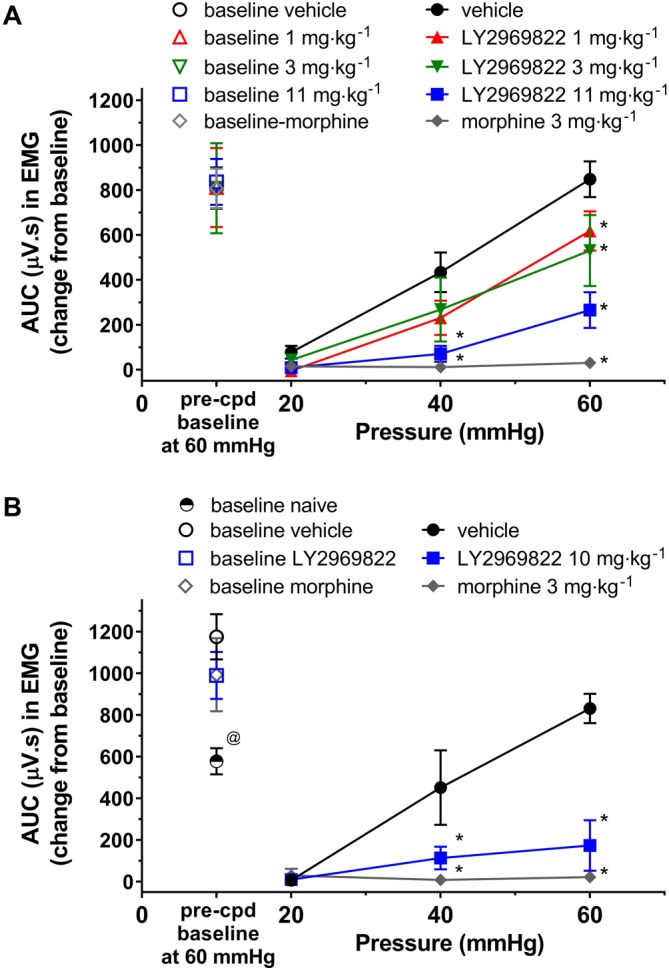
Effect of LY2969822 in the rat CRD model of visceral pain. (A) Subsequent to recovery from EMG electrode abdominal muscle implantation, baseline EMG readings with a 60 mmHg pressure stimulus were taken and animals randomized to treatment groups. Vehicle or LY2969822 1 to 11 mg·kg−1 (active equivalent) was dosed p.o. 90 min prior to beginning the CRD protocol. Note that due to an error in formulation an 11 mg·kg−1 dose rather than a 10 mg·kg−1 dose was administered to the naïve animals. Morphine (3 mg·kg−1, s.c. 30 min pretreatment) was utilized as a positive control. (B) Twenty‐four hours after sensitization of the colon by exposure to 1% acetic acid (2 mL), baseline EMG readings with a 60 mmHg pressure were determined and animals randomized to treatment groups. Vehicle or 10 mg·kg−1 LY2969822 was dosed p.o. 90 min prior to beginning the CRD protocol. Morphine (3 mg·kg−1, s.c. 30 min pretreatment) was utilized as a positive control. Values are mean ± SEM, n = 6–7 per group. *Indicates significantly different versus vehicle response at the same pressure (P < 0.05, two‐way ANOVA followed by Dunnett's test). @Indicates significantly different versus baseline vehicle response (P < 0.05, one‐way ANOVA followed by Dunnett's test).
Inhibition of acetic acid‐induced writhing in mice
Two hours following pretreatment with vehicle, 1–10 mg·kg−1 (active equivalent) of LY2969822 or 3 mg·kg−1 morphine, dilute acetic acid was injected i.p. and the number of writhings recorded for 20 min. As seen in Figure 9, a significant and dose‐dependent decrease in the numbers of writhings was evident with LY2969822 pretreatment. The magnitude of the response at the highest dose of LY2969822 tested was less than that seen with morphine in this pain model.
