

Abstract
Human podoplanin (hPDPN), a platelet aggregation‐inducing transmembrane glycoprotein, is expressed in different types of tumors, and it binds to C‐type lectin‐like receptor 2 (CLEC‐2). The overexpression of hPDPN is involved in invasion and metastasis. Anti‐hPDPN monoclonal antibodies (mAbs) such as NZ‐1 have shown antitumor and antimetastatic activities by binding to the platelet aggregation‐stimulating (PLAG) domain of hPDPN. Recently, we developed a novel mouse anti‐hPDPN mAb, LpMab‐2, using the cancer‐specific mAb (CasMab) technology. In this study we developed chLpMab‐2, a human–mouse chimeric anti‐hPDPN antibody, derived from LpMab‐2. chLpMab‐2 was produced using fucosyltransferase 8‐knockout (KO) Chinese hamster ovary (CHO)‐S cell lines. By flow cytometry, chLpMab‐2 reacted with hPDPN‐expressing cancer cell lines including glioblastomas, mesotheliomas, and lung cancers. However, it showed low reaction with normal cell lines such as lymphatic endothelial and renal epithelial cells. Moreover, chLpMab‐2 exhibited high antibody‐dependent cellular cytotoxicity (ADCC) against PDPN‐expressing cells, despite its low complement‐dependent cytotoxicity. Furthermore, treatment with chLpMab‐2 abolished tumor growth in xenograft models of CHO/hPDPN, indicating that chLpMab‐2 suppressed tumor development via ADCC. In conclusion, chLpMab‐2 could be useful as a novel antibody‐based therapy against hPDPN‐expressing tumors.
Keywords: Antibody‐dependent cellular cytotoxicity, chimeric antibody, human podoplanin, monoclonal antibody
Introduction
Human podoplanin (hPDPN), also known as hT1α, hAggrus, or gp36, is expressed in many types of cancers, such as malignant brain tumors, malignant mesotheliomas, lung cancers, esophageal cancers, testicular tumors, bladder cancers, osteosarcomas, and fibrosarcomas 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15. hPDPN expression is observed only in squamous cell carcinomas of lung and esophageal cancers, indicating that hPDPN exhibits histological type‐specific expression. The expression of hPDPN in cancer‐associated fibroblasts contributes to a poor prognosis 16, 17, 18, 19, 20, 21. C‐type lectin‐like receptor 2 (CLEC‐2) is an endogenous receptor of hPDPN 22, 23. CLEC‐2 binds to hPDPN through residues Glu47 and Asp48 within its platelet aggregation‐stimulating (PLAG) domain and to α‐2,6‐linked sialic acid, which is attached to Thr52 24.
Membrane proteins could be targeted by antibody‐based therapy if (1) they possess cancer‐specific mutations outside the membrane 25, (2) they are overexpressed in cancers rather than in normal tissues 26, 27, 28, or (3) they are posttranslationally modified by phosphorylation or glycosylation 29, 30. Although many anti‐hPDPN monoclonal antibodies (mAbs) are commercially available, most of them react with the N‐terminus of hPDPN, and they do not fulfill the above‐mentioned criteria 6, 31, 32, 33, 34, 35. hPDPN is highly expressed in normal lymphatic endothelial cells (LECs) and normal lung type‐I alveolar cells at the same level as in cancer cells. We previously produced a rat anti‐hPDPN mAb (NZ‐1), which detects hPDPN with high specificity and sensitivity 6, 10, 31. NZ‐1 is efficiently internalized by glioma cell lines and accumulates in tumors in vivo; therefore, it has been suggested to be a suitable candidate for therapy against malignant gliomas 5, 10. Moreover, NZ‐1 inhibits tumor cell‐induced platelet aggregation and tumor metastasis 23. NZ‐1 mediates antibody‐dependent cellular cytotoxicity (ADCC) and complement‐dependent cytotoxicity (CDC) against tumor cells that express hPDPN 36. Furthermore, human–rat chimeric antibodies, such as NZ‐8 and NZ‐12, exhibit high ADCC and CDC in vitro, and they show very high antitumor activities and neutralizing capabilities 36, 37. Nevertheless, these chimeric mAbs are not cancer‐specific; therefore, cancer‐specific anti‐hPDPN chimeric or humanized antibodies should be developed to prevent unfavorable side effects.
We have developed the original technology to produce cancer‐specific mAbs (CasMabs) against hPDPN 29, 38, 39, 40, 41, 42, 43, 44, 45, 46. The established LpMab‐2 recognized the aberrant O‐glycosylation and Thr55–Leu64 peptide of hPDPN 29. LpMab‐2 reacted with hPDPN‐expressing cancer cells but not with normal cells, as shown by flow cytometry and immunohistochemistry. Therefore, LpMab‐2 is an anti‐hPDPN CasMab that can be used for molecular‐targeted therapy against hPDPN‐expressing cancers. In immunohistochemistry, less than 10% of hPDPN‐expressing cancer cells were recognized by LpMab‐2. In contrast, another anti‐hPDPN mAb LpMab‐7 reacted with both hPDPN‐expressing cancer cells and normal cells 42. We identified the minimum epitope of LpMab‐7 as Arg79–Leu83 of hPDPN using ELISA, Western‐blot, and flow cytometry. We further produced a human–mouse chimeric anti‐hPDPN mAb, chLpMab‐7 40. chLpMab‐7 showed ADCC and CDC, and inhibited the growth of hPDPN‐expressing tumors in vivo.
In this study, we developed and characterized chLpMab‐2, a human–mouse chimeric anti‐hPDPN antibody derived from LpMab‐2.
Materials and Methods
Antibodies
LpMab‐2, a mouse anti‐hPDPN mAb (IgG1, kappa), was developed as previously described 29. Human IgG was purchased from Beckman Coulter, Inc. (Fullerton, CA). To generate human–mouse chimeric anti‐hPDPN (chLpMab‐2), appropriate VH and VL cDNAs of mouse LpMab‐2 and CH and CL of human IgG1 were subcloned into pCAG‐Ble and pCAG‐Neo vectors (Wako Pure Chemical Industries, Ltd., Osaka, Japan), respectively. CHO‐S/fucosyltransferase 8 (FUT8)‐KO (PDIS‐5) cell lines were generated by transfecting CRISPR/Cas9 plasmids that targeted FUT8 (Target ID: HS0000547010; Sigma‐Aldrich Corp., St. Louis, MO) into CHO‐S cells (Thermo Fisher Scientific Inc., Waltham, MA) using a Gene Pulser Xcell electroporation system. PDIS‐5 cells were screened using Aleuria aurantia lectin. Antibody expression vectors were transfected into PDIS‐5 using the Lipofectamine LTX reagent (Thermo Fisher Scientific Inc.). Stable transfectants of PDIS‐5/chLpMab‐2 were selected by cultivating the transfectants in a medium containing 0.5 mg/mL of both geneticin and zeocin (InvivoGen, San Diego, CA). PDIS‐5/chLpMab‐2 cells were cultivated in CHO‐S‐SFM II medium (Thermo Fisher Scientific Inc.). chLpMab‐2 was purified using Protein G‐Sepharose (GE healthcare Bio‐Sciences, Pittsburgh, PA).
