

Abstract
Transforming growth factor (TGF) β2 and fibroblast growth factor (FGF) 2 are involved in regulation of posterior capsule opacification (PCO) and other processes of epithelial–mesenchymal transition (EMT) such as cancer progression, wound healing and tissue fibrosis as well as normal embryonic development. We previously used an in vivo rodent PCO model to show the expression of tropomyosin (Tpm) 1/2 was aberrantly up‐regulated in remodelling the actin cytoskeleton during EMT. In this in vitro study, we show the Tpms family of cytoskeleton proteins are involved in regulating and stabilizing actin microfilaments (F‐actin) and are induced by TGFβ2 during EMT in lens epithelial cells (LECs). Importantly, we found TGFβ2 and FGF2 played contrasting roles. Stress fibre formation and up‐regulation of α‐smooth muscle actin (αSMA) induced by TGFβ2 could be reversed by Tpm1/2 knock‐down by siRNA. Expression of Tpm1/2 and stress fibre formation induced by TGFβ2 could be reversed by FGF2. Furthermore, FGF2 delivery to TGFβ‐treated LECs perturbed EMT by reactivating the mitogen‐activated protein kinase (MAPK)/ extracellular signal‐regulated kinase (ERK) pathway and subsequently enhanced EMT. Conversely, MEK inhibitor (PD98059) abated the FGF2‐mediated Tpm1/2 and αSMA suppression. However, we found that normal LECs which underwent EMT showed enhanced migration in response to combined TGFβ and FGF2 stimulation. These findings may help clarify the mechanism reprogramming the actin cytoskeleton during morphogenetic EMT cell proliferation and fibre regeneration in PCO. We propose that understanding the physiological link between levels of FGF2, Tpm1/2 expression and TGFβs‐driven EMT orchestration may provide clue(s) to develop therapeutic strategies to treat PCO based on Tpm1/2.
Keywords: epithelial–mesenchymal transition, tropomyosin, lens epithelial cells, FGF2, TGFβ2
Introduction
Age‐related cataract, a chronic disorder of ageing, is the main cause of blindness worldwide. PCO is a common, significant complication following cataract surgery. Advances in surgical techniques, intraocular lens materials and designs have reduced the PCO rate, but it remains a significant problem worldwide, even in young and infant patients 1, 2. After cataract surgery, aberrant cell growth across the lens capsule often leads to fibrosis and secondary visual loss, known as PCO, so‐called secondary or after cataracts 3. EMT of LECs is the main cause 4, 5, 6, 7. EMT is related to other eye diseases such as pterygium and glaucoma and the wound healing process after eye surgery 8, 9, 10. To regulate EMT is important for treatment of many eye diseases including PCO. The current treatment of PCO is YAG laser capsulotomy. However, this method can lead to uveitis, cystoid maculae oedema, elevation of intraocular pressure, retinal detachment and intraocular lens damage. Further, in paediatric cataract, it is clear that cataract extraction and correction of aphakia should be performed as soon as possible during the key period of vision 11. However, PCO is a common complication of the surgery and also leads to amblyopia. YAG laser capsulotomy is not difficult to perform, but it needs patients’ cooperation and eye fixation 11. Secondary capsulotomy surgery is sometimes required in children 12. We believe that it is important to study on the prevention of PCO for child patients. Further, it is important to regulate EMT and PCO in LECs for the clinical treatments using accommodative lens refilling13 and for the regeneration of clear lens in vivo in future.
Aberrant TGF β signalling plays a central role in the pathobiology of cells or tissues by dysregulating extracellular matrix (ECM)‐related genes in LECs, akin to the development of human anterior subcapsular cataract 14, 15, 16, 17 and PCO 5, 17, 18, 19, 20. Moreover, TGFβs are involved in induction of tissue fibrosis, myofibroblast formation and apoptosis 21, 22, 23 by up‐regulating genes encoding ECM proteins including αSMA, types I and III collagens. Previous studies suggest FGF may contribute to PCO development. FGF2 is expressed in human LECs 24 and involved in lens development 25 regulating cell proliferation and migration. This molecule is involved in stimulation of lens fibre differentiation in a dose‐dependent manner 26, 27 and also activates LEC mitosis increasing the formation of collagen 28. FGF2 was shown to reduce the contraction of a collagen gel in bovine LECs and the proportion of cells expressing αSMA 29, indicating that FGF2 acts contrary to TGFβ. Moreover, EMT in PCO was reported to be regulated by ECM components and soluble growth factors or cytokines, including epidermal growth factor, FGFs and TGFβs 26, 28, 29, 30, 31, 32, of which TGFβ and FGF are key mediators of EMT and are frequently and abundantly expressed in PCO tissues. Despite many studies on the activities and roles of TGFβ in lens, it remains unknown how TGFβs and FGF 2 synergistically act in EMT and how these molecules affect gene expressions including αSMA and tropomyosins (Tpms) during PCO development.
A number of target genes of FGF2 and TGFβ have been identified whose expression is activated by them, some of which are overly stimulated and implicated in EMT process including αSMA, fibronectin and Tpms, and thereby PCO formation 2, 33, 34, 35. More recently, our group identified modulation in expression of Tpms, specifically high molecular weight Tm isoforms from Tpm1 and Tpm2 genes, in a rodent model of PCO and in LECs obtained from cataractous humans of various ages 36. We previously reported expression of Tpm1/2 was minimal in rat LECs, and expression of Tpm1/2 that increased selectively during EMT was linked to fibrosis in PCO 36. Other cellular abnormalities, particularly in aberrant expression of cytoskeleton and ECM proteins, are induced because of overshooting of cellular signalling mediated by reactive oxygen species (ROS) 37. It is known that ROS‐induced damage to cells is related to ROS‐driven overstimulation TGF‐β1‐mediated signalling 38, 39 leading to over‐modulation of certain genes expression, including αSMA and TGF‐β‐induced protein (βig‐h3). Overexpression of those genes was involved in cataractogenesis, PCO and pathophysiological disorders of cells and tissues 31, 39, 40.
Previously, we showed that LECs deficient in peroxiredoxin 6 (Prdx6) display increased expression of ROS, phenotypic changes, a characteristic of terminal cell differentiation and EMT 39. Prdx6 provides cytoprotection against internal and external environmental stresses and plays a role in cellular signalling by detoxifying ROS thereby controlling gene regulation 39, 41, 42, 43. Using proteomic analysis of Prdx6‐deficient (Prdx6 −/−) mouse LECs, we found that such cells displayed elevated expression of cytoskeleton proteins Tpm1, Tpm2 and vimentin 44. Therefore, we posit that because Tpms are implicated in regulation of cellular activities by stabilizing ECM proteins (specifically actin microfilaments), aberrant expression of Tpm1 and Tpm2 genes is likely to be involved in the phenotypic alteration of Prdx6 −/− LECs in mice.