Figure 9.
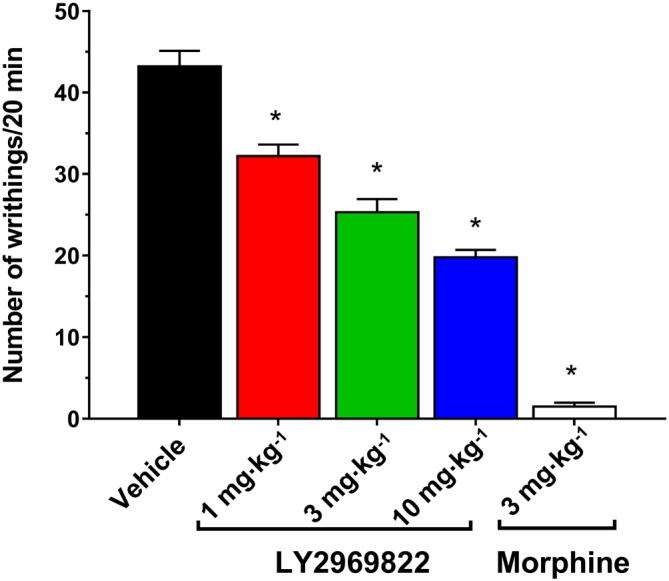
Effect of LY2969822 on the mouse acetic acid writhing model of visceral pain. Vehicle or LY2969822, 1 to 10 mg·kg−1 (active equivalent), was dosed p.o. 2 h prior to i.p. injections of 0.5% acetic acid. The number of writhing responses over a 20 min period was recorded. Morphine (3 mg·kg−1 s.c., 1 h pretreatment) was included as a positive control. Values are mean ± SEM, n = 10 per group. *Indicates significantly different versus vehicle response (P < 0.05, one‐way ANOVA followed by Dunnett's test).
Discussion
Previously, we reported the discovery and selective mGlu2/3 receptor agonist pharmacology of LY2934747 along with its rat pharmacokinetics and mGlu2‐mediated pharmacodynamic actions, including the inhibition of PCP‐induced ambulations and formalin‐induced nocifensive behaviours following either s.c. or i.p. dosing. However, LY2934747 was found to have a poor oral bioavailability, most likely due to poor absorption (Monn et al., 2015). Previous work with mGlu2/3 receptor agonists has demonstrated that in some cases, a prodrug approach can be employed to increase oral absorption. Specifically by coupling the parent amino acid to naturally occurring amino acids such as L‐alanine or L‐methionine, the resulting dipeptide‐like molecule can be recognized and actively transported by intestinal peptide transporters (e.g. PepT1) and subsequently undergo metabolic conversion to liberate the active mGlu2/3 receptor agonist (Bueno et al., 2005). Thus, the alanine amide prodrug of LY2934747, (Figure 1A, LY2969822) was synthesized and the resulting systemic and CSF levels of the active drug LY2937474 measured. As seen in Figure 1B, p.o. dosing of the prodrug LY2969822 resulted in rapid and dose‐dependent increases in plasma levels of the active mGlu2/3 receptor agonist LY2934747. CSF levels of the active agent after dosing with the prodrug at 1–10 mg·kg−1 (doses corrected for equivalence to LY2934747) exceeded the potency reported for inhibition of forskolin‐stimulated cAMP from rat cortical synaptosomes (EC50 = 14 nM, Monn et al., 2015), suggesting this would be an effective range to test for a pharmacodynamic response. Indeed as seen in Figure 2, treatment p.o. with the prodrug LY2969822 in the 1–10 mg·kg−1 (active equivalents) was able to block the nocifensive behaviours induced by formalin intraplantar injection as previously reported with an i.p. injection of 1–10 mg·kg−1 LY2934747.