Cell lines
The cell lines LN229, HEK‐293T, NCI‐H226, Met‐5A, Chinese hamster ovary (CHO)‐K1, and P3U1 were obtained from the American Type Culture Collection (Manassas, VA). The LN319 cell line was provided by Prof. Kazuhiko Mishima (Saitama Medical University, Saitama, Japan) 47. Human LECs and PC‐ 10 cells were purchased from Cambrex Corp. (Walkersville, MD) and Immuno‐ Biological Laboratories Co., Ltd. (Gunma, Japan), respectively. LN229 and CHO‐K1 cells were transfected with hPDPN plasmids using Lipofectamine 2000 (Thermo Fisher Scientific Inc.) according to the manufacturer's instructions 29. The LN319/hPDPN‐KO cell line (PDIS‐6) was generated by transfection using CRISPR/Cas9 plasmids (Target ID: HS0000333287) that targeted PDPN (Sigma‐Aldrich, St. Louis, MO), as previously described 48. The cell lines CHO‐S/GnT‐1‐KO (PDIS‐9) and CHO‐S/SLC35A1‐KO (PDIS‐14) were generated by transfecting TALEN and CRISPR/Cas9 plasmids, respectively. The former plasmid‐targeted hsMgat1 (Wako Pure Chemical Industries Ltd.) and the latter targeted SLC35A1 (Target ID: HS0000168432; Sigma‐Aldrich) 49.
The CHO‐K1, CHO/hPDPN, NCI‐H226, PC‐10, and P3U1 cells were cultured in RPMI 1640 medium containing L‐glutamine (Nacalai Tesque, Inc., Kyoto, Japan). The LN229, LN229/hPDPN, LN319, HEK‐293T, and PDIS‐6 cells were cultured at 37°C in a humidified atmosphere containing 5% CO2 in Dulbecco's Modified Eagle's Medium containing L‐glutamine (Nacalai Tesque, Inc.) and 10% heat‐inactivated fetal bovine serum (FBS) (Thermo Fisher Scientific Inc.). CHO‐S, PDIS‐9, and PDIS‐14 were cultured in CHO‐S‐SFMII medium (Thermo Fisher Scientific Inc.). LECs were cultured in the endothelial cell medium EGM‐2MV supplemented with 5% FBS (Cambrex Corp.). All media contained 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 25 μg/mL of amphotericin B (Nacalai Tesque, Inc.).
Flow cytometry
The cell lines were harvested after brief exposure to 0.25% trypsin/1 mmol/L EDTA (Nacalai Tesque, Inc.). After washing with 0.1% BSA in PBS, the cells were treated with primary mAbs for 30 min at 4°C, followed by treatment with FITC‐labeled goat anti‐human IgG (Thermo Fisher Scientific Inc.). Fluorescence data were acquired using Cell Analyzer EC800 (Sony Corp., Tokyo, Japan).
Preparation of effector cells
Effector cells were prepared as previously described 9. Human peripheral blood mononuclear cells (MNCs) were obtained from leukocytes, which were separated from the peripheral blood of healthy donors. The study with human subjects was approved by the Ethics Committee of Tokushima University.
ADCC
ADCC was determined with the 51Cr release assay 9. Target cells were incubated with 0.1 μCi of 51Cr‐sodium chromate at 37°C for 1 h. After washing three times with RPMI 1640 supplemented with 10% FBS, 51Cr‐labeled target cells were seeded in 96‐well plates in triplicate. Human peripheral blood MNCs and chLpMab‐2 or control human IgG were added to the cells. After 6 h of incubation, 51Cr released from cells into the supernatant (100 μL) was measured using a gamma counter (PerkinElmer, Waltham, MA). The percentage of cytotoxicity was calculated using the following formula: % specific lysis = (E − S)/(M − S) × 100, where E is the release in the test sample, S is the spontaneous release, and M is the maximum release.
CDC
CDC was evaluated by the 51Cr release assay as previously described 9. The CHO/hPDPN cells (target cells) were incubated with 51Cr‐sodium chromate (0.1 μCi) for 1 h at 37°C. After incubation, the cells were washed with RPMI 1640 supplemented with 10% FBS. The 51Cr‐labeled cells were incubated with baby rabbit complement (Cedarlane, Ontario, Canada) at a dilution of 1:32 and chLpMab‐2 (0.01–10 μg/mL) or control human IgG (0.01–10 μg/mL) for 3 h in 96‐well plates. After incubation, 51Cr in the supernatant was measured using a gamma counter. The percentage of cytotoxicity was calculated using the following formula: % specific lysis = (E − S)/(M − S) × 100, where E is the release in the test sample, S is the spontaneous release, and M is the maximum release.
Antitumor activity of anti‐hPDPN antibodies
CHO/hPDPN cells were trypsinized and washed with PBS. The cell density was adjusted with PBS to 5.0 × 107 cells/mL, and 100 μL/animal of the cell suspension was subcutaneously inoculated into BALB/c nude mice. After 1 day, 100 μL of 1 mg/mL of chLpMab‐2 and human IgG were injected into the peritoneal cavity of mice, once a week for 4 weeks (control group, n = 6; chLpMab‐2 group, n = 6). Human NK cells (5.0 × 105 cells, Takara Bio Inc., Shiga, Japan) were injected around the tumors 4 and 11 days after cell inoculation. The tumor diameter was measured every 3–4 days and was calculated using the following formula: volume = W2 × L/2, where W is the short diameter and L is the long diameter. The mice were euthanized 21 days after cell implantation.
Statistical analysis
All data were expressed as means ± SEMs. Student's t‐test, Mann–Whitney U‐test, one‐way ANOVA followed by Tukey–Kramer multiple comparisons, and two‐way ANOVA were performed as appropriate. P values less than 0.05 were considered to be statistically significant. All statistical tests were two‐sided.