Furthermore, Tpms are recognized as actin filament stabilizing proteins, regulating the dynamics and structural properties of the filaments by controlling the interaction of the filaments with actin‐binding proteins 45, 46, 47. The human tropomyosin genes should be known as TPM1 through TPM4 (Tpm1 through Tpm4 for mouse and rat tropomyosin) to be consistent with other gene nomenclatures 48. The various isoforms generated via alternative exon splicing are listed under each gene 47. The balance between levels of isoforms in a given cell determines the cell's Tpm functions 49, 50, 51, 52. Several TGFβ target genes, including Tpm1, Tpm2, α‐actinin1 and calponin2‐encoding actin‐binding proteins, were implicated in the assembly of stress fibres 51, 53, and Tpms played a crucial role in stabilizing actin filaments 54. TGFβ specifically up‐regulates expression of Tpm1 and Tpm2 genes but has no effect on regulation of Tpm3 and Tpm4 genes, which encode low molecular weight Tpms 51, 53. In addition, our group demonstrated an increased abundance of Tpm1/2 in differentiating LECs, suggesting involvement of TGFβ‐induced deleterious signalling in the induction of Tpm1/2 55. Given the above scenario, we surmised that over expression and activation of TGFβ induced by both surgical stress and ROS during cataract surgery induces and accelerates EMT by up‐regulating Tpm1/2 genes, leading to PCO 55 and the process may be modulated by FGF2 in cellular microenvironment. In this study, we determined the role and potential of FGF2 to act as an antagonist of aberrant TGFβ signalling and its inducible genes/proteins such as Tpm1/2 and αSMA that play a part in EMT process and stress fibre formation in mouse and human LECs. In addition, we show the involvement of FGF2‐mediated MAPK/ERK 1/2 signalling on TGF‐β2‐induced EMT. Loss of Tpms in TGFβ‐evoked EMT by FGF2 was significantly linked to MAPK/ERK1/2 pathway. We provide evidence for a regulatory role(s) of Tpms in the EMT process leading to PCO, and how the process may be restored by FGF2 in a concentration‐dependent manner. Understanding the underlying mechanisms of EMT, a cause of PCO, and its regulation by TGFβ, FGF2 and Tpms is important to develop new treatments to inhibit EMT and postpone PCO, secondary cataract.
Materials and methods
Cell culture
Primary cultured LECs were generated from 6W BalbC mice (n = 8) as described previously 56. Mice LECs (MLECs) were maintained in Dulbecco's modified Eagle's medium (DMEM; WAKO, Osaka, Japan) with 10% foetal bovine serum (FBS; Sigma‐Aldrich, St. Louis, MO, USA) at 37°C in an air/CO2 (19:1) atmosphere as described 39. Cells from 3 to 5 passages were used for the experiments. Simian virus 40‐transformed Huma LECs (HLECs) (SRA01/04) were kindly gifted by Dr. Nobuhiro Ibaraki (Ibaraki Eye Clinic, Tochigi, Japan). Human LECs were cultured in DMEM supplemented with 20% FBS.
To examine the effects of FGF2 (PEPROTECH, RockyHill, NJ, USA) and/or TGF‐β2 (HumanZyme, Chicago, IL, USA), MLECs or HLECs were plated in triplicate into 35 mm culture dishes (TPP® Techno Plastic Products AG, Trasadingen, Switzerland). Cells growing in DMEM containing 0.1% bovine serum albumin (BSA) (WAKO) in the presence or absence of various test growth regulators received 0.001–10.0 ng/ml FGF‐2 or 0–10 ng/ml TGF‐β2 every other day for up to 4 days.
RNA interference
siRNAs were transfected into cells according to the protocol recommended for Lipofectamine® RNAi MAX reagent (ThermoFisher Scientific Japan Ltd., Tokyo, Japan). MLEC and HLECs were transiently transfected with siRNAs against a mixture of mouse, and human Tpm1 (Silencer® Select pre‐designed siRNA, ID:s75390, ThermoFisher Scientific Japan Ltd.) and mouse and human Tpm2 (Silencer® Select pre‐designed siRNA, ID:s75393) or negative control (Silencer® Select Negative control#1 siRNA: NC‐siRNA). The final concentrations of the siRNAs were 10 nM.
Western blot analysis
Protein lysates of MLECs or HLECs were prepared in ice‐cold radioimmune precipitation (RIPA) buffer, and SDS‐PAGE and protein blot analysis was performed as described 38, 57, 58. The membranes were probed with antimouse Tpm1/2 monoclonal antibody (Ab) (TM311) (Abcam®, Cambridge, MA, USA), antimouse αSMA monoclonal Ab (Sigma‐Aldrich), anti‐rabbit p44/42 MAPK (Erk1/2) monoclonal Ab (Cell Signaling Technology (CST) Japan, K.K., Tokyo, Japan), and anti‐rabbit phospho‐p44/42 MAPK (Erk1/2) monoclonal Ab (CST Japan). Anti‐rabbit glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) polyclonal Ab (Sigma‐Aldrich) was used to demonstrate that equal amounts of protein were loaded onto each lane.
Real‐time reverse transcriptase‐polymerase chain reaction (RT‐PCR)
Total RNA from the MLECs or HLECs was extracted using an RNeasy Mini Kit (Qiagen, Valencia, CA, USA) per manufacturer's instructions. To measure the expression of mouse and human Tpm mRNAs, we conducted relative quantification of mRNA using a Prism7300 (Applied Biosystems®, ThermoFisher Scientific Japan Ltd., Tokyo Japan). PCR amplification was performed using a TaqMan Universal Master Mix and pre‐developed mouse Tpm2 probe mix (Applied Biosystems®), which recognize Tpm1 isoform, and mouse Tpm1 probe mix, which recognizes Tpm2, 3 and 5 isoforms. The relative quantity of Tpm1/2 and Tpm2 mRNA was determined using the comparative Ct method and then normalized using a pre‐developed TaqMan ribosomal RNA control reagent VIC probe as an endogenous control (Applied Biosystems®).
ERK‐MAPK signalling pathway assay
The activities of ERK kinase were examined by Western blot analyses using antibodies against their phosphorylated forms. For the analysis of ERK activity, MLECs were treated with TGFβ2 (10 ng/ml) and/or FGF2 (10 ng/ml) for 10 and 60 min. To analyse the effect of a pharmacological inhibitor of FGF receptor (FGFR) (SU5402: SU, Sigma‐Aldrich) and a potent and selective inhibitor of MAP kinase (also known as MAPK/ERK kinase or MEK kinase) (PD98059: PD, Sigma‐Aldrich) on FGF2‐induced ERK phosphorylation, MLECs were pretreated with PD or SU with/without TGFβ2 (10 ng/ml) and/or FGF2 (10 ng/ml) for 60 min and 24 hrs.
Immunofluorescence labelling and F‐actin staining
HLECs were grown on collagen‐coated eight‐well culture slides (Matsunami Glass Ind., Ltd., Osaka, Japan) and treated with the presence or absence of 10 ng/ml TGFβ2 and 10 ng/ml FGF2 in DMEM containing 2% FBS for 24 hrs. The cells were fixed with 4% paraformaldehyde and permeabilized with 0.05% Triton X‐100 (Sigma‐Aldrich). Tpm1/2 was stained with antimouse Tpm1/2 monoclonal antibody (Ab) (TM311) (Abcam®) and goat antimouse IgG (H+L) secondary Ab, Alexa Fluor® 594 conjugate (ThermoFisher Scientific Japan Ltd.). F‐actin filaments were stained with CytoPainter phalloidin‐iFluor 488 reagent (Abcam®). The cell nucleus was stained with 4,6‐Diamidino‐2‐phenylindole, dihydrochloride (DAPI; Fluoroshield Mounting Medium with DAPI, ImmunoBioScience Corp., Mukilteo, WA, USA). Fluorescent images were captured using EVOS® FLoid® Cell Imaging Station (ThermoFisher Scientific Japan Ltd.).
Cell migration assay
Cell migration assays were performed using Radius™ 24‐Well Cell Migration Assay Kit (Cell Biolabs, Inc., San Diego, CA, USA) per manufacturers’ protocol. Briefly, MLECs were grown on pre‐treated Radius™ 24‐well cell migration plate (Cell Biolabs, Inc.) at 2.0 × 105/well for 9 hrs. Cells do not attach in the Radius™ gel spot area (~0.68 mm). Plates were treated with Radius™ gel removal solution to expose the cell‐free area to cell migration. After removal of gel, cells were treated with the presence or absence of 10.0 ng/ml FGF2 or 10 ng/ml TGFβ2 in DMEM containing 0.1% BSA for 24 hrs to observe their effect on cell migration.
