

Abstract
Golgi phosphoprotein 3 (GOLPH3), a newly recognized oncogene, is associated with tumor growth, metastasis, and poor prognosis in several types of cancer. However, its biological role and underlying mechanism in epithelial ovarian cancer (EOC) remain poorly understood. Here, we found that GOLPH3 was overexpressed in EOC tissues and cell lines. This overexpression promoted the migration and invasion of EOC cells. Moreover, GOLPH3 upregulated the expression of epithelial–mesenchymal transition (EMT) markers, such as N‐cadherin and Snail, and the Wnt/β‐catenin‐related genes cyclin‐D1 and c‐Myc, which were restored via silencing of GOLPH3 expression. Furthermore, the inhibitor and activator of the Wnt/β‐catenin pathway, XAV939 and LiCl, enhanced or decreased, respectively, the effect of GOLPH3 on EMT, which further confirmed that GOLPH3 promoted EMT progression via activation of Wnt/β‐catenin signaling. In addition, we found that EDD, the human hyperplastic discs gene, was consistent with GOLPH3 expression and also promoted the EMT process and activated Wnt/β‐catenin signaling. These findings demonstrate that EDD might be a downstream factor of GOLPH3. Taken together, our findings demonstrate the existence of a GOLPH3–Wnt/β‐catenin–EMT axis in EOC and provide a new therapeutic target to treat EOC.
Keywords: Epithelial ovarian cancer, epithelial–mesenchymal transition, GOLPH3, metastasis, Wnt signaling pathway
Introduction
Ovarian cancer (OC) is a lethal gynecological malignant tumor and ranks fifth in cause of death for female cancer patients. In 2015, there were 14,180 deaths and 21,290 patients newly diagnosed with OC in the United States. However, the expected 5‐year survival rate of OC is only 45% 1. Of all reported OC subtypes, approximately 90% are epithelial ovarian cancer (EOC), which usually is present at advanced stage 2. For most advanced stage patients, cytoreductive surgery followed by platinum/taxol chemotherapy is regarded as the standard therapy 3. Although the majority of patients are sensitive to this treatment, over the past two decades, the overall cure rate hovers around 30% 4. The lack of effective early detection markers and tumor metastasis are major factors for poor outcomes and high death rates. Thus, improving targeting therapies and studying the mechanisms underlying tumor invasion and metastasis are necessary for reducing the OC mortality.
Golgi phosphoprotein 3 (GOLPH3), also known as GPP34/GMx33/MIDAS, is a newly identified 34‐kDa phosphorylated matrix protein, which localizes to the transface of the Golgi complex and plays a critical role in the Golgi secretory pathway and DNA damage 5, 6, 7, 8, 9. GOLPH3 resides on human chromosome 5p13, where it is amplified in multiple tumor types 10. Recent studies indicate that GOLPH3 is involved in cancer progression and correlates with clinical stages and poor prognosis in several types of tumors 11, 12, 13, 14, 15. It has been reported that high GOLPH3 expression promotes tumorigenicity and aggressive behavior of EOC 16, 17. However, its role in cell migration and invasion, as well as the molecular mechanism, remains unclear.
Metastasis is a process in which cancer cells spread from the primary tissue to surrounding tissues because cells lose cell–cell adhesion ability and gain migratory and invasive capability. Epithelial–mesenchymal transition (EMT) is defined as a dynamic process in which epithelial cells acquire the mesenchymal phenotype, which has motile and invasive characteristics 18. In recent years, accumulating evidence suggests that EMT is a crucial step in the cancer‐related metastatic cascade 19, 20. Various signaling pathways regulate EMT, including the HGF, EGF, TGF‐β, Notch, and Wnt/β‐catenin signaling pathways 19. As an important regulator of EMT, activation of the Wnt/β‐catenin pathway is common in many malignant tumors including EOC 21, 22, 23. Furthermore, it is proposed that the Wnt secretory pathway depends on endosome‐to‐Golgi transport 24, 25. However, the molecular mechanism of GOLPH3 in cancer process, especially in migration and invasion, remains poorly understood. As a “first‐in‐class Golgi oncoprotein,” we speculate that GOLPH3 may regulate Wnt/β‐catenin signaling pathway to promote EMT process in EOC.
Here, we provide evidences to confirm that overexpression of GOLPH3 stimulates EMT via the Wnt/β‐catenin signaling pathway, which further promotes metastasis of EOC. In addition, we find that EDD, as a DNA damage‐related factor, and oncogene, like GOLPH3, might play an important role in GOLPH3–Wnt/β‐catenin–EMT axis of EOC. To our knowledge, these findings indicate the molecular mechanisms of GOLPH3‐mediated oncogenesis in EOC for the first time.
Materials and Methods
Patient information and tissue samples
The study was approved by medical ethical committee of Shanghai General Hospital affiliated to Medical School of Shanghai Jiao Tong University. All patients in our study provided written informed consent. In total, 58 EOC tissues and 15 benign tumor tissues were acquired from patients treated at our hospital between January 2014 and December 2016. Two gynecological pathologists confirmed all collected tissues based on the WHO classification.
Immunohistochemistry
Paraffin‐embedded tissues were cut into 4 μm sections. The immunohistochemistry (IHC) procedure to determine GOLPH3 and EDD expression was performed as described previously 17. Briefly, the sections were incubated with mouse monoclonal anti‐GOLPH3 antibody (1:200 dilution; Proteintech, Chicago, IL) and rabbit monoclonal anti‐EDD antibody (1:1000 dilution; Abcam, Cambridge, UK) overnight at 4°C. Negative control slides replaced the primary antibody with phosphate‐buffered saline (PBS). To detect the antigen, sections were incubated with biotinylated anti‐mouse or anti‐rabbit secondary antibody. Slides were evaluated at 200× magnification, and 10 different staining fields of each section were assessed independently by two trained observers who were blinded to patient information. A score criteria was assigned to evaluate the percentage of positively stained carcinoma cells, as previously reported 26.