Results
Production of chLpMab‐2
We developed chLpMab‐2 from a mouse mAb, LpMab‐2. chLpMab‐2 reacted with LN229/hPDPN cells as revealed by flow cytometry (Fig. 1A). chLpMab‐2 detected endogenous hPDPN in glioblastoma cell line LN319 and not in the LN319/hPDPN‐KO cells (PDIS‐6) (Fig. 1B). Our results showed that chLpMab‐2 was specific against hPDPN.
Figure 1.
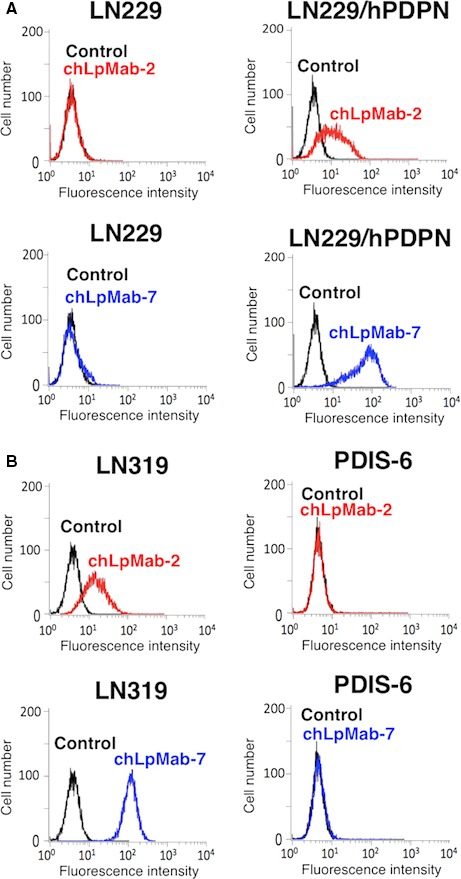
Flow cytometric analysis using chLpMab‐2 to detect hPDPN expression. (A) LN229 and LN229/hPDPN cells were treated with chLpMab‐2 (1 μg/mL, red), chLpMab‐7 (1 μg/mL, blue), and PBS (black) for 30 min at 4°C, followed by treatment with antihuman IgG‐FITC. (B) LN319 and LN319/hPDPN‐KO cells (PDIS‐6) were treated with chLpMab‐2 (1 μg/mL, red), chLpMab‐7 (1 μg/mL, blue), and PBS (black) for 30 min at 4°C, followed by treatment with antihuman IgG‐FITC. Fluorescence data were collected using Cell Analyzer EC800.
Flow cytometric analyses of chLpMab‐2 in cancer and normal cell lines
hPDPN is expressed in cancers such as brain tumors and mesotheliomas. By flow cytometry, chLpMab‐2 detected endogenous PDPN in human cancer cell lines such as PC‐10 of lung squamous cell carcinoma and NCI‐H226 of mesothelioma (Fig. 2A). A positive control, chLpMab‐7, detected PDPN expression in two cancer cell lines (Fig. 2A). Because of low hPDPN expression in NCI‐H226 29, the reaction of chLpMab‐2 against NCI‐H226 was much lower than that of chLpMab‐7. In contrast, chLpMab‐2 failed to detect the expression of hPDPN in normal cells such as renal epithelial cells (HEK‐293T) and the mesothelial cell line Met‐5A (Fig. 2B). However, chLpMab‐7 reacted with these cells (Fig. 2B), indicating that chLpMab‐2 was cancer‐specific. These results are consistent with those of our previous study 29.
Figure 2.
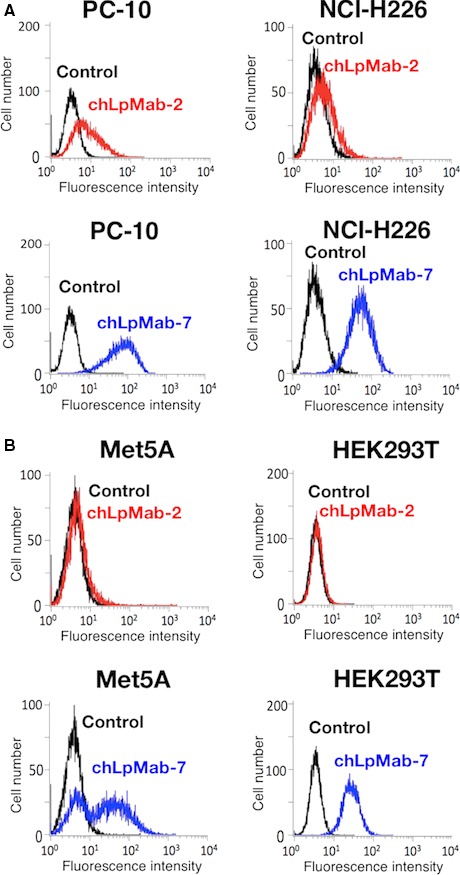
Flow cytometric analysis using chLpMab‐2 to detect PDPN expression in human cancer and normal cells. (A) Human cancer cell lines such as lung squamous cell carcinoma (PC‐10) and mesothelioma (NCI‐H226) were treated with chLpMab‐2 (1 μg/mL, red), chLpMab‐7 (1 μg/mL, blue), and PBS (black) for 30 min at 4°C, followed by treatment with antihuman IgG‐FITC. (B) Normal human cell lines such as mesothelial cells (Met5A) and renal epithelial cells (HEK‐293T) were treated with chLpMab‐2 (1 μg/mL, red), chLpMab‐7 (1 μg/mL, blue), and PBS (black) for 30 min at 4°C, followed by treatment with antihuman IgG‐FITC. Fluorescence data were acquired using Cell Analyzer EC800.
Characterization of chLpMab‐2 using glycan‐deficient cell lines
Previous studies have shown that hPDPN is O‐glycosylated and not N‐glycosylated 6, 24, 50, 51, 52. We used a GnT‐1‐KO cell line (CHO‐S/GnT‐1‐KO, PDIS‐9) and a CMP‐sialic acid transporter (SLC35A1)‐KO cell line (CHO‐S/SLC35A1‐KO, PDIS‐14) 49 to characterize chLpMab‐2. As shown in Figure 3B, chLpMab‐7 reacted with CHO‐S/hPDPN, PDIS‐9/hPDPN, and PDIS‐14/hPDPN cells transfected with the hPDPN expression vector. In contrast, chLpMab‐2 reacted with the CHO‐S/hPDPN and PDIS‐9/hPDPN cells and not with the PDIS‐14/hPDPN cells (Fig. 3A). This result agrees with that of our previous study 29 that the epitope of chLpMab‐2 includes sialic acid.
Figure 3.