Statistical analysis
Data were reported as means ± S.D.s and analysed by one‐way anova, followed by a t‐test when appropriate, with P < 0.05 deemed significant.
Results
Effect of TGFβ and/or FGF2 on morphological changes and the expression of Tpm1/2 and αSMA, EMT markers in MLECs in vitro
To examine whether MLECs treated with TGFβ or TGFβ plus FGF2 showed phenotypic change, we selected TGFβ2 as the major isoform expressed in eyes, which plays a role in PCO 59, and FGF2 which is the most potent in FGF family. We performed phase contrast microscopic observation to analyse phenotypes of MLECs with/without addition of TGFβ2 (10 ng/ml) and/or FGF2 (10 ng/ml) for 48 hrs. Untreated MLECs were cuboidal shaped and organized in compact islets (Fig. 1A). After 48 hrs with TGF‐β, cells in these islets showed a spindle‐shaped morphology, elongated and underwent EMT‐like change (Fig. 1B) in response to TGFβ2 compared to untreated control (Fig. 1A). Many rounded dead cells were observed. FGF2‐treated MLECs were more proliferated than control (Fig. 1C). MLECs treated with TGFβ2 plus FGF2 (10 ng/ml each) were elongated showing fibroblastic‐like changes (Fig. 1D). These results prompted us to examine the contribution of TGFβ alone or in combination with FGF2 in mobilizing LECs towards EMT or vice versa.
Figure 1.
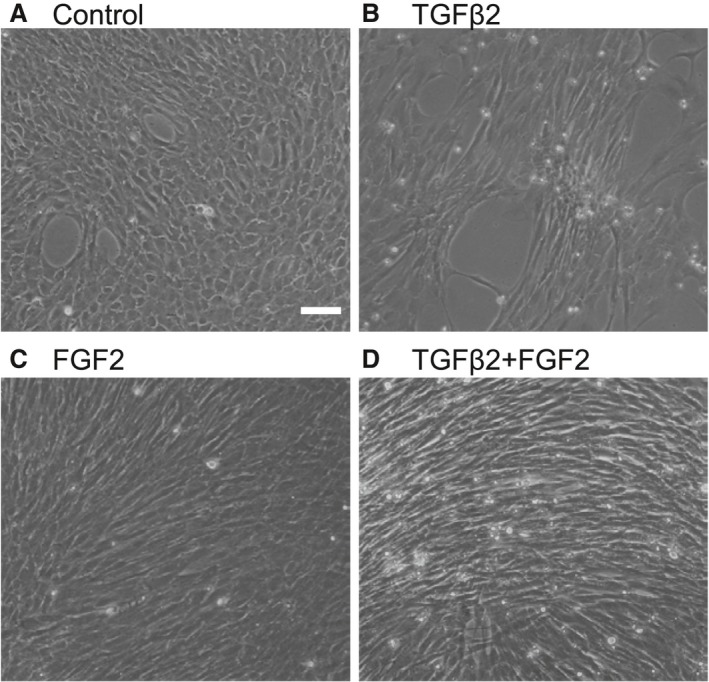
TGFβ2‐ and/or FGF2‐mediated phenotypic changes of LECs in vitro. Cultured MLECs were plated in 35 mm dishes at a density of 1 × 105 in DMEM with 10%FBS for 24 hrs. LECs were treated with 10 ng/ml of TGFβ2 and/or FGF2 in DMEM containing 0.1% BSA for 2 days. Phase contrast photomicrographs were taken with a digital camera. Data were from three experiments. Scale bar, 90 μm.
Figure 1 shows TGFβ2, and TGFβ2 in combination with FGF2 differentially affected LECs phenotype. Next we examined levels of genes which have been reported to be involved in regulation of shape and geometric features of cells including αSMA and Tpms. Western blotting analysis of levels of αSMA and Tpm1/2 protein in MLECs in response to TGFβ1 and β2 (10 ng/ml, each) using antibody specific to αSMA and Tpm1/2 revealed that TGFβ1‐treated cells displayed enhanced expression of Tpm1 and αSMA proteins on day 4 (Fig. 2A; **P < 0.016, ****P < 0.05). Similarly, TGFβ2‐treated MLECs showed increased abundance of Tpm1 and αSMA proteins (Fig. 2A; *P < 0.0002, ***P < 0.0015, Fig. 3A and B; *P < 0.0042, ***P < 0.0028). TGFβ2 was more potent in up‐regulating expression. To determine whether increased expression of αSMA and Tpm1 by TGFβ was transcriptional level, we performed real‐time RT‐PCR. We found the expressions of Tpm1/2 mRNA were significantly elevated in MLECs following TGFβ1 and TGFβ2 treatment (Fig. 2B, *P < 0.0001; **P < 0.05). Further, levels of Tpm1/2 protein in Tpm1/2 siRNA‐transfected MLECs were reduced after treatment with/without TGFβ2 on day 2 (Fig. 3A and B; **P < 0.0061). TGFβ2‐treated MLECs showed increased abundance of Tpm1 protein after transfection with NC‐siRNA (control) (Fig. 3A and B; *P < 0.0042). Similarly, TGFβ2‐treated MLECs showed increased abundance of αSMA protein after transfection with NC‐siRNA (control) and siRNA against Tpm1/2 (siTpm1/2) (Fig. 3A and B; ***P < 0.0028, ****P < 0.0241). Levels of αSMA protein in TGFβ2‐treated MLECs were reduced in siTpm1/2 group in comparison with control group suggesting EMT induced by TGFβ2 may be inhibited by Tpm1/2 knock‐down (Fig. 3A and B; ****P < 0.0024).
Figure 2.
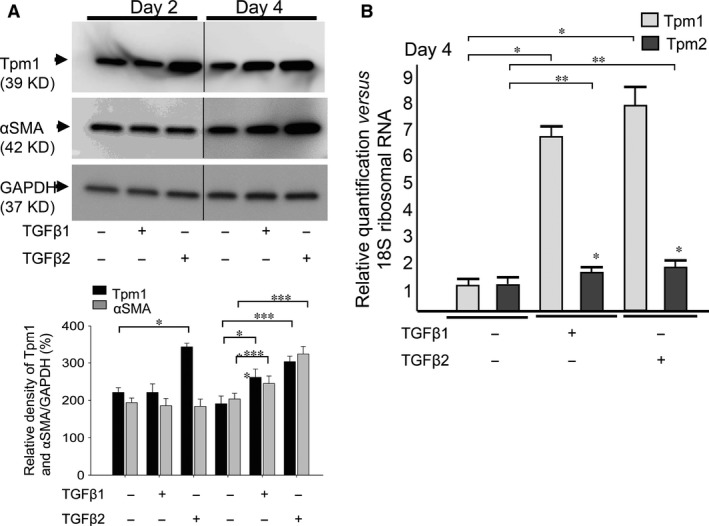
Expression of Tpm1/2 and αSMA in response to TGFβ1 and TGFβ2 in MLECs. Cultured MLECs were plated in 35 mm dishes at a density of 1 × 105 in DMEM with 10%FBS for 24 hrs. LECs were treated with 10 ng/ml of TGFβ1 or TGFβ2 in DMEM containing 0.1% BSA for 2 days. (A) Cell lysates were prepared, and Western blotting analysis was performed. αSMA was used as the marker of EMT. GAPDH was used for control of protein concentration on Western blot analysis. Relative densities of Tpm1/2, αSMA and GAPDH were determined using the Image Quant LAS 4000 (GE Healthcare UK Ltd. Buckinghamshire, England). Data are representative of three experiments. (B) Total RNA was prepared, and real‐time PCR analysis was performed. 18s ribosomal RNA was used for control of cDNA concentration on real‐time PCR analysis. Relative quantity of Tpm1/2 was determined using Prism 7300 System SDS RQ Study Software (Applied Biosystems®). Data were from three experiments and were reported as means ± S.D.s.