Cell culture
Human epithelial ovarian cancer cell lines, including HEY, SKOV3, HO8910, HO8910‐PM, and ES‐2 cell lines, were purchased from The Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The normal ovarian cell line (MOODY) was kindly provided by Dr. Wenxin Zheng (Department of Pathology, University of Texas Southwestern Medical Center, USA). All of the cells were grown in DMEM/F‐12 supplemented with 10% FBS and cultured in a sterile incubator maintained with 5% CO2 at 37°C.
Western blot analysis
The western blot procedure was performed as described previously 27. Briefly, treated cells were lysed in RIPA lysis buffer containing protease inhibitor (1:1000). Approximately 30 μg of the protein samples was separated by 7.5–12.5% SDS‐PAGE gels and then transferred to PVDF membranes (Millipore, Bedford, MA). After being blocked in 5% skim milk at room temperature for 1 h, the membranes were incubated with the corresponding specific primary antibodies (1:1000 dilution) overnight at 4°C. Then, the bands were robed with the appropriate secondary antibody (1:5000 dilution; Proteintech, Chicago, IL) at room temperature for 1 h. Enhanced chemiluminescence reagents ((Pierce, Rockford, IL) were used to detect antibody complexes. The primary antibodies used in our study included GOLPH3, EDD (Abcam, Cambridge, UK), cyclin‐D1, c‐Myc (Proteintech, Chicago, IL), E‐cadherin, N‐cadherin, Snail, and β‐catenin (Cell Signaling Technology, Danvers, MA). To determine the effect of Wnt/β‐catenin signaling, the pathway agonist LiCl (20 mmol/L; Sigma, St. Louis) and antagonist XAV939 (10 μmol/L; Sigma) were used to treat cells for 24 h after transfection. β‐actin (Proteintech, Chicago, IL) was used as a loading control. Each experiment was performed in triplicate.
Transient transfection
Cells were transiently transfected using Lipofectamine 2000 (Invitrogen, Grand Island, NY) following the manufacturer's protocol. Briefly, cells were seeded into six‐well plates at a density of 2 × 104 cells/well. When cultured to 50–60% confluency, cells were serum starved for 24 h to minimize the influence of FBS. Then, cells were transfected with siRNA or plasmid using Lipofectamine 2000. After 6–8 h of incubation, the treated cells were cultured in DMEM/F‐12 with 10% FBS. The GOLPH3 siRNA and negative control were constructed by GeneChen (Shanghai, China). The pcDNA3.1‐GOLPH3 and pcDNA3.1‐vector plasmids were designed and purchased from Genera Biotechnology (Shanghai, China). The sequences of the GOLPH3 and EDD siRNA are listed in Table 1.
Table 1.
GOLPH3 and EDD siRNA sequences
| Name | Sequence |
|---|---|
| GOLPH3 | |
| Sense | 5′‐GGUGUAUUGACAACAGAGA‐3′ |
| Antisense | 5′‐UCUCUGUUGUCAAUACACC‐3′ |
| EDD | |
| Sense | 5′‐GCGUGAACGUGAAUCCGUU‐3′ |
| Antisense | 5′‐AACGGAUUCACGUUCACGC‐3′ |
Quantitative real‐time PCR analysis
Total RNA was extracted from cells using Trizol Reagent (Invitrogen), following the manufacturer's protocol. Reverse transcription was performed in a 20‐μL reaction system with 1 μg of total RNA via the Prime‐Script RT reagent kit (Takara, Kyoto, Japan). The complimentary DNA (cDNA) was then synthesized with SYBR Premix Ex Taq (Takara, Dalian, China) for quantitative real‐time PCR. β‐actin served as the internal control gene. The amplification was performed for 40 cycles including 5 min at 95°C, 5 sec at 95°C, and 30 sec at 60°C. The data were analyzed using the 2−ΔΔCT method to determine the relative gene expression levels. Each experiment was repeated three independent times. The PCR primers for GOLPH3, EDD, and β‐actin were synthesized by Sangon Biotech (Shanghai, China) and are listed in Table 2.
Table 2.
PCR primer sequences
| Name | Sequence |
|---|---|
| GOLPH3 | |
| Forward primer | 5′‐ACC TGT TTT GGG TTT CTG GT‐3′ |
| Reverse primer | 5′‐TGT GCG TAT GAG GAG GCT G‐3′ |
| EDD | |
| Forward primer | 5′‐CCA TAC AAA CGA CGA CGG T‐3′ |
| Reverse primer | 5′‐GCC AAC AGG AAC ATT CTT GAC‐3′ |
| β‐actin | |
| Forward primer | 5′‐AAG GTG ACA GCA GTC GGT T‐3′ |
| Reverse primer | 5′‐TGT GTG GAC TTG GGA GAG G‐3′ |
Cell invasion and migration assays
Cell migration and invasion were assayed using Transwell plates with 8‐μm pore filters (Corning, NY). The procedure was performed according to the manufacturer's protocol. Briefly, for the cell migration assay, transfected HEY, SKOV3, and HO8910 (2 × 105) cells were suspended in 200 μl of serum‐free DMEM and plated into the upper chambers. Then, 600 μl of DMEM/F‐12 supplemented with 10% FBS was added to the lower chamber. After incubation for 12–14 h at 37°C, the tumor cells were fixed with 4% cold paraformaldehyde and stained with crystal violet (Beyotime, Shanghai, China). For the invasion assay, the procedures were conducted as described earlier, except the filter inserts were coated with BD Matrigel and the plates were incubated for 16–20 h at 37°C. Cells that had migrated were counted at 100× magnification under an inverted microscope.
Scratch migration assay
For the scratch migration assay, treated cells were seeded onto six‐well plates to form cell monolayer (near 90% confluence). Subsequently, the cell layer was scratched with a 200‐μL pipette tip, washed three times with PBS to remove floating cells, and photographed (time 0 h). Later, the wounded cultures were incubated in conditioned medium at 37°C. At 0, 24, and 48 h, images were captured using an inverted microscope to assess wound closure and then compared to determine differences in cell migration. Three fields (×100) were randomly selected from each scratch wound.
Statistical analysis
Statistical analyses were performed using the Statistical Package for the Social Sciences (SPSS) 20.0 (IBM, Armonk, NY). The data are presented as the mean ± standard deviation (SD). The statistical difference between the test and control group was assessed using Student's t‐test. The association between GOLPH3 and EDD expression was analyzed by using Spearman's correlation analysis. A P < 0.05 was considered statistically significant.