The present work demonstrates an impressive breadth and depth of analgesic activity for the mGlu2/3 receptor agonist (LY2934747) and its corresponding prodrug (LY2969822) in a number of rodent models measuring nocifensive or pain related behaviours. This includes efficacy similar to that seen with positive controls in spontaneous measures such as formalin‐induced nocifensive responses and acetic‐acid‐induced writhing and evoked measures of sensitivity such as tactile allodynia or mechanical hyperalgesia. The ability to block both spontaneous and evoked painful responses is further strengthened by the work examining WDR neuronal responses after i.v. injection of LY2934747. After SNL, an increase in spontaneous activity of WDR neurons within the receptive field of the affected paw is well documented (Chu et al., 2004). Treatment with the mGlu2/3 receptor agonist decreased this spontaneous activity. Similarly, treatment with LY2934747 inhibited the increased firing seen with a protocol of electrically stimulation (wind‐up). Thus in both observational measures of painful behaviours and in the correlated measures of neuronal firing in spinal cord nociceptive neurons, the mGlu2/3 receptor agonist blocks spontaneous and evoked responses.
It is also worth noting that the mGlu2/3 receptor agonist prodrug is equally efficacious with both blockade of acute sensitization after intraplantar capsaicin or treatment of more prolonged inflammatory‐induced sensitization after CFA. Furthermore, the efficacy is not limited to skin, as LY2969822 was as effective as morphine in blocking the painful responses induced with CRD, that is, it showed effects in a visceral organ. However, LY2969822 was not robustly effective in all pain measurements. For instance, there was neither a clear dose‐dependent nor duration of action consistent with exposures, analgesic response seen when testing for the thermal hypersensitivity following capsaicin. It is possible that this may be due to interference with the measurement, as an increase in startle response was observed at the highest dose in these experiments. However, this may also be an indication of varying degrees of analgesic response depending upon the means utilized to induce the painful behaviour and/or the evoked stimulus (i.e. thermal vs. tactile). Although the less robust, but statistically significant, efficacious response to tactile allodynia in the rat paw incision model would argue against differential efficacy based on evoked stimulus as an explanation. The robust effect on tactile allodynia after SNL or capsaicin further suggests it is not simply a differential efficacy based on the sensitization method. The reason for these small but notable differences between models is unknown, but this highlights the importance of profiling potential analgesic agents in several models and considering the weight of evidence prior to advancing a molecule into human clinical trials.
Prolonged activation of GPCRs can result in receptor desensitization and behavioural tolerance, and repeated daily dosing of another mGlu2/3 receptor agonist, LY379268 (Monn et al., 1999), given once daily over 4 to 14 days resulted in the loss of efficacy in both pain (Jones et al., 2005) and psychomotor stimulant (Galici et al., 2005) assays. In the current study, we found that LY2969822 given p.o. once daily over 3 days at doses of 3 and 10 mg·kg−1 exhibited efficacy in the formalin assay that was indistinguishable from efficacy responses when these doses were acutely administered. The question of whether longer‐term dosing of LY2969822 might lead to tachyphylaxis in rodents was not explored; however, it bears mentioning that the development of tolerance following chronic exposure to mGlu2/3 receptor agonists in rodents is not consistently observed (see for instance Cartmell et al., 2000b; Schobel et al., 2013; Anderson et al., 2014; Battaglia et al., 2015) and may be a function of several interacting factors including species/strain, assay, dose, route of administration and specific agonist ligand. Importantly, there is no evidence that loss of efficacy on repeat dosing of mGlu2/3 receptor agonists in rodents translates to humans. For instance, in spite of evidence that repeated daily dosing of the mGlu2/3 receptor agonist LY354740 (eglumegad) (Monn et al., 1997) led to tolerance after only 3 days of dosing (Ahnaou et al., 2015), statistically significant anxiolytic efficacy observed in generalized anxiety disorder patients following 5 weeks dosing of LY354740 (unpublished data) or 8 weeks dosing of LY544344 (Dunayevich et al., 2007), an oral prodrug of LY354740 (Bueno et al., 2005) was either maintained or enhanced over the course of treatment. Similarly, in schizophrenia patients (Patil et al., 2007) or schizophrenia patient subgroups (Kinon et al., 2015; Nisenbaum et al., 2016), statistically significant antipsychotic efficacy was maintained or enhanced following up to 6 weeks dosing of LY2140023, an oral prodrug of the mGlu2/3 receptor agonist LY404039 (Monn et al., 2007).