Figure 3.
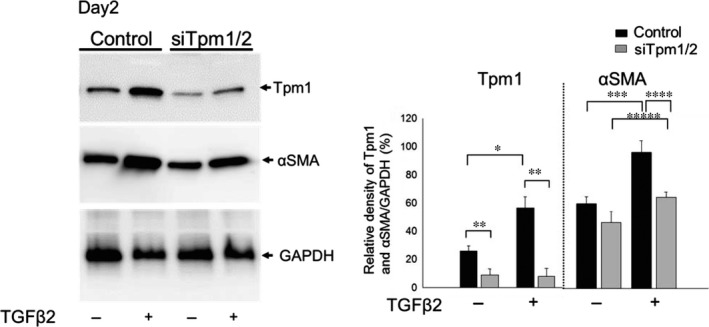
Expression of Tpm1/2 and αSMA in response to TGFβ2 in MLECs transfected with siRNA against Tpm1/2 and negative control. Cultured MLECs were plated in 35 mm dishes at a density of 1 × 105 in DMEM with 10%FBS for 24 hrs. MLECs were transfected with siRNA against Tpm1/2 and negative control. At 24 hrs after transfection, LECs were treated with 10 ng/ml of TGFβ2 in DMEM containing 2%FBS for 2 days. A: Cell lysates were prepared, and Western blotting analysis was performed. αSMA was used as the marker of EMT. GAPDH was used for control of protein concentration on Western blot analysis. Data were from three experiments and were reported as means ± S.D.s.
FGF2‐treated cells also exhibited changes as shown in Figure 1. MLECs treated with FGF2 for 2 and 4 days showed a marked reduction in the expressions of Tpm1 and αSMA protein (Fig. 4A; *P < 0.0004, **P < 0.002, *** P < 0.00001). To investigate the effect of FGF2 concentration on levels of Tpm1 or αSMA expression in MLECs, cells were treated with different concentrations of FGF2 for 2 days. We found that reduction of Tpm1 expression in MLECs was inversely related to the concentration of FGF2 (Fig. 4B; *P < 0.007, **P < 0.0005, ***P < 0.0000001). Unlike TGFβ, FGF2 suppressed the expression of Tpm1 and αSMA in MLECs suggesting that FGF2 acts as an antagonist of TGFβ signalling.
Figure 4.
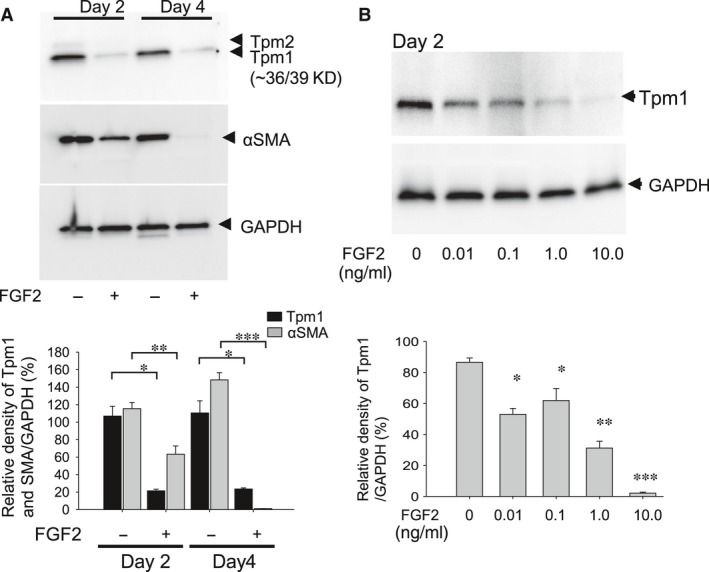
Expression of Tpm1 and αSMA in MLECs in response to FGF2. Cultured MLECs were plated in 35 mm dishes at a density of 1 × 105 in DMEM with 10%FBS for 24 hrs. (A) MLECs were treated with 0 or 10 ng/ml of FGF2 in DMEM containing 0.1% BSA for 2 days. αSMA was used as the marker of EMT. (B) MLECs were treated with 0, 0.01, 0.1, 1.0 or 10 ng/ml of FGF2 in DMEM containing 0.1% BSA for 2 days. A and B: Cell lysates were prepared, and Western blotting analysis was performed, with GAPDH used for control of protein concentration. Data were from three experiments and were reported as means ± S.D.s.
Next, to examine the combined effect of TGFβ2 and FGF2 on Tpm1 and αSMA expression in MLECs, cells were stimulated with FGF2 (10 ng/ml) and TGFβ2 (10 ng/ml) for 2 and 4 days. Based on the observed EMT‐like changes in MLECs (Fig. 1D), we speculated that FGF2 and TGFβ2 may induce Tpm1 or Tpm2 expression. However, FGF2 showed an opposing action against TGFβ2′s adverse effects, with significantly reduced expression of Tpm1 and αSMA proteins (Fig. 5A; *P < 0.0006, **P < 0.002). Similarly, in HLECs treated with TGFβ2 and FGF2 in combination, the expression of Tpm1 and Tpm2 protein was increased in response to TGFβ2 and decreased in response to FGF2 and TGFβ2 at day 4 (Fig 5B; P < 0.0008, **P < 0.013, ***P < 0.005). Taken together, these findings indicate that TGFβ2 induces expression of Tpm1, Tpm2 and αSMA, and in contrast, FGF2 acts as antagonist and reverses the TGFβ2‐mediated aberrant activation of Tpm1, Tpm2 and αSMA expression in MLECs.
Figure 5.
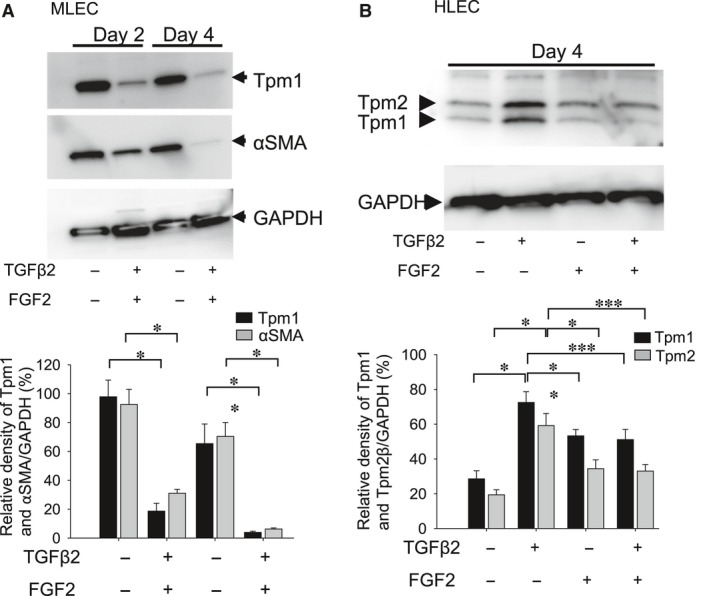
Expression of Tpm1 and αSMA proteins in MLECs and HLECs stimulated with FGF2 and TGFβ2. Cultured MLECs (A) and HLECs (B) were plated in 35 mm dishes at a density of 1 × 105 in DMEM with 10%FBS for 24 hrs. (A) MLECs were treated with 0 or 10 ng/ml of TGFβ2 and FGF2 in DMEM containing 0.1% BSA for 2 and 4 days. αSMA was used for marker of EMT. (B) HLECs were treated with 0 or 10 ng/ml of TGFβ2 and/or FGF2 in DMEM containing 0.1% BSA for 4 days. A and B: Cell lysates were prepared, and Western blotting analysis was performed, with GAPDH used for control of protein concentration. Data were from three experiments and were reported as means ± S.D.s.