Results
High GOLPH3 expression in epithelial ovarian cancer cells and tissues
To investigate the oncogenic role of GOLPH3 during EOC progression, we examined the expression level of GOLPH3 in EOC tissues and cell lines. As shown in Table 3, ovarian tissue samples from 73 patients were used in this study. There were 2 (13.33%) of the 15 cases of benign tumors, 3 (60%) of the five cases of borderline tumors, and 45 (84.91%) of the 53 cases of epithelial ovarian cancer that showed high expression of GOLPH3 protein. Clearly, GOLPH3 expression was higher in epithelial ovarian cancer than in the borderline tumors and benign cystadenomas (Fig. 1A, P < 0.05).
Table 3.
GOLPH3 expression in different ovarian tissues
| Tissue type | Total | High level (%) | Low level | P value |
|---|---|---|---|---|
| Benign tumor | 15 | 2 (13.33) | 13 | |
| Borderline tumor | 5 | 3 (60.00) | 2 | <0.051 |
| Epithelial cancer | 53 | 45 (84.91) | 8 | <0.052 |
An IHC score of <3 was considered low expression and a score of 4–12 was considered high expression.
1Benign ovarian tumor versus borderline ovarian tumor.
2Benign ovarian tumor versus epithelial ovarian cancer.
Figure 1.
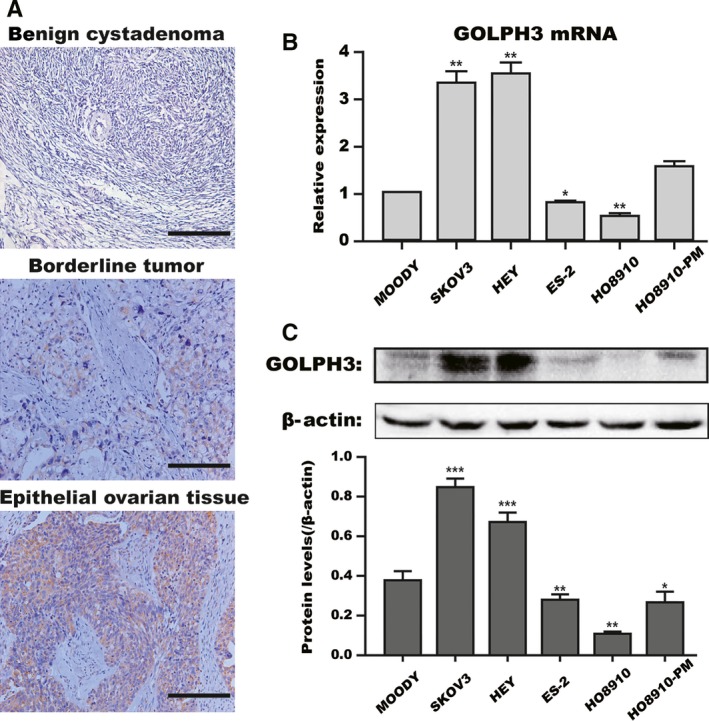
High GOLPH3 expression in epithelial ovarian cancer cells and tissues. (A) Immunohistochemical staining showed that GOLPH3 expression was higher in epithelial ovarian tissues than in benign cystadenoma and borderline tumors. Scale bar: 200 μm. Original magnification 200×. GOLPH3 mRNA (B) and protein (C) expression in five ovarian cancer cell lines (HEY, SKOV3, ES‐2, HO8910, HO8910‐PM) and one normal ovarian cell line (Moody) were detected by quantitative real‐time PCR and western blot analyses. β‐Actin served as a loading control. The data were expressed as the mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.01, *** P < 0.001 compared with control cells.
In addition, to further explore the potential effect of GOLPH3 in EOC, we measured the GOLPH3 mRNA and protein expression in ovarian cell lines (Fig. 1B and C). As our data shows, GOLPH3 was upregulated in two of five ovarian cancer cell lines (SKOV3 and HEY) at both the mRNA and protein level compared with the normal immortalized cell line, Moody (P < 0.05). These findings suggest that GOLPH3 is overexpressed in human ovarian carcinoma and may contribute to some of the tumor behaviors.
GOLPH3 promotes the migration and invasion capacity of ovarian cancer cells
Notably, we found that SKOV3 and HEY had a higher level of GOLPH3 expression than the other cell lines, whereas HO8910 had the lowest expression. To examine the role of GOLPH3 in EOC, we knocked down its expression in SKOV3 and HEY cells and overexpressed it in HO8910 cells. As our data shows, compared to the control group, the relative expression of GOLPH3 mRNA and protein was significantly decreased in the siRNA‐transfected group and increased in the plasmid‐treated group (P < 0.01, Fig. 2A and B).
Figure 2.
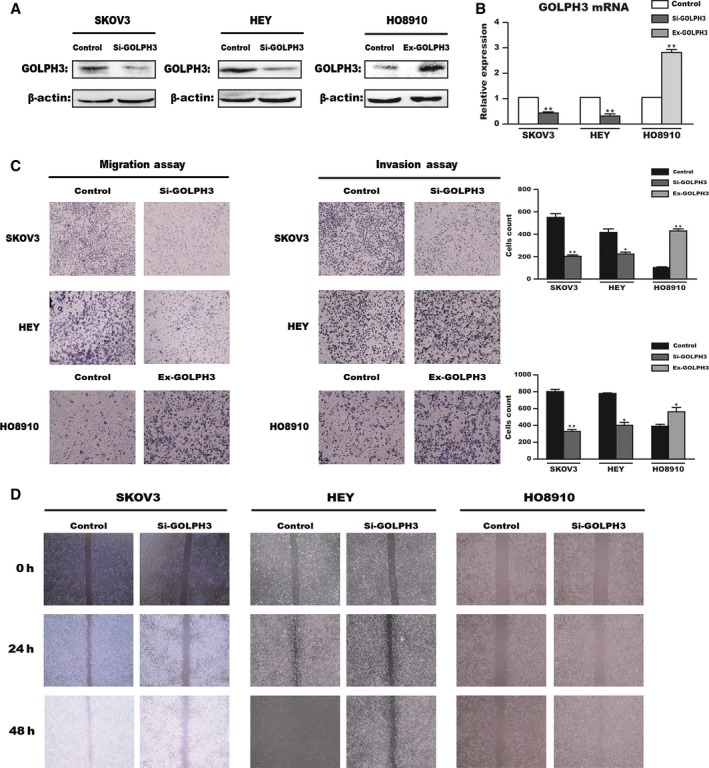
GOLPH3 promotes the migration and invasion capacity of ovarian cancer cells. GOLPH3 expression was downregulated by siRNA in HEY and SKOV3 cell lines and overexpressed by plasmid in the HO8910 cell line. The transfection efficiencies were measured by western blot (A) and quantitative real‐time PCR (B) analyses. β‐Actin served as a loading control. Transwell (C) and scratch migration (D) analyses were used to determine the function of GOLPH3 in the migratory and invasive capability of EOC cells. Original magnification 100×. *P < 0.05, **P < 0.01 compared with the control group.