In conclusion, oral administration of the prodrug LY2969822 in rodents results in rapid absorption and conversion to the active mGlu2/3 receptor agonist LY2934747. LY2934747 could suppress the evoked and spontaneous activity on WDR neurons in neuropathic animals, and its efficacy in preventing formalin‐induced nocifensive behaviours was mGlu2/3 receptor dependent. Dosing the mGlu2/3 receptor agonist prodrug resulted in a broad spectrum of analgesic actions in nociceptive sensitization, neuropathic, inflammatory and visceral pain models. Thus, treatment with this mGlu2/3 receptor agonist prodrug may demonstrate efficacy in controlled clinical chronic pain trial(s).
Author contributions
M.P.J. and J.A.M. contributed to experimental design and data analysis and wrote the manuscript. K.L.K. and J.D.K. contributed to the experimental design and data analysis of in vivo behavioural data. M.A.M. and E.S.N. contributed to the experimental design and data analysis of the in vivo electrophysiological data. R.M.A.S., B.M.F., L.Y., D.M. and D.L.L. contributed to experimental design and performed experiments. S.S. contributed to the experimental design and analysis of the pharmacokinetic experiments.
Conflict of interest
The authors were full time employees of Lilly Research Laboratories, Eli Lilly and Company, while undertaking this research. The work was funded by Eli Lilly and Company.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
The authors would like to acknowledge Melanie Owens and Xin Fang (Neurosolutions LTD, Coventry, UK) for performing the WDR neuron experiments under a contract with Eli Lilly and Company.
Johnson, M. P. , Muhlhauser, M. A. , Nisenbaum, E. S. , Simmons, R. M. A. , Forster, B. M. , Knopp, K. L. , Yang, L. , Morrow, D. , Li, D. L. , Kennedy, J. D. , Swanson, S. , and Monn, J. A. (2017) Broad spectrum efficacy with LY2969822, an oral prodrug of metabotropic glutamate 2/3 receptor agonist LY2934747, in rodent pain models. British Journal of Pharmacology, 174: 822–835. doi: 10.1111/bph.13740.
References
- Ago Y, Araki R, Yano K, Kawasaki T, Chaki S, Nakazato A et al. (2012). The selective metabotropic glutamate 2/3 receptor agonist MGS0028 reverses isolation rearing‐induced abnormal behaviors in mice. J Pharmacol Sci 118: 295–298. [DOI] [PubMed] [Google Scholar]
- Ahnaou A, Lavreysen H, Tresadern G, Cid JM, Drinkenburg WH (2015). MGlu2 receptor agonism, but not positive allosteric modulation, elicits rapid tolerance towards their primary efficacy on sleep measures in rats. PLoS One 10: e0144017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson PM, Pinault D, O'Brien TJ, Jones NC (2014). Chronic administration of antipsychotics attenuates ongoing and ketamine‐induced increases in cortical γoscillations. Int J Neuropsychopharmacol 17: 1895–1904. [DOI] [PubMed] [Google Scholar]
- Arendt‐Nielsen L, Petersen‐Felix S (1995). Wind‐up and neuroplasticity: is there a correlation to clinical pain? Eur J Anaesthesiol Suppl 10: 1–7. [PubMed] [Google Scholar]
- Battaglia G, Riozzi B, Bucci D, Di Menna L, Molinaro G, Pallottino S et al. (2015). Activation of mGlu3 metabotropic glutamate receptors enhances GDNF and GLT‐1 formation in the spinal cord and rescues motor neurons in the SOD‐1 mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 74: 126–136. [DOI] [PubMed] [Google Scholar]