Effect of TGFβ2 and/or FGF2 on stress fibre formation in Tpm1/2‐siRNA‐transfected HLECs
The actin cytoskeleton plays a crucial role in regulation of cellular processes including proliferation, apoptosis, cell migration and invasion 54, 60. TGFβ induces rapid reorganization of the actin cytoskeleton, whereas prolonged incubation with TGFβ induces stress fibres 61, 62. Stress fibres, which are contractile bundles of actin filaments and actomyosin, are essential for cell adhesion, migration and maintenance of cell shape 63. To examine stress fibre formation in HLECs, we performed F‐actin staining with phalloidin as described in the ‘Materials and Methods’ section. Microscopic examination revealed an absence of stress fibres (Fig. 6A‐a, left) in untreated HLECs, and Tpm1/2 was faintly immunolabelled in the perinuclear area of untreated HLECs (Fig. 6A‐a, right). In contrast, after 48 hrs with TGF‐β, F‐actin was assembled into thick parallel bundles, or actin stress fibres, traversing the ventral cell surface (Fig. 6A‐b, left) and Tpm1/2 was localized at these stress fibres indicating involvement of TGFβ2 in their formation (Fig. 6A‐b, right). In HLECs treated with FGF2, stress fibre formation was markedly reduced (Fig. 6A‐c, left) and levels of Tpm1/2 were reduced. In HLECs treated with TGFβ2 and FGF2, stress fibre formation was reduced and located in the cellular periphery (Fig. 6A‐d, left) and Tpm1/2 was immunolabelled in the perinuclear area of HLECs. This result suggests that stress fibre formation in TGFβ2‐treated HLECs was blocked because of the presence of FGF2. Further, siRNA against Tpm1/2 was transfected in HLECs. Immunolabelling of Tpm1/2 was reduced in Tpm1/2‐siRNA‐transfected HLECs (Fig. 6B a–d, right). In Tpm1/2‐siRNA‐treated HLECs, stress fibres were not induced after treatment with or without TGFβ2 and/or FGF2 (Fig. 6B b–d, right). These results suggest that FGF2 and knock‐down of Tpm1/2 may inhibit TGFβ2‐mediated stress fibre formation in LECs. Higher expression of Tpm1/2 may induce stress fibre formation in LECs.
Figure 6.
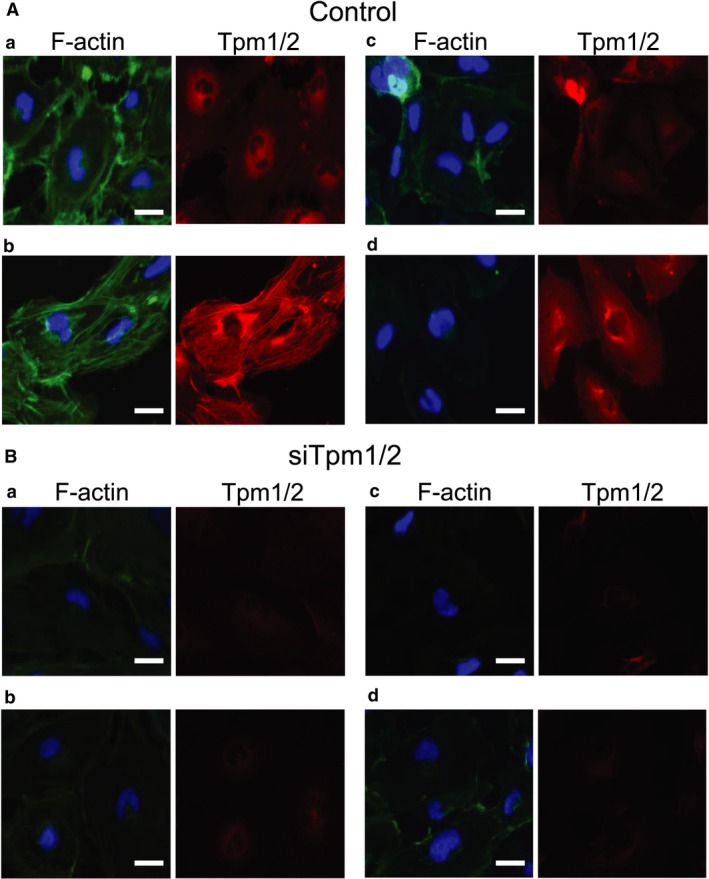
F‐actin staining with phalloidin in HLECs stimulated with 10 ng/ml TGFβ2 and/or 10 ng/ml FGF2. Cultured HLECs were plated in collagen‐coated eight‐well chamber slides at a density of 4 × 104 in DMEM with 10%FBS for 24 hrs. MLECs were transfected with siRNA against Tpm1/2 (A) and negative control (B). At 24 hrs after transfection, LECs were treated with 10 ng/ml of TGFβ2 and/or FGF2 in DMEM containing 2% FBS for 24 hrs. To observe immunolocalization of Tpm1/2 and stress fibre formation, immunolabelling of Tpm1/2 (A and B) and co‐labelling of F‐actin (A and B) were performed in HLEC treated without TGFβ2 (A‐a and B‐a), with TGFβ2 (A‐b and B‐b), with FGF2 (A‐c and B‐c) or TGFβ2 + FGF2 (A‐d and B‐d) treatment for 2 days. The cell nucleus was stained with DAPI (A and B; Blue colour). Data are representative of three experiments. Scale bar, 15 μm.
Effect of TGFβ2 and/or FGF2 on cell migration in MLECs
To determine the effect of TGFβ2 with/without FGF2 on migration, we conducted a cell migration assay as described in the ‘Materials and Methods’ section. We found that treatment with TGFβ2 induced MLECs migration (Fig. 7A and B; *P < 0.01), which was further augmented, when MLECs were treated with both TGFβ2 and FGF2 in combination (Fig. 7A and B; *P < 0.01), indicating TGFβ2 in combination with FGF2 accelerated the migration process of MLECs. Further, treatment with FGF2 alone did not induce the migration of MLECs (Fig. 7A and B).
Figure 7.
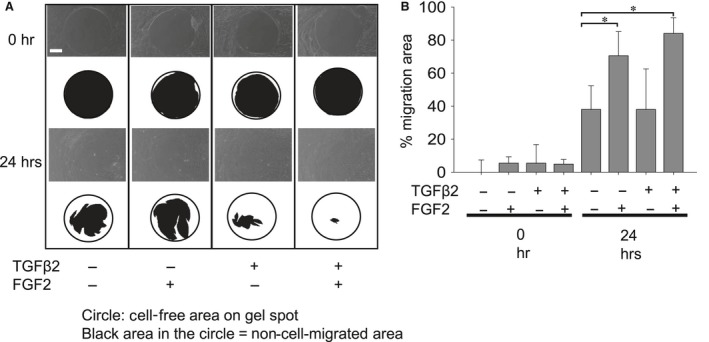
Migration of MLECs treated with/without TGFβ2 and/or FGF2. MLECs were plated in 24‐well plates pre‐coated with collagen type I, at a density of 1 × 105 in DMEM with 10%FBS for 24 hrs. Each plate contained 0.68 mm non‐toxic biocompatible hydrogel spot (Radius™ Gel) where cells cannot attach. After hydrogel removal to expose the cell‐free region, MLECs were treated with 0 or 10 ng/ml of TGFβ2 and/or FGF2 in DMEM containing 0.1% BSA for 24 hrs. Phase contrast micrographs were then taken with a digital camera. Data shown are representative of three experiments. The cell‐free area was analysed using MultiGauge Software (Fuji Film, Tokyo, Japan). Data were from three experiments and were reported as means ± S.D.s. Scale bar, 180 μm.