We then studied the impact of GOLPH3 on cell migration and invasion in vitro. The results of the Transwell assay revealed that silencing GOLPH3 significantly reduced the number of cells on the membrane filters for control, while overexpressing GOLPH3 increased the number of cells present (P < 0.05, Fig. 2C). Moreover, scratch migration assays also showed that knockdown of GOLPH3 in SKOV3 and HEY cells resulted in reduced wound healing ability compared with control cells, which was restored by increasing GOLPH3 expression in HO8910 cells (Fig. 2D). Collectively, these results suggest that GOLPH3 contributes to the migration and invasion capacity of EOC cells.
GOLPH3 is involved in EMT‐related protein expression in vitro
Collectively, to further support the proposal that GOLPH3 plays an important role in the migration and invasion of EOC cells, we examined the expression of EMT‐related proteins in treated cells through western blot analysis. As shown in Figure 3, compared to the control group, knockdown of GOLPH3 in HEY and SKOV3 cells inhibited the expression of the mesenchymal marker, N‐cadherin, and stimulated the expression of the epithelial marker, E‐cadherin, and the EMT‐associated transcription factor, Snail, which was in contrast to GOLPH3 overexpression in HO8910 cells. These results indicate that GOLPH3 promotes migration and invasion via EMT in vitro.
Figure 3.
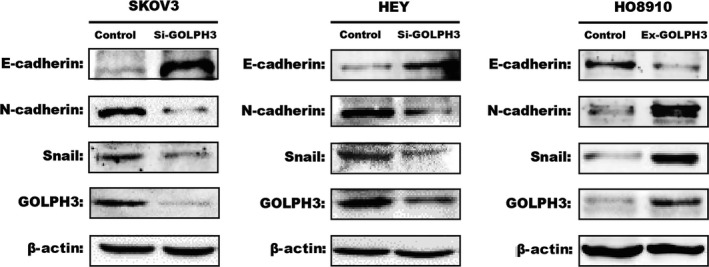
GOLPH3 induces EMT through the Wnt/β‐catenin signaling pathway. Silencing GOLPH3 expression in HEY and SKOV3 cell lines increased the expression of E‐cadherin and decreased N‐cadherin and Snail expression, which was restored via increasing GOLPH3 expression in HO8910 cell line. β‐Actin was served as the loading control.
GOLPH3 induces EMT through the Wnt/β‐catenin signaling pathway
Due to the important role of the Wnt/β‐catenin signaling pathway in tumorigenesis and metastasis of tumors, we wondered whether GOLPH3 participated in the EMT process by activating Wnt/β‐catenin signaling. To address this question, we examined the expression of β‐catenin, c‐Myc, and cyclin‐D1, which are related to Wnt/β‐catenin signaling. In our results, we found that GOLPH3 overexpression in HO8910 cells significantly increased the expression of β‐catenin, c‐Myc, and cyclin‐D1. Conversely, knockdown of GOLPH3 in HEY and SKOV3 cells suppressed the expression of these genes (Fig. 4A). The results above demonstrate that the Wnt/β‐catenin signaling pathway plays a functional role in GOLPH3‐induced EMT in EOC.
Figure 4.
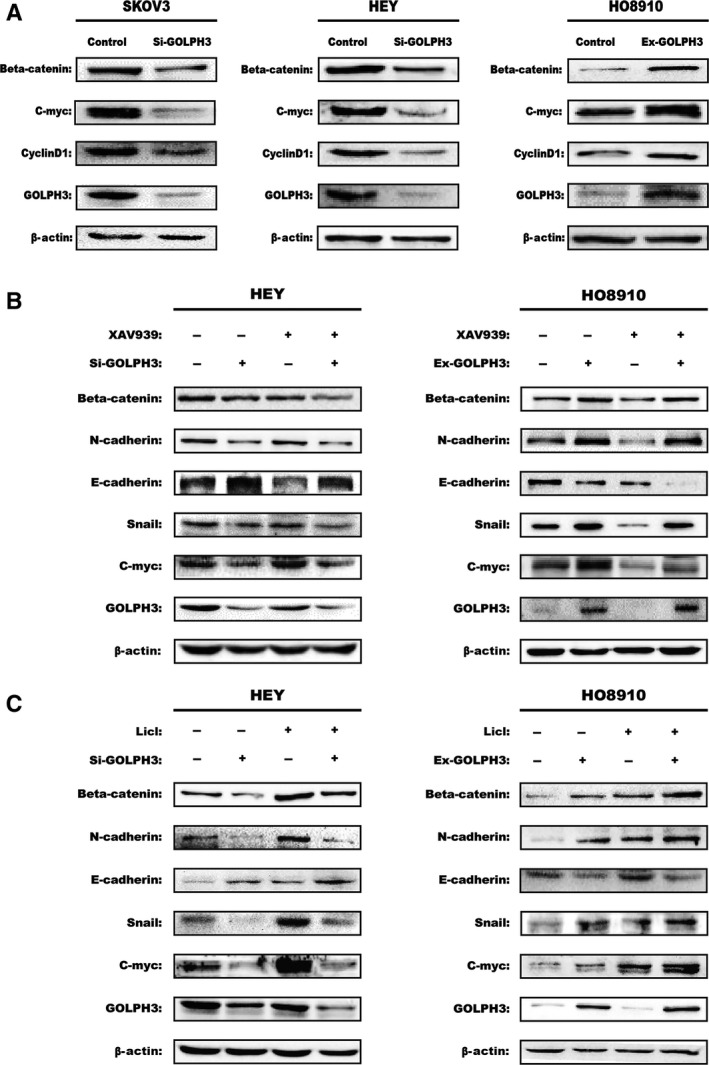
GOLPH3 induces EMT through the Wnt/β‐catenin signaling pathway. (A) Western blot analysis showed that silencing GOLPH3 expression in HEY and SKOV3 cell lines decreased β‐catenin, cyclin D1, and C‐myc expression, which was restored via increasing GOLPH3 expression in HO8910 cell line. Expression levels of GOLPH3, EMT‐related markers, and Wnt/β‐catenin target genes in the different cells following treatment with 10 μmol/L XAV939 (B) or 20 mmol/L LiCl (C) for 24 h were measured using western blotting. The β‐actin was used as the loading control.