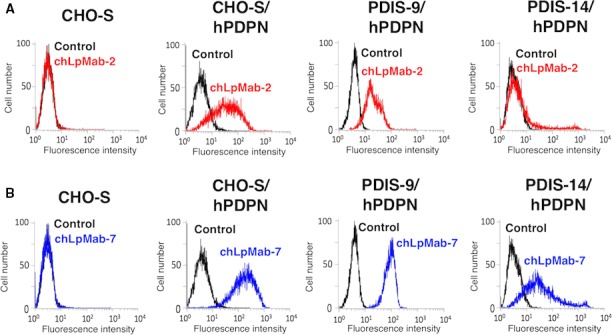
Flow cytometric analysis using chLpMab‐2 to detect hPDPN expression in N‐glycan and sialic acid‐deficient cells. CHO‐S, CHO‐S/hPDPN, PDIS‐9/hPDPN, and PDIS‐14/hPDPN cells were reacted with chLpMab‐2 (A, 1 μg/mL; red), chLpMab‐7 (B, 1 μg/mL; blue), and PBS (black) for 30 min at 4°C, followed by treatment with antihuman IgG‐FITC. Fluorescence data were acquired using Cell Analyzer EC800.
ADCC and CDC
LpMab‐2 was previously determined to belong to the mouse IgG1 subclass that failed to induce ADCC and CDC 29. Therefore, in this study, we converted LpMab‐2 to chLpMab‐2 of human IgG1 to study ADCC and CDC 40. As shown in Figure 4A, chLpMab‐2 showed ADCC against the LN319, PC‐10, and NCI‐H226 cell lines. The CHO/hPDPN and CHO‐K1 cells were used to study the specificity of chLpMab‐2 against hPDPN. As shown in Figure 4B, chLpMab‐2 showed ADCC against the CHO/hPDPN cells and not against the hPDPN‐negative CHO‐K1 cells. Furthermore, chLpMab‐2‐induced ADCC against CHO/hPDPN in a dose‐dependent manner (Fig. 5A). In contrast, chLpMab‐2 shows weak CDC against CHO/hPDPN cells (Fig. 5B) and against LN319, PC‐10, and NCI‐H226 cell lines (data not shown).
Figure 4.
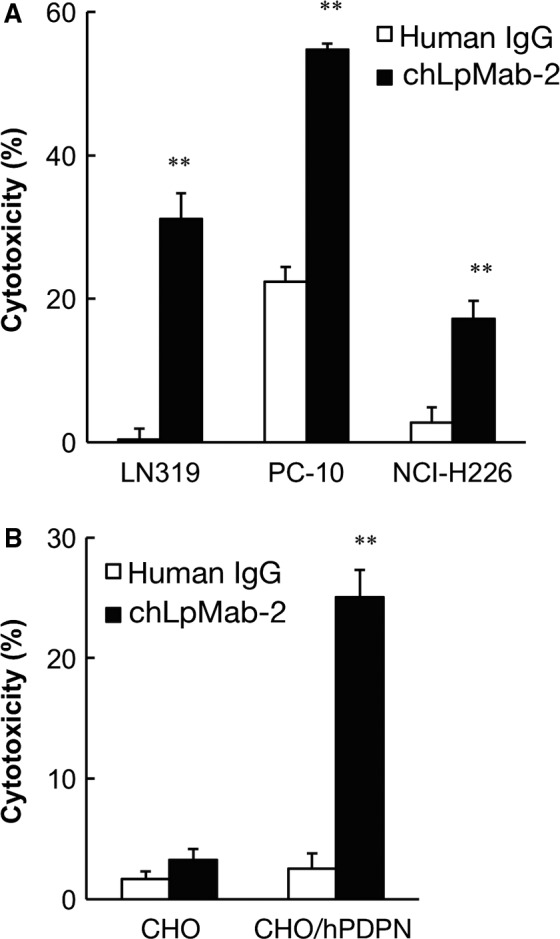
ADCC of chLpMab‐2 against hPDPN‐expressing cell lines. (A) ADCC of chLpMab‐2 using human MNCs against (A) LN319, PC‐10, and NCI‐H226 and (B) CHO and CHO/hPDPN were evaluated by a 6‐h 51Cr release assay in the presence of 1 μg/mL of antibody with an E/T ratio of 100. **P < 0.01
Figure 5.
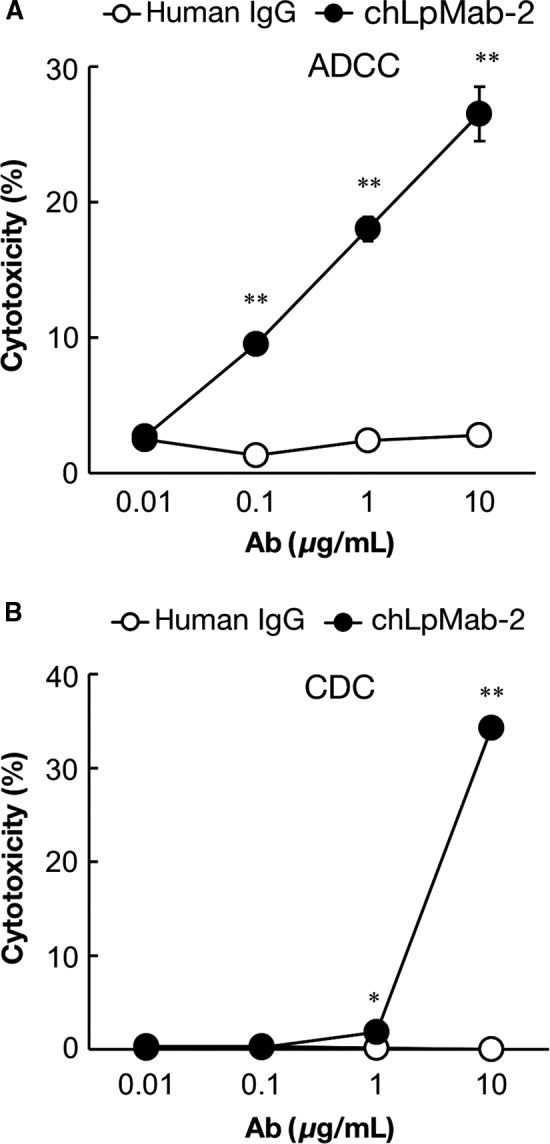
ADCC and complement‐dependent cytotoxicity (CDC) of chLpMab‐2 against CHO/hPDPN cells. (A) ADCC of chLpMab‐2 using human MNCs against CHO/hPDPN was evaluated by a 6‐h 51Cr release assay in the presence of 0.01–10 μg/mL of antibody with an E/T ratio of 100. (B) CDC of chLpMab‐2 using baby rabbit complement against CHO/hPDPN was evaluated by 51Cr release assay in the presence of 0.01–10 μg/mL of antibody. *P < 0.05; **P < 0.01
Antitumor activity of chLpMab‐2
To study the antitumor activity of chLpMab‐2 on primary tumor growth in vivo, the CHO/hPDPN cells were subcutaneously implanted into the flanks of nude mice. The LN319, PC‐10, and NCI‐H226 cells were not tested because they were not suitable for xenograft models. chLpMab‐2 and control human IgG were injected into the peritoneal cavity of mice, once weekly for 4 weeks (n = 6 each), and human NK cells were injected twice around the tumors. Tumor formation was observed in mice from the control and treated groups. However, chLpMab‐2 significantly reduced tumor development compared with control human IgG (Fig. 6A and B). The tumor volume was significantly reduced by chLpMab‐2 treatment on day 15, 18, and 21 (Fig. 6C). These results indicate that the administration of chLpMab‐2 with the NK cells inhibited the primary tumor growth of the CHO/hPDPN cells in vivo.