- Bernabucci M, Notartomaso S, Zappulla C, Fazio F, Cannella M, Motolese M et al. (2012). N‐Acetyl‐cysteine causes analgesia by reinforcing the endogenous activation of type‐2 metabotropic glutamate receptors. Mol Pain 8: 77. doi:10.1186/1744‐8069‐8‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxall SJ, Berthele A, Laurie DJ, Sommer B, Zieglgansberger W, Urban L et al. (1998). Enhanced expression of metabotropic glutamate receptor 3 messenger RNA in the rat spinal cord during ultraviolet irradiation induced peripheral inflammation. Neuroscience 82: 591–602. [DOI] [PubMed] [Google Scholar]
- Brennan TJ, Vandermeulen EP, Gebhart GF (1996). Characterization of a rat model of incisional pain. Pain 64: 493–501. [DOI] [PubMed] [Google Scholar]
- Bueno AB, Catlow JT, Clay MP, Coffey S, Collado I, Dantzig AH et al. (2005). Dipeptides as effective prodrugs of the unnatural amino acid LY354740, a selective group II mGlu receptor agonist. J Med Chem 48: 5305–5320. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Zhou S, Govea R, Du J (2009). Group II/III metabotropic glutamate receptors exert endogenous activity‐dependent modulation of TRPV1 receptors on peripheral nociceptors. J Neurosci 31: 12727–12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartmell J, Monn JA, Schoepp DD (1999). The metabotropic glutamate 2/3 receptor agonists LY354740 and LY379268 selectively attenuate phencyclidine versus d‐amphetamine motor behaviors in rats. J Pharmacol Exp Ther 291: 161–170. [PubMed] [Google Scholar]
- Cartmell J, Monn JA, Schoepp DD (2000a). The mGlu(2/3) receptor agonist LY379268 selectively blocks amphetamine ambulations and rearing. Eur J Pharmacol 400: 221–224. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Monn JA, Schoepp DD (2000b). Tolerance to the motor impairment, but not to the reversal of PCP‐induced motor activities by oral administration of the mGlu2/3 receptor agonist, LY379268. Naunyn Schmiedebergs Arch Pharmacol 361: 39–46. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53: 55–63. [DOI] [PubMed] [Google Scholar]
- Chiechio S, Caricasole A, Barletta E, Storto M, Catania MV, Copani A et al. (2002). L‐acetylcarnitine induces analgesia by selectively up‐regulating mGlu2 metabotropic glutamate receptors. Mol Pharmacol 61: 989–996. [DOI] [PubMed] [Google Scholar]
- Chiechio S, Nicoletti F (2012). Metabotropic glutamate receptors and the control of chronic pain. Curr Opin Pharmacol 12: 28–34. [DOI] [PubMed] [Google Scholar]
- Chu KL, Faltynek CR, Jarvis MF, McGaraughty S (2004). Increased WDR spontaneous activity and receptive field size in rats following a neuropathic or inflammatory injury: implications for mechanical sensitivity. Neurosci Lett 371: 123–126. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin J‐P (1997). Pharmacology and functions of metabotropic glutamate receptors. Ann Rev Pharmacol Toxicol 37: 205–237. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson S, Golden JP, Copitsa BA, Rayb PR, Vogta SK, Valtchevaa MV et al. (2016). Group II mGluRs suppress hyperexcitability in mouse and human nociceptors. Pain 157: 2081–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon WJ (1980). Efficient analysis of experimental observations. Ann Rev Pharmacol Toxicol 20: 441–462. [DOI] [PubMed] [Google Scholar]
- Du J, Zhou S, Carlton SM (2008). Group II metabotropic glutamate receptor activation attenuates peripheral sensitization in inflammatory states. Neuroscience 154: 754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunayevich E, Erickson J, Levine L, Landbloom R, Schoepp DD, Tollefson GD (2007). Efficacy and tolerability of an mGlu2/3 agonist in the treatment of generalized anxiety disorder. Neuropsychopharmacology 33: 1–8. [DOI] [PubMed] [Google Scholar]