FGF2 inhibits TGFβ2‐induced Tpm1 and αSMA gene, but promotes TGFβ2‐induced cell migration via activation of MAPK/ERK pathway
We next examined the involvement of signalling pathway(s) responsible for FGF2‐mediated down‐regulation of Tmp1α expression and promotion of cell migration in the presence or absence of FGF2 with TGFβ2. To investigate the effect of FGF2 with/without TGFβ2 stimulation on phosphorylation of ERK1/2 in MLECs, MLECs were treated with FGF2 and/or TGFβ2. Cellular extracts were immunoblotted using antibody as indicated in Fig. 8. FGF2 induced the ERK phosphorylation within/at 10 min and remained for 60 min with/without TGFβ2 stimulation. These results suggest that FGF2 activated ERK pathway, as it stimulated ERK phosphorylation, and wherein treatment of TGFβ2 had no effect on ERK phosphorylation (Fig. 8; *P < 0.00002, **P < 0.0003). For validation, we used MECK inhibitor (PD) to block the ERK pathway, and FGFR antagonist (SU) to inhibit FGF2 stimulation. As shown in Fig. 9, FGF2‐stimulated phosphorylation of ERK was inhibited by PD and SU at 60 min (*P < 0.0005) confirming the existence of ERK pathway in MLECs, which was activated by FGF2.
Figure 8.
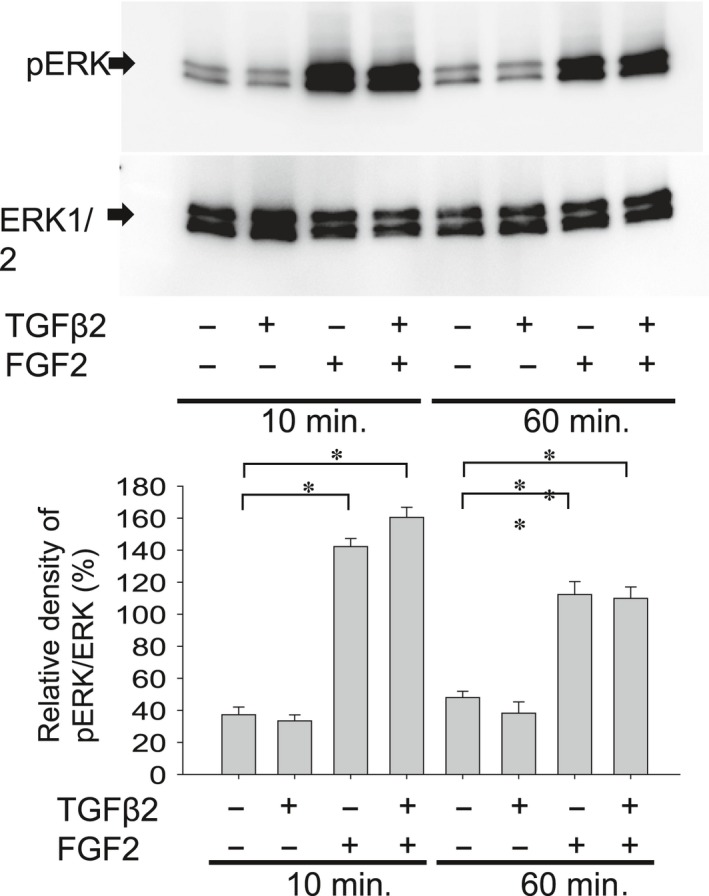
Effect of FGF2 and/or TGFβ2 stimulation on activation of ERK1/2 pathway in MLECs. To evaluate the effect of FGF2 with/without TGFβ2 stimulation on phosphorylation of ERK1/2 in MLECs, MLECs were treated with 0 or 10 ng/ml of TGFβ2 and/or FGF2 in DMEM containing 0.1% BSA for 10 or 60 min. Cell lysates were prepared, and Western blotting analysis was performed using anti‐rabbit p44/42 MAPK (Erk1/2) monoclonal Ab or anti‐phospho‐rabbit p44/42 MAPK (Erk1/2) monoclonal Ab. Data were from three experiments and were reported as means ± S.D.s.
Figure 9.
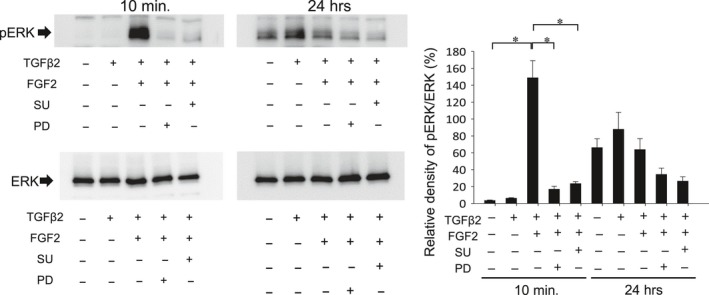
Effect of FGFR antagonist (SU) and MECK inhibitor (PD) on FGF2‐induced activation of ERK pathway. MECK inhibitor (PD) to block ERK pathway and FGFR antagonist (SU) to inhibit FGF2 stimulation were used. To evaluate the effect of FGF2 with/without TGFβ2 stimulation on phosphorylation of ERK1/2, MLECs were treated with 0 or 10 ng/ml of TGFβ2 and/or FGF2 in DMEM containing 0.1% BSA with/without SU or PD for 10 min or 24 hrs. Cell lysates were prepared, and Western blotting analysis was performed using anti‐rabbit p44/42 MAPK (Erk1/2) monoclonal Ab or anti‐phospho‐rabbit p44/42 MAPK (Erk1/2) monoclonal Ab. Data were from three experiments and were reported as means ± S.D.s.
MAPK has been reported to be involved in regulation of Tpms 64. To investigate the MAPK/ERK pathway involved in the repression of Tpm gene, we tested the effects of MECK inhibitor and FGF2 antagonist on FGF2‐induced repression of Tpm1 and αSMA expression (Fig. 10). We observed PD inhibited the repression of Tpm1 induced by FGF2 at 24 hrs of treatment (Fig. 10; *P < 0.05), and both PD and SU inhibited the repression of αSMA expression induced by FGF2 at 24 hrs of treatment (Fig. 10; *P < 0.05, **P < 0.002).
Figure 10.
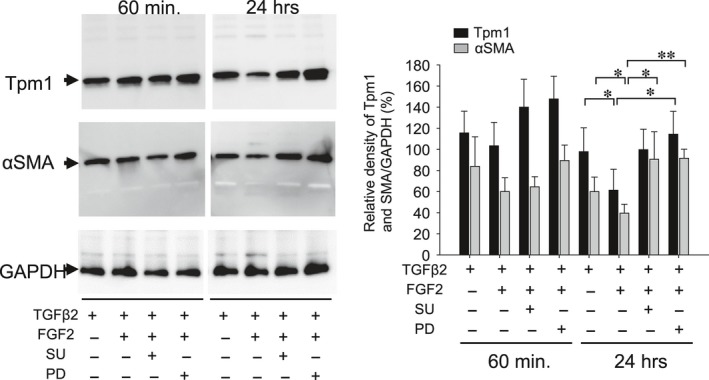
Effect of MECK inhibitor (PD) and FGF2 antagonist (SU) on repression of Tpm1 and αSMA expression in response to FGF2. Cultured MLECs were plated in 35 mm dishes at a density of 1 × 105 in DMEM with 10%FBS for 24 hrs. MLECs were treated with 0 or 10 ng/ml of TGFβ2 plus FGF2 in DMEM containing 0.1% BSA with/without SU or PD for 60 min and 24 hrs. Cell lysates were prepared, and Western blotting analysis was performed using anti‐Tpm1/2 Ab and anti αSMA Ab, with GAPDH used for control of protein concentration. Data were from three experiments and were reported as means ± S.D.s.