Figure 6.
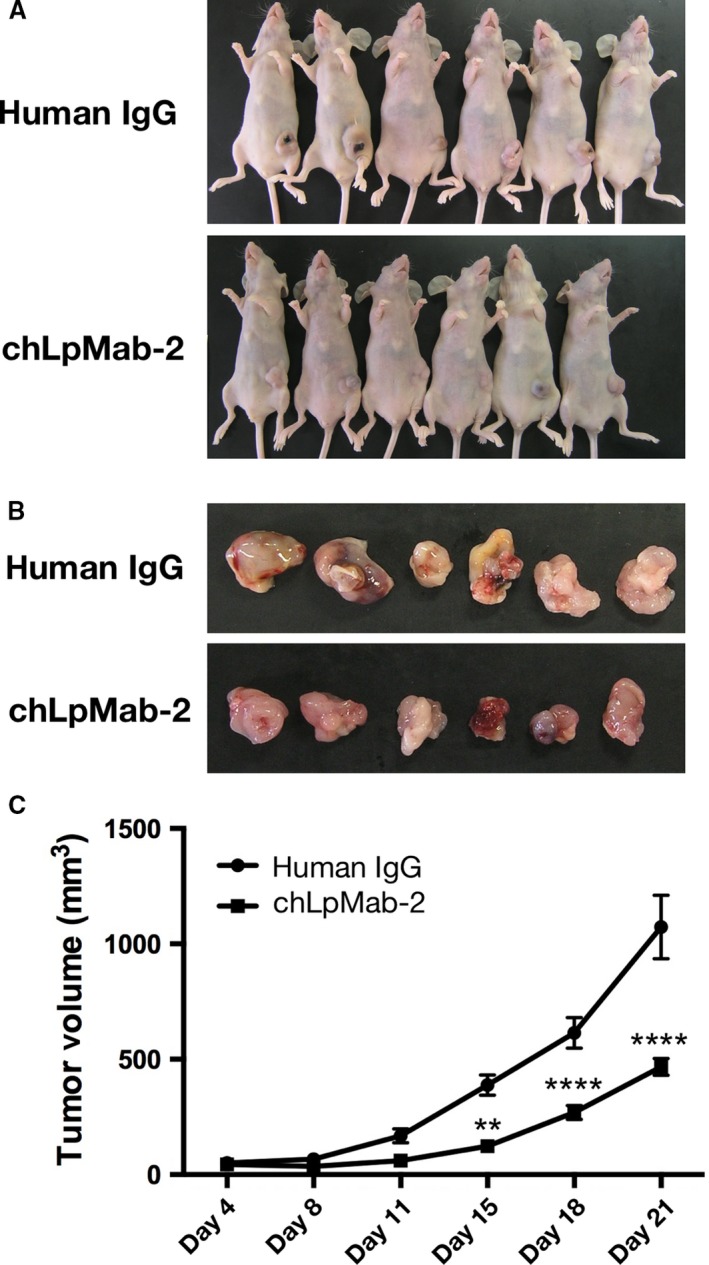
Antitumor effects of chLpMab‐2 on CHO/hPDPN tumor development. CHO/hPDPN cells (3 × 106 cells/100 μL) were subcutaneously inoculated into BALB/c nude mice. After 1 day, 100 μg of chLpMab‐2 or control human IgG was injected into the peritoneal cavity of the mice. The antibodies were injected once weekly for 4 weeks (control group: n = 6; chLpMab‐2 group: n = 6). The tumor diameter was measured every 3‐4 days and was calculated using the following formula: tumor volume = W2 × L/2, where W is the short diameter and L is the long diameter. (A) Comparison of the tumor size and tumor incidence in nude mice (day 21). (B) Comparison of the tumor size (day 21). (C) Primary tumor growth in human IgG and chLpMab‐2‐treated mice. **P < 0.01; ****P < 0.001 with two‐way ANOVA.
Discussion
We previously produced LpMab‐2 (mouse IgG1, kappa), one of the CasMabs against hPDPN 29. LpMab‐2 recognized not only the Thr55–Leu64 peptide of hPDPN but also an aberrant O‐glycosylated hPDPN, which is attached to Thr55 or Ser56 of hPDPN 48. LpMab‐2 reacted with hPDPN‐expressing cancer cells and not with normal cells, as revealed by flow cytometry and immunohistochemistry 29; therefore, LpMab‐2 is an anti‐hPDPN CasMab that is potentially advantageous for antibody‐based molecular‐targeted therapy against hPDPN‐expressing cancers. However, LpMab‐2 is a mouse IgG1; therefore, it cannot be used to study ADCC and CDC against hPDPN‐expressing cancers.
To our knowledge, herein we developed the first cancer‐specific human–mouse chimeric anti‐hPDPN antibody (chLpMab‐2) from LpMab‐2. Previously, we had developed a human–mouse chimeric anti‐hPDPN antibody (chLpMab‐7) 40 and human–rat chimeric anti‐hPDPN antibodies such as NZ‐8 36 and NZ‐12 37. Other groups have reported the development of human–mouse chimeric anti‐hPDPN/hAggrus antibodies that were not cancer‐specific 32, 33. Because hPDPN is expressed in many normal organs such as the lung and kidney, non‐CasMabs against hPDPN are not suitable for antibody‐based molecular targeting therapy.