- Fabricius K, Helboe L, Fink‐Jensen A, Wortwein G, Steiniger‐Brach B (2011). Pharmacological characterization of social isolation‐induced hyperactivity. Psychopharmacology (Berl) 215: 257–266. [DOI] [PubMed] [Google Scholar]
- Galici R, Echemendia NG, Rodriguez AL, Conn PJ (2005). A selective allosteric potentiator of metabotropic glutamate (mGlu) 2 receptors has effects similar to an orthosteric mGlu2/3 receptor agonist in mouse models predictive of antipsychotic activity. J Pharmacol Exp Ther 315: 1181–1187. [DOI] [PubMed] [Google Scholar]
- Gu G, Lorrain DS, Wei H, Cole RL, Zhang X, Daggett LP et al. (2008). Distribution of metabotropic glutamate 2 and 3 receptors in the rat forebrain: implication in emotional responses and central disinhibition. Brain Res 1197: 47–62. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988). A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77–88. [DOI] [PubMed] [Google Scholar]
- Helton DR, Tizzano JP, Monn JA, Schoepp DD, Kallman MJ (1998). Anxiolytic and side‐effect profile of LY354740: a potent, highly selective, orally active agonist for group II metabotropic glutamate receptors. J Pharmacol Exp Ther 284: 651–660. [PubMed] [Google Scholar]
- Jett MF, Michelson S (1996). The formalin test in rat: validation of an automated system. Pain 64: 19025. [DOI] [PubMed] [Google Scholar]
- Jones CK, Eberle EL, Peters SC, Monn JA, Shannon HE (2005). Analgesic effects of the selective group II (mGlu2/3) metabotropic glutamate receptor agonists LY379268 and LY389795 in persistent and inflammatory pain models after acute and repeated dosing. Neuropharmacology 49: 206–218. [DOI] [PubMed] [Google Scholar]
- Jones CA, Brown AM, Auer DP, Fone KCF (2011). The mGluR2/3 agonist LY379268 reverses post‐weaning social isolation‐induced recognition memory deficits in the rat. Psychopharmacology (Berl) 214: 269–283. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM (1992). An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50: 355–363. [DOI] [PubMed] [Google Scholar]
- Kinon BJ, Millen BA, Zhang L, McKinzie DL (2015). Exploratory analysis for a targeted population responsive to the metabotropic glutamate 2/3 agonist pomaglumetad methionil in schizophrenia. Biol Psychiatry 78: 754–762. [DOI] [PubMed] [Google Scholar]
- Kłodzińska A, Chojnacka‐Wójcik E, Pałucha A, Brański P, Popik P, Pilc A (1999). Potential anti‐anxiety, anti‐addictive effects of LY 354740, a selective group II glutamate metabotropic receptors agonist in animal models. Neuropharmacology 38: 1831–1839. [DOI] [PubMed] [Google Scholar]
- Kumar N, Laferriere A, Yu JSC, Poon T, Coderre TJ (2010). Metabotropic glutamate receptors (mGluRs) regulate noxious stimulus‐induced glutamate release in the spinal cord dorsal horn of rats with neuropathic and inflammatory pain. J Neurochem 114: 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B, Adams BW (1998). Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonists in rats. Science 281: 1349–1352. [DOI] [PubMed] [Google Scholar]
- Monn JA, Valli MJ, Massey SM, Wright RA, Salhoff CR, Johnson BG et al. (1997). Design, synthesis and pharmacological characterization of (+)‐2‐aminobicyclo[3.1.0]hexane‐2,6‐dicarboxylic Acid (LY354740): a potent, selective and orally active group 2 metabotropic glutamate receptor agonist possessing anticonvulsant and anxiolytic properties. J Med Chem 40: 528–537. [DOI] [PubMed] [Google Scholar]