To determine whether MAPK/ERK pathway was involved in the FGF2 and TGFβ2‐induced cell migration, we examined the effect of PD on migration of MLECs treated with TGFβ2 and FGF2. Cell migration was induced by treatment with TGFβ2 and FGF2, and PD inhibited such migration (Fig. 11; *P < 0.02, **P < 0.05) suggesting that both factors are functionally involved in LECs’ migration.
Figure 11.
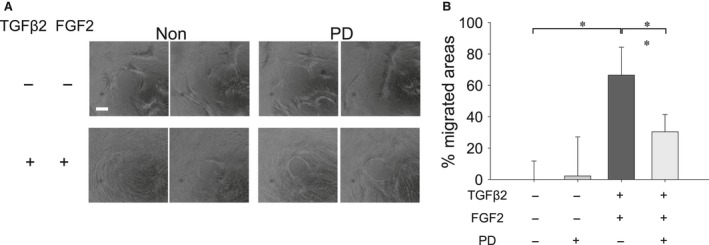
Effect of MECK inhibitor (PD) on migration of MLECs treated with TGFβ2 and FGF2. MLECs were plated in 24‐well plates, pre‐coated with collagen type I, at a density of 1 × 105 in DMEM with 10%FBS for 24 hrs. After hydrogel removal to expose the cell‐free region, MLECs were treated with 0 or 10 ng/ml of TGFβ2 plus FGF2 with/without PD in DMEM containing 0.1% BSA for 24 hrs. Phase contrast micrographs were then taken with a digital camera. Data were from three experiments and were reported as means ± S.D.s. Scale bar, 180 μm.
Discussion
This study provides evidence that Tpm1/2 plays an important role in TGFβ2‐induced EMT, stress fibre formation and cell migration in LECs. Knock‐down of Tpm1/2 by siRNA blocks the elevation of αSMA and formation of actin stress fibre. The process of EMT is implicated in cancer progression, wound healing and tissue fibrosis as well as normal embryonic development 65, 66, 67 (Thiery, 2003; Lee et al., 2006)68. In cancer, EMT leads to generation of more aggressive and invasive carcinoma cells as well as cancer stem cells 68. EMT involves disassembly of the polarized epithelial architecture and remodelling of the cell cytoskeleton, including intermediate and actin filaments. The present study revealed a dynamic process in the initiation and progression of EMT that occurs in PCO. We demonstrated that the effect of aberrant TGFβ2 signalling on LECs migration, proliferation, stress fibre and EMT is influenced by FGF2 growth factor in the cellular microenvironment. Undoubtedly, this effect of FGF2 can be associated with its concentration. Interestingly, we found that FGF2 and knock‐down of Tpm1/2 antagonize the effect of TGFβ2 on EMT features (Figs 2, 5 and 6) by repressing the expression of Tpms, αSMA and stress fibres. In contrast, this molecule synergistically acted with TGFβ2 and promoted cell migration (Fig. 7). This function of FGF2 may be related to FGF2 concentration in the cellular microenvironment. We posit that during the development of EMT, FGF2 and TGFβ2 differentially affect the EMT process in a manner linked to their differential concentrations. Tpms are actin‐stabilizing proteins that play a major role in maintaining cellular integrity. By assessing expression of Tpm1 and Tpm2 genes, in MLECs and HLECs, we found that growth and differentiation of LECs were differentially regulated by TGFβ and FGF2 according to differential expression of Tpm genes (Figs 2–5, 7). TGFβ induces epithelial to myofibroblastic transition (EMyoT) which was accompanied with Tpm and αSMA expression (a maker for EMT). Importantly, Tpms may promote the formation of stress fibres undergoing EMyoT which expresses F‐actin. However, we found FGF2 suppresses the TGFβ2‐induced up‐regulation of Tpm and αSMA (Fig. 5). Reduction of Tpm by FGF2 suppresses the formation of stress fibres and thereby activates fibroblastic LECs which induce cell migration as we observed (Figs 1 and 6). This result indicates that LECs in PCO that contain abundant FGF2 are less differentiated than those expressing predominant TGFβ2 in the cellular microenvironment.
We previously reported increased expression of Tpm1/2 during EMT and demonstrated that selective elevation of Tpm1/2 in rat LECs was correlated with fibrosis observed in PCO using in vivo rat model 55. In addition, we showed that expression of Tpm1/2 was induced/elevated in transdifferentiated multi‐layered and spindle‐shaped LECs in a rat model of PCO and human cataracts with anterior subcapsular cataract including in differentiated HLECs in a dislocated lens capsule 55. Data from these previous results strongly suggested that expression of Tpm1/2 is linked with progression of PCO 55. Several TGF‐β target genes, including Tpm1 and Tpm2, have been implicated in the assembly of stress fibres 51, 53. Of these, Tpms in particular have been shown to play a crucial role in stabilizing actin filaments 54. Furthermore, we have also reported up‐regulation of Tpm1/2 expression in differentiating LECs (in the presence of TGFβ), demonstrating involvement of TGFβ‐induced deleterious signalling in the induction of Tpm1/2. In the past, several in vitro and in vivo studies have examined the role of TGFβ2 in EMT and wound healing processes using LECs 5, 6, 69. Transdifferentiation analogous to that observed in PCO can be induced by exposing LECs to TGFβ 5, 6, 70, 71.
Moreover, a major contribution of FGF2 in PCO development has been reported 1, 27, 72. FGF2 is a potent mitogenic growth factor which is present in the eye lens environment 27, and concentration of FGF2 may increase after cataract surgery 72. For instance, Wallentin et al. 73 showed that aqueous FGF2 was increased in rabbits up to 30 days following cataract extraction. Furthermore, in a study using a rabbit PCO model, the level of active TGFβ decreased and increased FGF2 stimulated cell proliferation, immediately after surgery, and at around 2 weeks after surgery, active TGFβ returned to normal level stimulating EMT 35. TGFβ2 has been shown to inhibit the proliferative effect of FGF2 on rabbit LECs growth 73. FGF2 reduced the contraction of a collagen gel by bovine LECs and the proportion of cells expressing αSMA 29, indicating that FGF2 has opposing actions to TGFβ2. Furthermore, simultaneous treatment of rat LECs explants with TGFβ2 and FGF2 were more affected by TGFβ2 than corresponding high cell coverage explants, showing greater cell loss, more marked formation of spindle cells and expression of αSMA 71. Excessive deposition of ECM and formation of plaques of swollen cells, also features of PCO 1, 71, occurred only when TGFβ was supplemented with FGF2 70, 71.
Our study found cotreatment with TGFβ2‐ and FGF2‐induced spindle‐shaped fibroblastic formation of HLECs and accelerated the EMT process. Furthermore, we found that TGFβ2 and FGF2 in combination suppressed the formation of stress fibres in LECs and the expression of Tpm and αSMA. Importantly, Tpm1/2 knock‐down inhibited the TGFβ2‐induced formation of stress fibres. Tpms have been shown to stabilize actin filaments 54. Thus, our study revealed that Tpms may induce stress fibre in response to TGFβ2 in the EMT process.