In this study, we used FUT8‐KO CHO‐S cells (PDIS‐5) to express afucosylated chLpMab‐2. Afucosylated antibodies are known to exhibit high ADCC 53. Consistent with the literature, the ADCC of afucosylated chLpMab‐2 was 3.4 times higher than that of fucosylated chLpMab‐2 (data not shown). In contrast, afucosylated chLpMab‐2 showed a lower CDC (Fig. 5B) than fucosylated chLpMab‐2 (data not shown). Gasdaska et al. have reported that the higher ADCC of afucosylated rituximab suggests an improvement in effectiveness and potency, however, its lower CDC may mitigate infusion toxicity 54. They concluded that afucosylated rituximab was clinically better than fucosylated rituximab. In contrast, Niwa et al. have reported that fucose depletion can provide a panel of IgGs (IgG1, IgG2, IgG3, and IgG4) with enhanced ADCC but that none of the IgGs affected CDC 53.
In our previous study, a human–mouse chimeric anti‐hPDPN mAb showed CDC for antitumor activity in the absence of human NK cells in a mouse xenograft model 36. In this study, chLpMab‐2 exhibited ADCC for antitumor activity in the xenograft model with added NK cells 9. Our results indicate that ADCC is important to induce the antitumor activity of anti‐hPDPN CasMab in the xenograft model for two reasons: i) fucosylated chLpMab‐2, which was expressed in CHO‐K1 cells, showed high CDC but failed to induce enough ADCC and antitumor activity against hPDPN‐expressing tumors (data not shown) and ii) afucosylated chLpMab‐2 did not show enough CDC in vitro (Fig. 5B) but exhibited higher ADCC in vitro (Fig. 5A) and antitumor activity against hPDPN‐expressing tumors (Fig. 6).
Taken together, chLpMab‐2 may be useful as a novel antibody‐based therapy against hPDPN‐expressing tumors with no unexpected side effects.
Conflict of Interest
None declared.
Acknowledgments
We thank Miyuki Yanaka, Noriko Saidoh, and Kanae Yoshida. for their excellent technical assistance. We also thank Takeshi Murata, Hiroaki Uchida, and Hideaki Tahara for their specialized advice. This work was supported in part by the Basic Science and Platform Technology Program for Innovative Biological Medicine from Japan Agency for Medical Research and Development, AMED (Y.K.), by the Translational Research Network Program from AMED (Y.K.), by the Platform for Drug Discovery, Informatics, and Structural Life Science (PDIS) from AMED (Y.K.), by Project for utilizing glycans in the development of innovative drug discovery technologies from AMED (Y.K.), by the Regional Innovation Strategy Support Program from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (Y.K.), by JSPS KAKENHI Grant Number 26440019 (M.K.K.), 16K08372 (S.A.), and 16K10748 (Y.K.). This work was performed in part under the Cooperative Research Program of Institute for Protein Research, Osaka University, CR‐16‐05 and by the Grant for Joint Research Project of the Institute of Medical Science, the University of Tokyo. The authors would like to thank Enago (www.enago.jp) for the English language review.
Cancer Medicine 2017, 6(4):768–777
References
- 1. Martin‐Villar, E. , Scholl F. G., Gamallo C., Yurrita M. M., Munoz‐Guerra M., Cruces J., et al. 2005. Characterization of human PA2.26 antigen (T1alpha‐2, podoplanin), a small membrane mucin induced in oral squamous cell carcinomas. Int. J. Cancer 113:899–910. [DOI] [PubMed] [Google Scholar]
- 2. Yuan, P. , Temam S., El‐Naggar A., Zhou X., Liu D., Lee J., et al. 2006. Overexpression of podoplanin in oral cancer and its association with poor clinical outcome. Cancer 107:563–569. [DOI] [PubMed] [Google Scholar]
- 3. Takagi, S. , Oh‐hara T., Sato S., Gong B., Takami M., and Fujita N.. 2014. Expression of Aggrus/podoplanin in bladder cancer and its role in pulmonary metastasis. Int. J. Cancer 134:2605–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kunita, A. , Kashima T. G., Ohazama A., Grigoriadis A. E., and Fukayama M.. 2011. Podoplanin is regulated by AP‐1 and promotes platelet aggregation and cell migration in osteosarcoma. Am. J. Pathol. 179:1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chandramohan, V. , Bao X., Kato Kaneko M., Kato Y., Keir S. T., Szafranski S. E., et al. 2013. Recombinant anti‐podoplanin (NZ‐1) immunotoxin for the treatment of malignant brain tumors. Int. J. Cancer 132:2339–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kato, Y. , Kaneko M. K., Kuno A., Uchiyama N., Amano K., Chiba Y., et al. 2006. Inhibition of tumor cell‐induced platelet aggregation using a novel anti‐podoplanin antibody reacting with its platelet‐aggregation‐stimulating domain. Biochem. Biophys. Res. Commun. 349:1301–1307. [DOI] [PubMed] [Google Scholar]
- 7. Mishima, K. , Kato Y., Kaneko M. K., Nakazawa Y., Kunita A., Fujita N., et al. 2006. Podoplanin expression in primary central nervous system germ cell tumors: a useful histological marker for the diagnosis of germinoma. Acta Neuropathol. (Berl). 111:563–568. [DOI] [PubMed] [Google Scholar]
- 8. Mishima, K. , Kato Y., Kaneko M. K., Nishikawa R., Hirose T., and Matsutani M.. 2006. Increased expression of podoplanin in malignant astrocytic tumors as a novel molecular marker of malignant progression. Acta Neuropathol. (Berl). 111:483–488. [DOI] [PubMed] [Google Scholar]
- 9. Abe, S. , Morita Y., Kaneko M. K., Hanibuchi M., Tsujimoto Y., Goto H., et al. 2013. A novel targeting therapy of malignant mesothelioma using anti‐podoplanin antibody. J. Immunol. 190:6239–6249. [DOI] [PubMed] [Google Scholar]
- 10. Kato, Y. , Vaidyanathan G., Kaneko M. K., Mishima K., Srivastava N., Chandramohan V., et al. 2010. Evaluation of anti‐podoplanin rat monoclonal antibody NZ‐1 for targeting malignant gliomas. Nucl. Med. Biol. 37:785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kato, Y. , Fujita N., Kunita A., Sato S., Kaneko M., Osawa M., et al. 2003. Molecular identification of Aggrus/T1alpha as a platelet aggregation‐inducing factor expressed in colorectal tumors. J. Biol. Chem. 278:51599–51605. [DOI] [PubMed] [Google Scholar]