- Monn JA, Valli MJ, Massey SM, Hansen MM, Kress TJ, Wepsiec JP et al. (1999). Synthesis, pharmacological characterization and molecular modeling of heterobicyclic amino acids related to LY354740: Identification of LY379268 and LY389795: two new potent, selective and systemically active agonists for group II metabotropic glutamate receptors. J Med Chem 42: 1027–1040. [DOI] [PubMed] [Google Scholar]
- Monn JA, Valli MJ, Massey SM, Henry SS, Stephenson GA, Bures M et al. (2007). Synthesis and metabotropic glutamate receptor activity of S‐oxidized variants of LY389795: Identification of potent, selective and orally bioavailable agonists for mGlu2/3 receptors. J Med Chem 50: 233–240. [DOI] [PubMed] [Google Scholar]
- Monn JA, Prieto L, Toboada L, Pedregal C, Hao J, Reinhard MR et al. (2015). Synthesis and pharmacological characterization of C4‐disubstituted analogs of 1S,2S,5R,6S‐2‐aminobicyclo[3.1.0]hexane‐2,6‐dicarboxylate: identification of a potent, selective metabotropic glutamate receptor agonist and determination of agonist‐bound human mGlu2 and mGlu3 amino terminal domain structures. J Med Chem 58: 1776–1794. [DOI] [PubMed] [Google Scholar]
- Nakazato A, Kumagai T, Sakagami K, Yoshikawa R, Suzuki Y, Shigeyuki C et al. (2000). Synthesis, SARs, and pharmacological characterization of 2‐amino‐3 or 6‐fluorobicyclo[3.1.0]hexane‐2,6‐dicarboxylic acid derivatives as potent, selective, and orally active group II metabotropic glutamate receptor agonists. J Med Chem 43: 4893–4909. [DOI] [PubMed] [Google Scholar]
- Neki A, Ohishi H, Kaneko T, Shigemoto R, Nakanishi S, Mizuno N (1996). Pre‐ and postsynaptic localization of a metabotropic glutamate receptor, mGluR2, in the rat brain: an immunohistochemical study with a monoclonal antibody. Neurosci Lett 202: 197–200. [DOI] [PubMed] [Google Scholar]
- Ness TJ, Gebhart GF (1988). Colorectal distension as a noxious visceral stimulus: physiologic and pharmacologic characterization of pseudo‐affective reflexes in the rat. Brain Res 450: 153–169. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Chen PS, Willis WD (2000). Groups II and III metabotropic glutamate receptors differentially modulate brief and prolonged nociception in primate STT cells. J Neurophysiol 84: 2998–3009. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD et al. (2011). Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology 60: 1017–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisenbaum LK, Downing ACM, Zhao F, Millen BA, Munsie L, Kinon BJ et al. (2016). Serotonin 2A receptor SNP rs7330461 association with treatment response to pomaglumetad methionil in patients with schizophrenia. J Pers Med 6. doi:10.3390/jpm6010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N (1993). Distribution of the mRNA for a metabotropic glutamate receptor (mGluR3) in the rat brain: an in situ hybridization study. J Comp Neurol 335: 252–266. [DOI] [PubMed] [Google Scholar]
- Osikowicz M, Skup M, Mika J, Makuch W, Czarkowska‐Bauch J, Przewlocka B (2009). Glial inhibitors influence the mRNA and protein levels of mGlu2/3, 5 and 7 receptors and potentiate the analgesic effects of their ligands in a mouse model of neuropathic pain. Pain 147: 175–186. [DOI] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV et al. (2007). Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med 13: 1102–1107. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang Y‐X, Niedzielski AS, Wenthold RJ (1996). The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience 71: 949–976. [DOI] [PubMed] [Google Scholar]
- Pin J‐P, Archer F (2002). The metabotropic glutamate receptors: structure, activation mechanism, and pharmacology. Curr Drug Target CNS Neurol Disord 1: 297–317. [DOI] [PubMed] [Google Scholar]