Moreover, the hypothesis was further confirmed by treating cells (HEY and HO8910) with the pathway‐specific antagonist XAV939 and agonist LiCl. In HEY and HO8910 cell lines, XAV939 significantly inhibited Wnt/β‐catenin signaling as manifested by the reduced expression of β‐catenin and c‐Myc, while LiCl activated this pathway (Fig. 4B and C). Moreover, we found that the effect on EMT‐related proteins was reversed by inhibiting the Wnt/β‐catenin pathway in GOLPH3 overexpression group, while it was promoted by activating the pathway in GOLPH3 interference group. Furthermore, as we expected, after adding XAV939 in GOLPH3 interference group, the expression of EMT‐related proteins were further decreased, while these proteins were further increased by adding LiCl in GOLPH3 overexpression group. Taken together, we concluded that GOLPH3 induces EMT through the Wnt/β‐catenin signaling pathway in EOC.
GOLPH3 might regulate EDD during Wnt signaling‐induced EMT in EOC cells
Furthermore, we were also interested in whether there is another factor affecting the function of GOLPH3 in EOC. Finally, we found that the expression levels of EDD were consistent with GOLPH3 in different ovarian tissues using immunohistochemistry assays (Fig. 5A). We examined the expression level of EDD or GOLPH3 in the same section of 23 different tissues. As shown in Table 4, there were 2 of 6 cases of benign tumors, all 2 borderline tumors, and 13 of 15 cases of epithelial ovarian cancer that showed high expression of EDD protein (P < 0.05), while 1 of 2 borderline tumors and 11 of 15 cases of epithelial ovarian cancer showed high GOLPH3 expression (P < 0.01). Correlation analysis then revealed that a positive correlation existed between GOLPH3 and EDD expression in ovarian tissues (r = 0.680, P < 0.001; Fig. 5B).
Figure 5.
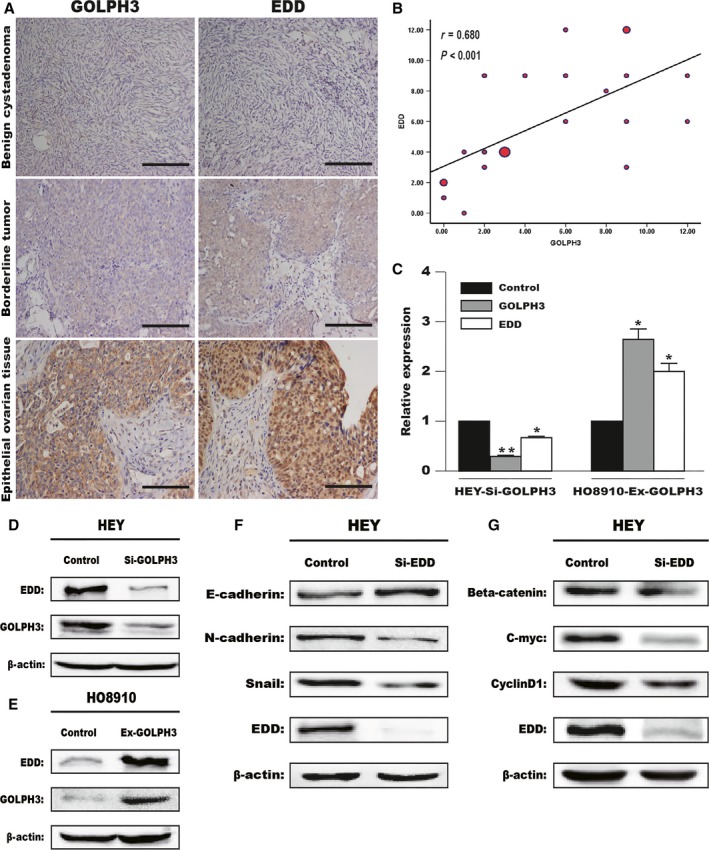
GOLPH3 enhanced EDD expression during Wnt signaling‐induced EMT in EOC cells. (A) The expression of GOLPH3 and EDD was measured using immunohistochemistry in the same section of different tissues. Expression of the nuclear protein EDD was consistent with GOLPH3 expression in benign cystadenoma, borderline tumors and epithelial ovarian tissues. Scale bar: 200 μm. Original magnification 200×. (B) Correlation analysis between GOLPH3 and EDD expression in ovarian tissues (r = 0.680, P < 0.001). The dot size represented the number of ovarian tissue, respectively. The mRNA (C) and protein levels (D and E) of GOLPH3 and EDD in treated cells were evaluated by quantitative real‐time PCR and western blot analyses. (F and G) Expression levels of EMT‐related markers and Wnt/β‐catenin target genes in low‐expressing EDD cells were measured using western blot analysis. β‐Actin served as a loading control. *P < 0.05, **P < 0.01 compared with control cells.
Table 4.