- 12. Kato, Y. , Sasagawa I., Kaneko M., Osawa M., Fujita N., and Tsuruo T.. 2004. Aggrus: a diagnostic marker that distinguishes seminoma from embryonal carcinoma in testicular germ cell tumors. Oncogene 23:8552–8556. [DOI] [PubMed] [Google Scholar]
- 13. Kato, Y. , Kaneko M., Sata M., Fujita N., Tsuruo T., and Osawa M.. 2005. Enhanced expression of Aggrus (T1alpha/podoplanin), a platelet‐aggregation‐inducing factor in lung squamous cell carcinoma. Tumor Biol. 26:195–200. [DOI] [PubMed] [Google Scholar]
- 14. Kunita, A. , Kashima T. G., Morishita Y., Fukayama M., Kato Y., Tsuruo T., et al. 2007. The platelet aggregation‐inducing factor aggrus/podoplanin promotes pulmonary metastasis. Am. J. Pathol. 170:1337–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaneko, M. K. , Kato Y., Kitano T., and Osawa M.. 2006. Conservation of a platelet activating domain of Aggrus/podoplanin as a platelet aggregation‐inducing factor. Gene 378:52–57. [DOI] [PubMed] [Google Scholar]
- 16. Pula, B. , Jethon A., Piotrowska A., Gomulkiewicz A., Owczarek T., Calik J., et al. 2011. Podoplanin expression by cancer‐associated fibroblasts predicts poor outcome in invasive ductal breast carcinoma. Histopathology 59:1249–1260. [DOI] [PubMed] [Google Scholar]
- 17. Kawase, A. , Ishii G., Nagai K., Ito T., Nagano T., Murata Y., et al. 2008. Podoplanin expression by cancer associated fibroblasts predicts poor prognosis of lung adenocarcinoma. Int. J. Cancer 123:1053–1059. [DOI] [PubMed] [Google Scholar]
- 18. Hoshino, A. , Ishii G., Ito T., Aoyagi K., Ohtaki Y., Nagai K., et al. 2011. Podoplanin‐positive fibroblasts enhance lung adenocarcinoma tumor formation: podoplanin in fibroblast functions for tumor progression. Cancer Res. 71:4769–4779. [DOI] [PubMed] [Google Scholar]
- 19. Schoppmann, S. F. , Jesch B., Riegler M. F., Maroske F., Schwameis K., Jomrich G., et al. 2013. Podoplanin expressing cancer associated fibroblasts are associated with unfavourable prognosis in adenocarcinoma of the esophagus. Clin. Exp. Metastasis 30:441–446. [DOI] [PubMed] [Google Scholar]
- 20. Shindo, K. , Aishima S., Ohuchida K., Fujiwara K., Fujino M., Mizuuchi Y., et al. 2013. Podoplanin expression in cancer‐associated fibroblasts enhances tumor progression of invasive ductal carcinoma of the pancreas. Mol. Cancer. 12:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Inoue, H. , Tsuchiya H., Miyazaki Y., Kikuchi K., Ide F., Sakashita H., et al. 2014. Podoplanin expressing cancer‐associated fibroblasts in oral cancer. Tumour Biol. 35:11345–11352. [DOI] [PubMed] [Google Scholar]
- 22. Suzuki‐Inoue, K. , Kato Y., Inoue O., Kaneko M. K., Mishima K., Yatomi Y., et al. 2007. Involvement of the snake toxin receptor CLEC‐2, in podoplanin‐mediated platelet activation, by cancer cells. J. Biol. Chem. 282:25993–26001. [DOI] [PubMed] [Google Scholar]
- 23. Kato, Y. , Kaneko M. K., Kunita A., Ito H., Kameyama A., Ogasawara S., et al. 2008. Molecular analysis of the pathophysiological binding of the platelet aggregation‐inducing factor podoplanin to the C‐type lectin‐like receptor CLEC‐2. Cancer Sci. 99:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nagae, M. , Morita‐Matsumoto K., Kato M., Kaneko M. K., Kato Y., and Yamaguchi Y.. 2014. A Platform of C‐Type lectin‐like receptor CLEC‐2 for binding O‐glycosylated podoplanin and nonglycosylated rhodocytin. Structure. 22:1711–1721. [DOI] [PubMed] [Google Scholar]
- 25. Wikstrand, C. J. , Hale L. P., Batra S. K., Hill M. L., Humphrey P. A., Kurpad S. N., et al. 1995. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 55:3140–3148. [PubMed] [Google Scholar]
- 26. Baselga, J. , Norton L., Albanell J., Kim Y. M., and Mendelsohn J.. 1998. Recombinant humanized anti‐HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 58:2825–2831. [PubMed] [Google Scholar]
- 27. Goldstein, N. I. , Prewett M., Zuklys K., Rockwell P., and Mendelsohn J.. 1995. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res. 1:1311–1318. [PubMed] [Google Scholar]
- 28. Reff, M. E. , Carner K., Chambers K. S., Chinn P. C., Leonard J. E., Raab R., et al. 1994. Depletion of B‐Cells in‐Vivo by a Chimeric Mouse‐Human Monoclonal‐Antibody to Cd20. Blood 83:435–445. [PubMed] [Google Scholar]
- 29. Kato, Y. , and Kaneko M. K.. 2014. A cancer‐specific monoclonal antibody recognizes the aberrantly glycosylated podoplanin. Sci. Rep. 4:5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lohmueller, J. J. , Sato S., Popova L., Chu I. M., Tucker M. A., Barberena R., et al. 2016. Antibodies elicited by the first non‐viral prophylactic cancer vaccine show tumor‐specificity and immunotherapeutic potential. Sci. Rep. 6:31740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ogasawara, S. , Kaneko M. K., Price J. E., and Kato Y.. 2008. Characterization of anti‐podoplanin monoclonal antibodies: critical epitopes for neutralizing the interaction between podoplanin and CLEC‐2. Hybridoma. 27:259–267. [DOI] [PubMed] [Google Scholar]
- 32. Takagi, S. , Sato S., Oh‐hara T., Takami M., Koike S., Mishima Y., et al. 2013. Platelets promote tumor growth and metastasis via direct interaction between Aggrus/podoplanin and CLEC‐2. PLoS ONE 8:e73609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakazawa, Y. , Takagi S., Sato S., Oh‐hara T., Koike S., Takami M., et al. 2011. Prevention of hematogenous metastasis by neutralizing mice and its chimeric anti‐Aggrus/podoplanin antibodies. Cancer Sci. 102:2051–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marks, A. , Sutherland D. R., Bailey D., Iglesias J., Law J., Lei M., et al. 1999. Characterization and distribution of an oncofetal antigen (M2A antigen) expressed on testicular germ cell tumours. Br. J. Cancer 80:569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kono, T. , Shimoda M., Takahashi M., Matsumoto K., Yoshimoto T., Mizutani M., et al. 2007. Immunohistochemical detection of the lymphatic marker podoplanin in diverse types of human cancer cells using a novel antibody. Int. J. Oncol. 31:501–508. [PubMed] [Google Scholar]