- Ren B‐X, Gu X‐P, Zheng Y‐G, Liu C‐L, Wang D, Sun Y‐E et al. (2012). Intrathecal injection of metabotropic glutamate receptor subtype 3 and 5 agonist/antagonist attenuates bone cancer pain by inhibition of spinal astrocyte activation in a mouse model. Anesthesiology 116: 122–132. [DOI] [PubMed] [Google Scholar]
- Richards G, Messer J, Malherbe P, Pink R, Brockhaus M, Stadler H et al. (2005). Distribution and abundance of metabotropic glutamate receptor subtype 2 in rat brain revealed by [3H]LY354740 binding in vitro and quantitative radioautoradiography: Correlation with the sites of synthesis, expression, and agonist stimulation of [35S]GTPγS binding. J Comp Neurol 487: 15–27. [DOI] [PubMed] [Google Scholar]
- Rorick‐Kehn LM, Johnson BG, Burkey JL, Wright RA, Calligaro DO, Marek GJ et al. (2007). Pharmacological and pharmacokinetic properties of a structurally novel, potent, and selective metabotropic glutamate 2/3 receptor agonist: In vitro characterization of agonist (−)‐(1R,4S,5S,6S)‐4‐Amino‐2‐sulfonylbicyclo[3.1.0]‐hexane‐4,6‐dicarboxylic acid (LY404039). J Pharmacol Exp Ther 321: 308–317. [DOI] [PubMed] [Google Scholar]
- Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I et al. (2013). Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron 78: 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA (1999). Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 38: 1431–1476. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Wright RA, Levine LR, Gaydos B, Potter WZ (2003). LY354740, an mGlu2/3 receptor agonist as a novel approach to treat anxiety/stress. Stress 6: 189–197. [DOI] [PubMed] [Google Scholar]
- Simmons RMA, Webster AA, Kalra AB, Iyengar S (2002). Group II mGluR receptor agonists are effective in persistent and neuropathic pain models in rats. Pharmacol Biochem Behav 73: 419–427. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Millan MJ, Herz A (1988). Unilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholds. Pharmacol Biochem Behav 31: 445–451. [DOI] [PubMed] [Google Scholar]
- Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA, Schoepp DD (2005). Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Discov 4: 131–144. [DOI] [PubMed] [Google Scholar]
- Takamori K, Hirota S, Chaki S, Tanaka M (2003). Antipsychotic action of selective group II metabotropic glutamate receptor agonist MGS0008 and MGS0028 on conditioned avoidance responses in the rat. Life Sci 73: 1721–1728. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62: 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truini A, Piroso S, Pasquale E, Notartomaso S, Di Stefano G, Lattanzi R et al. (2015). N‐acetyl‐cysteine, a drug that enhances the endogenous activation of group‐II metabotropic glutamate receptors, inhibits nociceptive transmission in humans. Mol Pain 11: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varney MA, Gereau RW (2002). Metabotropic glutamate receptor involvement in models of acute and persistent pain: prospects for the development of novel analgesics. Curr Drug Target CNS Neurol Disord 1: 283–296. [DOI] [PubMed] [Google Scholar]
- Whiteside GT, Harrison J, Boulet J, Mark L, Pearson M, Gottshall S et al. (2004). Pharmacological characterisation of a rat model of incisional pain. Br J Pharmacol 141: 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright RA, Johnson BG, Zhang C, Salhoff C, Kingston AE, Calligaro DO et al. (2013). CNS distribution of metabotropic glutamate 2 and 3 receptors: transgenic mice and [3H]LY459477 autoradiography. Neuropharmacology 66: 89–98. [DOI] [PubMed] [Google Scholar]