GOLPH3 or EDD expression in different ovarian tissues
| Tissue type | Total | GOLPH3 | P value | EDD | P value | ||
|---|---|---|---|---|---|---|---|
| High | Low | High | Low | ||||
| Benign tumor | 6 | 0 | 6 | <0.01 | 2 | 4 | <0.05 |
| Borderline tumor | 2 | 1 | 1 | 2 | 0 | ||
| Epithelial cancer | 15 | 11 | 4 | 13 | 2 | ||
An IHC score of <3 was considered low expression and a score of 4–12 was considered high expression.
To further investigate the relationship between EDD and GOLPH3, HEY or HO8910 cells were transfected with GOLPH3 siRNA or plasmid separately. Using RT‐PCR and western blot assays, we found that silencing or overexpressing GOLPH3 could significantly reduce or enhance, respectively, the expression of EDD (Fig. 5C–E). Furthermore, we examined the expression of EMT‐related markers (N‐cadherin, E‐cadherin, and Snail) and Wnt signaling‐related genes (β‐catenin, c‐Myc, and cyclin D1) in low‐expressing EDD cells (Fig. 5F and G). We found that silencing EDD decreased the expression of N‐cadherin, Snail, β‐catenin, c‐Myc, and cyclin D1, while increasing the level of E‐cadherin. These results indicate that GOLPH3 might regulate EDD in the GOLPH3–Wnt/β‐catenin–EMT axis.
Discussion
Tumor formation is a multiple step process that is related to abnormal activity of specific genes and particular pathways 28. Previous studies have demonstrated that GOLPH3, a newly recognized oncogene, is associated with poor prognosis and aggressive behavior in EOC tissues 16, 17. In this study, we focused on the underlying molecular mechanism of GOLPH3 in EOC. We found that GOLPH3 was overexpressed in EOC cell lines and tissues. Moreover, GOLPH3 promoted EMT progression by activating Wnt/β‐catenin signaling, which further influenced the metastatic capacity of EOC. Furthermore, our results showed that EDD, a DNA damage‐related factor and oncogene, might play an important role in the GOLPH3–Wnt/β‐catenin–EMT axis of EOC.
GOLPH3 was originally identified as a member of the trans‐Golgi matrix, which plays critical roles in maintaining Golgi traffic and structure and regulating the cellular response to DNA damage 5, 25. Scott et al. reported that GOLPH3, a target of 5p13 amplification, was a potent proto‐oncogene in human cancers 10. In this study, we found that the expression of GOLPH3 was significantly increased in EOC tissues, as well as in cell lines. Similar results have been reported in several types of cancers 12, 14, 15, which demonstrates the vital role of the oncogene GOLPH3 in tumorigenesis and metastasis.
Previous studies have verified that Golgi PI(4)P promotes tumor metastasis capacity and increases the EMT process by regulating its effector GOLPH3 29, 30. Mengke et al. reported that GOLPH3 promoted cell migration by driving Golgi reorientation to the leading edge 31. Recently, some scholars have proposed that high levels of GOLPH3 enhance the migration and invasion in several cancers 12, 32, 33. Based on these findings, we hypothesized that GOLPH3 might be associated with migration and invasion capability in EOC cells. Transwell and scratch migration analyses showed that knocking down GOLPH3 significantly decreased the migration and invasion capacity of EOC cells. Moreover, the opposite results were observed in GOLPH3‐overexpressing cells, which further confirmed our presumption that GOLPH3 promoted the metastatic capability of EOC cells. Cancer cells in the mesenchymal state can acquire the ability to migrate to distant tissues and form metastases 34. Thus, EMT is regarded as a critical process in cancer metastasis 18, 19. However, there have been no reports exploring the association between GOLPH3 and the EMT process. In this study, we found that GOLPH3 clearly downregulated the expression of E‐cadherin and upregulated the expression of N‐cadherin, β‐catenin, and Snail compared with the control group, which demonstrated that GOLPH3 was a new inducer of EMT. In addition, an increasing number of studies have reported that DNA damage might induce EMT via the TGF‐β pathway 35, 36. Thus, our data also provides new insight into the association between the DNA damage response and metastasis.
As an important regulator of EMT, the Wnt signaling pathway plays a crucial role in EOC 23. Tatyana et al. reported that the retromer subunit Vps35, which interacts with GOLPH3, influenced Wnt secretion by recycling its receptor Wntless 10, 37. β‐catenin regulates the classic Wnt signaling pathway by escaping degradation and translocating into the nucleus. Moreover, when the Wnt/β‐catenin pathway is activated, specific target oncogenes, such as c‐Myc and cyclin‐D1, are aberrantly activated 24. High expression levels of these genes indicate activation of the Wnt/β‐catenin pathway, which promotes the invasion and metastasis ability of tumor cells. Based on our findings that GOLPH3 elevated the expression of β‐catenin, we suggest that GOLPH3 might promote the EMT process by activating the Wnt/β‐catenin signaling pathway. As we expected, the results of this study showed that high GOLPH3 expression enhanced the expression level of c‐Myc and cyclin‐D1. To further confirm our view, we treated cell lines with XAV939 (an inhibitor of the pathway) or LiCl (an activator of pathway). Our results demonstrated that XAV939 blocked and LiCl enhanced the EMT process induced by GOLPH3, which effectively support our hypothesis. In view of previous studies reporting that GOLPH3 modulated mTOR signaling 10, 26, our study may suggest new ideas for investigating the multiple functions of GOLPH3. However, whether GOLPH3 can regulate mTOR signaling in EOC or whether there is cross‐talk among GOLPH3, mTOR, and the Wnt signaling pathway remains to be investigated in future studies.
Hay‐Koren et al. reported that the human hyperplastic discs gene, EDD, upregulated the expression and enhanced the stability of β‐catenin in colorectal cancer 38. Meanwhile, similar to GOLPH3, EDD, a newly discovered mediator in DNA damage signaling, was overexpressed and modulated cisplatin resistance in ovarian cancer 39, 40, 41. Based on these findings, we first investigated the relationship between EDD and GOLPH3. Immunohistochemistry assays showed that the expression of the nuclear protein EDD was consistent with GOLPH3 expression in different types of ovarian tumors. Similar results were presented in the western blot and RT‐PCR assays. Furthermore, using western blotting, we found that EDD might promote the EMT process, as well as activate Wnt/β‐catenin signaling, which means that EDD might be regulated by GOLPH3 in a GOLPH3–Wnt/β‐catenin–EMT axis. However, Ohshima et al. reported that APC was upregulated by EDD, which inhibited β‐catenin, resulting in suppression of Wnt signaling 42. Thus, further studies are needed to confirm the hypothesis, and the molecular mechanisms among EDD and GOLPH3 need to be further evaluated.