- 36. Kaneko, M. K. , Kunita A., Abe S., Tsujimoto Y., Fukayama M., Goto K., et al. 2012. Chimeric anti‐podoplanin antibody suppresses tumor metastasis through neutralization and antibody‐dependent cellular cytotoxicity. Cancer Sci. 103:1913–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shiina, S. , Ohno M., Ohka F., Kuramitsu S., Yamamichi A., Kato A., et al. 2016. CAR T Cells Targeting Podoplanin Reduce Orthotopic Glioblastomas in Mouse Brains. Cancer Immunol Res. 4:259–268. [DOI] [PubMed] [Google Scholar]
- 38. Kaneko, M. K. , Oki H., Hozumi Y., Liu X., Ogasawara S., Takagi M., et al. 2015. Monoclonal antibody LpMab‐9 recognizes O‐glycosylated N‐terminus of human podoplanin. Monoclon. Antib. Immunodiagn. Immunother. 34:310–317. [DOI] [PubMed] [Google Scholar]
- 39. Kaneko, M. K. , Oki H., Ogasawara S., Takagi M., and Kato Y.. 2015. Anti‐podoplanin monoclonal antibody LpMab‐7 detects metastatic legions of osteosarcoma. Monoclon. Antib. Immunodiagn. Immunother. 34:154–161. [DOI] [PubMed] [Google Scholar]
- 40. Kato, Y. , Kunita A., Abe S., Ogasawara S., Fujii Y., Oki H., et al. 2015. The chimeric antibody chLpMab‐7 targeting human podoplanin suppresses pulmonary metastasis via ADCC and CDC rather than via its neutralizing activity. Oncotarget. 6:36003–36018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ogasawara, S. , Oki H., Kaneko M. K., Hozumi Y., Liu X., Honma R., et al. 2015. Development of monoclonal antibody LpMab‐10 recognizing non‐glycosylated PLAG1/2 domain including Thr34 of human podoplanin. Monoclon. Antib. Immunodiagn. Immunother. 34:318–326. [DOI] [PubMed] [Google Scholar]
- 42. Oki, H. , Kaneko M. K., Ogasawara S., Tsujimoto Y., Liu X., Sugawara M., et al. 2015. Characterization of a monoclonal antibody LpMab‐7 recognizing non‐PLAG domain of podoplanin. Monoclon. Antib. Immunodiagn. Immunother. 34:174–180. [DOI] [PubMed] [Google Scholar]
- 43. Oki, H. , Ogasawara S., Kaneko M. K., Takagi M., M. Yamauchi , and Kato Y.. 2015. Characterization of monoclonal antibody LpMab‐3 recognizing sialylated glycopeptide of podoplanin. Monoclon. Antib. Immunodiagn Immunother. 34:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kato, Y. , Ogasawara S., Oki H., Goichberg P., Honma R., Fujii Y., et al. 2016. LpMab‐12 Established by CasMab Technology Specifically Detects Sialylated O‐Glycan on Thr52 of Platelet Aggregation‐Stimulating Domain of Human Podoplanin. PLoS ONE 11:e0152912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kato, Y. , Ogasawara S., Oki H., Honma R., Takagi M., Fujii Y., et al. 2016. Novel monoclonal antibody LpMab‐17 developed by CasMab technology distinguishes human podoplanin from monkey podoplanin. Monoclon. Antib. Immunodiagn. Immunother. 35:109–116. [DOI] [PubMed] [Google Scholar]
- 46. Ogasawara, S. , Kaneko M. K., Honma R., Oki H., Y. Fujii , Takagi M., et al. 2016. Establishment of Mouse Monoclonal Antibody LpMab‐13 against Human Podoplanin. Monoclon Antib Immunodiagn Immunother 35:155–162. [DOI] [PubMed] [Google Scholar]
- 47. Hayatsu, N. , Ogasawara S., Kaneko M. K., Kato Y., and Narimatsu H.. 2008. Expression of highly sulfated keratan sulfate synthesized in human glioblastoma cells. Biochem. Biophys. Res. Commun. 368:217–222. [DOI] [PubMed] [Google Scholar]
- 48. Abe, S. , Kaneko M. K., Tsuchihashi Y., Izumi T., Ogasawara S., Okada N., et al. 2016. Antitumor effect of novel anti‐podoplanin antibody NZ‐12 against malignant pleural mesothelioma in an orthotopic xenograft model. Cancer Sci. 107:1198–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kaneko, M. K. , Nakamura T., Honma R., Ogasawara S., Fujii Y., Abe S., et al. 2017. Development and characterization of anti‐glycopeptide monoclonal antibodies against human podoplanin using glycan‐deficient cell lines generated by CRISPR/Cas9 and TALEN. Cancer Med. 6:382–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaneko, M. , Kato Y., Kunita A., Fujita N., Tsuruo T., and Osawa M.. 2004. Functional sialylated O‐glycan to platelet aggregation on Aggrus (T1alpha/podoplanin) molecules expressed in Chinese Hamster Ovary cells. J. Biol. Chem. 279:38838–38843. [DOI] [PubMed] [Google Scholar]
- 51. Kaneko, M. K. , Kato Y., Kameyama A., Ito H., A. Kuno , Hirabayashi J., et al. 2007. Functional glycosylation of human podoplanin: glycan structure of platelet aggregation‐inducing factor. FEBS Lett. 581:331–336. [DOI] [PubMed] [Google Scholar]
- 52. Kuno, A. , Kato Y., Matsuda A., Kaneko M. K., Ito H., Amano K., et al. 2009. Focused differential glycan analysis with the platform antibody‐assisted lectin profiling for glycan‐related biomarker verification. Mol. Cell Proteomics 8:99–108. [DOI] [PubMed] [Google Scholar]
- 53. Niwa, R. , Natsume A., Uehara A., Wakitani M., Iida S., Uchida K., et al. 2005. IgG subclass‐independent improvement of antibody‐dependent cellular cytotoxicity by fucose removal from Asn297‐linked oligosaccharides. J. Immunol. Methods 306:151–160. [DOI] [PubMed] [Google Scholar]
- 54. Gasdaska, J. R. , Sherwood S., Regan J. T., and Dickey L. F.. 2012. An afucosylated anti‐CD20 monoclonal antibody with greater antibody‐dependent cellular cytotoxicity and B‐cell depletion and lower complement‐dependent cytotoxicity than rituximab. Mol. Immunol. 50:134–141. [DOI] [PubMed] [Google Scholar]