In summary, this study provides evidence for the existence of a GOLPH3–Wnt/β‐catenin–EMT axis, which promotes the migration and invasion capability of EOC cells. Furthermore, we find that EDD may be a downstream factor of GOLPH3 in this axis. Given that chemoresistant tumor cells have an EMT phenotype 43, whether GOLPH3 regulates chemoresistance in EOC needs to be investigated in further studies. Furthermore, fully understanding the multiple functions of GOLPH3 may provide new guidance for developing targeted therapy to treat EOC.
Conflict of Interest
The authors have no conflict of interest to declare.
Acknowledgments
This study was supported by the National Natural Science Foundation (No. 30600674) and the Natural Science Foundation of Shanghai (12ZR1424300), and the National Key Clinical Specialist Construction Programs of China.
Cancer Medicine 2017, 6(4):834–844
This study was supported by the National Natural Science Foundation (No. 30600674) and the Natural Science Foundation of Shanghai (12ZR1424300), and the National Key Clinical Specialist Construction Programs of China.
References
- 1. Siegel, R. L. , Miller K. D., and Jemal A.. 2015. Cancer statistics, 2015. CA Cancer J. Clin. 65:5–29. [DOI] [PubMed] [Google Scholar]
- 2. Prat, J. 2015. Oncology FCoG. FIGO's staging classification for cancer of the ovary, fallopian tube, and peritoneum: abridged republication. J. Gynecol. Oncol. 26:87–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coleman, R. L. , Monk B. J., Sood A. K., and Herzog T. J.. 2013. Latest research and treatment of advanced‐stage epithelial ovarian cancer. Nat. Rev. Clin. Oncol. 10:211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bast, R. C. Jr ., Hennessy B., and Mills G. B.. 2009. The biology of ovarian cancer: new opportunities for translation. Nat. Rev. Cancer 9:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Snyder, C. M. , Mardones G. A., Ladinsky M. S., and Howell K. E.. 2006. GMx33 associates with the trans‐Golgi matrix in a dynamic manner and sorts within tubules exiting the Golgi. Mol. Biol. Cell 17:511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li, T. , You H., Zhang J., Mo X., He W., Chen Y., et al. 2014. Study of GOLPH3: a potential stress‐inducible protein from Golgi apparatus. Mol. Neurobiol. 49:1449–1459. [DOI] [PubMed] [Google Scholar]
- 7. Dippold, H. C. , Ng M. M., Farber‐Katz S. E., Lee S. K., Kerr M. L., Peterman M. C., et al. 2009. GOLPH3 bridges phosphatidylinositol‐4‐ phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 139:337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buschman, M. D. , Rahajeng J., and Field S. J.. 2015. GOLPH3 links the Golgi, DNA damage, and cancer. Cancer Res. 75:624–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu, C. C. , Taylor R. S., Lane D. R., Ladinsky M. S., Weisz J. A., and Howell K. E.. 2000. GMx33: a novel family of trans‐Golgi proteins identified by proteomics. Traffic 1:963–975. [PubMed] [Google Scholar]
- 10. Scott, K. L. , Kabbarah O., Liang M.‐C., Ivanova E., Anagnostou V., Wu J., et al. 2009. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 459:1085–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hu, G. S. , Li Y. Q., Yang Y. M., Shi W., Liao A. J., Yao Y. H., et al. 2014. High expression of Golgi phosphoprotein‐3 is associated with poor survival in patients with hepatocellular carcinoma. Tumour Biol. 35:8625–8632. [DOI] [PubMed] [Google Scholar]
- 12. Xue, Y. , Wu G., Liao Y., Xiao G., Ma X., Zou X., et al. 2014. GOLPH3 is a novel marker of poor prognosis and a potential therapeutic target in human renal cell carcinoma. Br. J. Cancer 110:2250–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu, B. S. , Hu H., Zhu C. Y., Gu Y. L., and Li J. P.. 2013. Overexpression of GOLPH3 is associated with poor clinical outcome in gastric cancer. Tumour Biol. 34:515–520. [DOI] [PubMed] [Google Scholar]
- 14. Zhu, K. , Zhao Q., Yue J., Shi P., Yan H., Xu X., et al. 2016. GOLPH3 overexpression correlates with poor response to neoadjuvant therapy and prognosis in locally advanced rectal cancer. Oncotarget 7:68328–68338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zeng, Z. , Lin H., Zhao X., Liu G., Wang X., Xu R., et al. 2012. Overexpression of GOLPH3 promotes proliferation and tumorigenicity in breast cancer via suppression of the FOXO1 transcription factor. Clin. Cancer Res. 18:4059–4069. [DOI] [PubMed] [Google Scholar]
- 16. Ma, Y. , Ren Y., Zhang X., Lin L., Liu Y., Rong F., et al. 2014. High GOLPH3 expression is associated with a more aggressive behavior of epithelial ovarian carcinoma. Virchows Arch. 464:443–452. [DOI] [PubMed] [Google Scholar]
- 17. Ma, Y. , Wang X., Wu Y., Sun B., Lv H., Rong F., et al. 2014. Overexpression of GOLPH3 protein is associated with worse prognosis in patients with epithelial ovarian cancer. Tumour Biol. 35:11845–11849. [DOI] [PubMed] [Google Scholar]
- 18. Nieto, M. A. 2011. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 27:347–376. [DOI] [PubMed] [Google Scholar]
- 19. Polyak, K. , and Weinberg R. A.. 2009. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer 9:265–273. [DOI] [PubMed] [Google Scholar]
- 20. Brabletz, T. 2012. EMT and MET in metastasis: where are the cancer stem cells? Cancer Cell 22:699–701. [DOI] [PubMed] [Google Scholar]
- 21. Li, X. , Xu Y., Chen Y., Chen S., Jia X., Sun T., et al. 2013. SOX2 promotes tumor metastasis by stimulating epithelial‐to‐mesenchymal transition via regulation of WNT/beta‐catenin signal network. Cancer Lett. 336:379–389. [DOI] [PubMed] [Google Scholar]
- 22. Huang, J. , Xiao D., Li G., Ma J., Chen P., Yuan W., et al. 2014. EphA2 promotes epithelial‐mesenchymal transition through the Wnt/beta‐catenin pathway in gastric cancer cells. Oncogene 33:2737–2747. [DOI] [PubMed] [Google Scholar]
- 23. Nagaraj, A. B. , Joseph P., Kovalenko O., Singh S., Armstrong A., Redline R., et al. 2015. Critical role of Wnt/beta‐catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget 6:23720–23734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clevers, H. 2006. Wnt/beta‐catenin signaling in development and disease. Cell 127:469–480. [DOI] [PubMed] [Google Scholar]
- 25. Sechi, S. , Frappaolo A., Belloni G., Colotti G., and Giansanti M. G.. 2015. The multiple cellular functions of the oncoprotein Golgi phosphoprotein 3. Oncotarget 6:3493–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang, Q. , Zhuang J., Deng Y., Zhao X., Tang B., Yao D., et al. 2015. GOLPH3 is a potential therapeutic target and a prognostic indicator of poor survival in bladder cancer treated by cystectomy. Oncotarget 6:32177–32192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feng, Y. , He F., Wu H., Huang H., Zhang L., Han X., et al. 2015. GOLPH3L is a novel prognostic biomarker for epithelial ovarian cancer. J. Cancer 6:893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ponder, B. A. 2001. Cancer genetics. Nature 411:336–341. [DOI] [PubMed] [Google Scholar]
- 29. Tokuda, E. , Itoh T., Hasegawa J., Ijuin T., Takeuchi Y., Irino Y., et al. 2014. Phosphatidylinositol 4‐phosphate in the Golgi apparatus regulates cell‐cell adhesion and invasive cell migration in human breast cancer. Cancer Res. 74:3054–3066. [DOI] [PubMed] [Google Scholar]
- 30. Wood, C. S. , Schmitz K. R., Bessman N. J., Setty T. G., Ferguson K. M., and Burd C. G.. 2009. PtdIns4P recognition by Vps74/GOLPH3 links PtdIns 4‐kinase signaling to retrograde Golgi trafficking. J. Cell Biol. 187:967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xing, M. , Peterman M. C., Davis R. L., Oegema K., Shiau A. K., and Field S. J.. 2016. GOLPH3 drives cell migration by promoting Golgi reorientation and directional trafficking to the leading edge. Mol. Biol. Cell 27:3828–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou, X. , Zhan W., Bian W., Hua L., Shi Q., Xie S., et al. 2013. GOLPH3 regulates the migration and invasion of glioma cells though RhoA. Biochem. Biophys. Res. Commun. 433:338–344. [DOI] [PubMed] [Google Scholar]
- 33. Zhang, X. , Ding Z., Mo J., Sang B., Shi Q., Hu J., et al. 2015. GOLPH3 promotes glioblastoma cell migration and invasion via the mTOR‐YB1 pathway in vitro. Mol. Carcinog. 54:1252–1263. [DOI] [PubMed] [Google Scholar]
- 34. Thiery, J. P. , Acloque H., Huang R. Y., and Nieto M. A.. 2009. Epithelial‐mesenchymal transitions in development and disease. Cell 139:871–890. [DOI] [PubMed] [Google Scholar]
- 35. Andarawewa, K. L. , Erickson A. C., Chou W. S., Costes S. V., Gascard P., Mott J. D., et al. 2007. Ionizing radiation predisposes nonmalignant human mammary epithelial cells to undergo transforming growth factor beta induced epithelial to mesenchymal transition. Cancer Res. 67:8662–8670. [DOI] [PubMed] [Google Scholar]
- 36. Chiba, N. , Comaills V., Shiotani B., Takahashi F., Shimada T., Tajima K., et al. 2012. Homeobox B9 induces epithelial‐to‐mesenchymal transition‐associated radioresistance by accelerating DNA damage responses. Proc. Natl. Acad. Sci. USA 109:2760–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Belenkaya, T. Y. , Wu Y., Tang X., Zhou B., Cheng L., Sharma Y. V., et al. 2008. The retromer complex influences Wnt secretion by recycling wntless from endosomes to the trans‐Golgi network. Dev. Cell 14:120–131. [DOI] [PubMed] [Google Scholar]
- 38. Hay‐Koren, A. , Caspi M., Zilberberg A., and Rosin‐Arbesfeld R.. 2011. The EDD E3 ubiquitin ligase ubiquitinates and up‐regulates beta‐catenin. Mol. Biol. Cell 22:399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Henderson, M. J. , Munoz M. A., Saunders D. N., Clancy J. L., Russell A. J., Williams B., et al. 2006. EDD mediates DNA damage‐induced activation of CHK2. J. Biol. Chem. 281:39990–40000. [DOI] [PubMed] [Google Scholar]
- 40. O'Brien, P. M. , Davies M. J., Scurry J. P., Smith A. N., Barton C. A., Henderson M. J., et al. 2008. The E3 ubiquitin ligase EDD is an adverse prognostic factor for serous epithelial ovarian cancer and modulates cisplatin resistance in vitro. Br. J. Cancer 98:1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bradley, A. , Zheng H., Ziebarth A., Sakati W., Branham‐O'Connor M., Blumer J. B., et al. 2014. EDD enhances cell survival and cisplatin resistance and is a therapeutic target for epithelial ovarian cancer. Carcinogenesis 35:1100–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ohshima, R. , Ohta T., Wu W., Koike A., Iwatani T., Henderson M., et al. 2007. Putative tumor suppressor EDD interacts with and up‐regulates APC. Genes Cells 12:1339–1345. [DOI] [PubMed] [Google Scholar]
- 43. Chiu, W. T. , Huang Y. F., Tsai H. Y., Chen C. C., Chang C. H., Huang S. C., et al. 2015. FOXM1 confers to epithelial‐mesenchymal transition, stemness and chemoresistance in epithelial ovarian carcinoma cells. Oncotarget 6:2349–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]