We found that TGFβ2 induced differentiation of LECs into two types of mesenchymal cells: one was Tpm1‐ and αSMA‐positive myofibroblastic cells generated through EMyoT by TGFβ2 alone, and the other was activated Tpm1‐ and αSMA‐negative fibroblastic cells generated through EMT by both TGFβ2 and FGF2 in combination. Furthermore, our result also indicated that TGFβ2 enhanced cell migration compared with non‐treated cells, and the co‐addition of FGF2 further promoted cell migration (Fig. 7). Based on these data, we believe that Tpm abundance may inhibit cell migration and reduced level of Tpm may induce cell migration as we showed that FGF2 markedly suppressed the expression of Tpms in LECs.
Moreover, FGF has been shown to be a major activator of the ERK‐MAPK pathway in the eye lens in vivo 74, 75. Further, FGF2 activation of ERK1/2 signalling could influence TGFβ2 induction of Tpm1 gene expression. The present study showed the inducible expression of Tpm1 was attenuated in the presence of PD, a specific inhibitor of MAPK/ERK signalling, and SU, an inhibitor of FGFR2. Also, cell migration induced by TGFβ2 and FGF2 was inhibited in the presence of PD. These data support the notion that ERK signalling affects cell migration and the formation of stress fibres by suppressing the expression of Tpm.
In summary, we demonstrated both independent and combined roles of TGFβ2 and FGF2 in the differential regulation of EMT and how this process is associated with Tpm expression including the implications for PCO development. Importantly, we found that FGF2 acts as an antagonist against TGFβ‐mediated EMT progression. We demonstrated that FGF2‐induced ERK signalling attenuates aberrant expression of Tpm1 and cell migration. Our results provide a novel insight into the regulation and function of Tpms during LEC differentiation, which is influenced profoundly by growth factors. These findings reveal that opposing effects of FGF2 and TGFβ2 on Tpm1/2 gene expression control the phenotypic plasticity of LECs on PCO progression. Further, Tpm1/2 may regulate other eye diseases such as pterygium and glaucoma and wound healing processes related to EMT.
We hope this study provides clues to develop new therapies of PCO and other EMT‐related diseases targeting the balance regulation of Tpms by growth factors. However, further in‐depth studies will be required to fully clarify the underlying mechanism of Tpm1/2 involvement in EMT process.
Conflict of interest
This study was partly funded by Ono Pharmaceutical Co. Ltd (Osaka Japan).
Acknowledgements
This work was supported by grants from Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Numbers JP23592588 (to E.K.), National Eye Institute, National Institute of Health (NIH) (EY024589) (to D.P.S.) and Research for Preventing Blindness (to D.P.S.). Their support is gratefully acknowledged.
References
- 1. Apple DJ, Solomon KD, Tetz MR, et al Posterior capsule opacification. Surv Ophthalmol. 1992; 37: 73–116. [DOI] [PubMed] [Google Scholar]
- 2. Awasthi N, Guo S, Wagner BJ. Posterior capsular opacification: a problem reduced but not yet eradicated. Arch Ophthalmol. 2009; 127: 555–62. [DOI] [PubMed] [Google Scholar]
- 3. McDonnell PJ, Rowen SL, Glaser BM, et al Posterior capsule opacification. An in vitro model. Arch Ophthalmol. 1985; 103: 1378–81. [DOI] [PubMed] [Google Scholar]
- 4. Lovicu FJ, McAvoy JW. Structural analysis of lens epithelial explants induced to differentiate into fibres by fibroblast growth factor (FGF). Exp Eye Res. 1989; 49: 479–94. [DOI] [PubMed] [Google Scholar]
- 5. Wormstone IM. Posterior capsule opacification: a cell biological perspective. Exp Eye Res. 2002; 74: 337–47. [DOI] [PubMed] [Google Scholar]
- 6. Saika S. Relationship between posterior capsule opacification and intraocular lens biocompatibility. Prog Retin Eye Res. 2004; 23: 283–305. [DOI] [PubMed] [Google Scholar]
- 7. Marcantonio JM, Syam PP, Liu CS, et al Epithelial transdifferentiation and cataract in the human lens. Exp Eye Res. 2003; 77: 339–46. [DOI] [PubMed] [Google Scholar]
- 8. Kim KW, Park SH, Kim JC. Fibroblast biology in pterygia. Exp Eye Res. 2016; 142: 32–9. [DOI] [PubMed] [Google Scholar]
- 9. Vranka JA, Kelley MJ, Acott TS, et al Extracellular matrix in the trabecular meshwork: intraocular pressure regulation and dysregulation in glaucoma. Exp Eye Res. 2015; 133: 112–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kowtharapu BS, Stahnke T, Wree A, et al Corneal epithelial and neuronal interactions: role in wound healing. Exp Eye Res. 2014; 125: 53–61. [DOI] [PubMed] [Google Scholar]
- 11. Luo Y, Lu Y, Lu G, et al Primary posterior capsulorhexis with anterior vitrectomy in preventing posterior capsule opacification in pediatric cataract microsurgery. Microsurgery. 2008; 28: 113–6. [DOI] [PubMed] [Google Scholar]
- 12. BenEzra D, Cohen E. Posterior capsulectomy in pediatric cataract surgery: the necessity of a choice. Ophthalmology. 1997; 104: 2168–74. [DOI] [PubMed] [Google Scholar]
- 13. Nibourg LM, Sharma PK, van Kooten TG, et al Changes in lens stiffness due to capsular opacification in accommodative lens refilling. Exp Eye Res. 2015; 134: 148–54. [DOI] [PubMed] [Google Scholar]
- 14. de Iongh RU, Lovicu FJ, Overbeek PA, et al Requirement for TGFbeta receptor signaling during terminal lens fiber differentiation. Development. 2001; 128: 3995–4010. [DOI] [PubMed] [Google Scholar]
- 15. de Iongh RU, Wederell E, Lovicu FJ, et al Transforming growth factor‐beta‐induced epithelial‐mesenchymal transition in the lens: a model for cataract formation. Cells, tissues, organs. 2005; 179: 43–55. [DOI] [PubMed] [Google Scholar]
- 16. Lovicu FJ, Schulz MW, Hales AM, et al TGFbeta induces morphological and molecular changes similar to human anterior subcapsular cataract. Br J Ophthalmol. 2002; 86: 220–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee EH, Seomun Y, Hwang KH, et al Overexpression of the transforming growth factor‐beta‐inducible gene betaig‐h3 in anterior polar cataracts. Invest Ophthalmol Vis Sci. 2000; 41: 1840–5. [PubMed] [Google Scholar]
- 18. Srinivasan Y, Lovicu FJ, Overbeek PA. Lens‐specific expression of transforming growth factor beta1 in transgenic mice causes anterior subcapsular cataracts. J Clin Invest. 1998; 101: 625–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Medvedovic M, Tomlinson CR, Call MK, et al Gene expression and discovery during lens regeneration in mouse: regulation of epithelial to mesenchymal transition and lens differentiation. Mol Vis. 2006; 12: 422–40. [PubMed] [Google Scholar]
- 20. Saika S, Miyamoto T, Ishida I, et al TGFbeta‐Smad signalling in postoperative human lens epithelial cells. Br J Ophthalmol. 2002; 86: 1428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Border WA, Noble NA, Yamamoto T, et al Natural inhibitor of transforming growth factor‐beta protects against scarring in experimental kidney disease. Nature. 1992; 360: 361–4. [DOI] [PubMed] [Google Scholar]
- 22. Seomun Y, Kim J, Lee EH, et al Overexpression of matrix metalloproteinase‐2 mediates phenotypic transformation of lens epithelial cells. Biochem J. 2001; 358: 41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mincione G, Esposito DL, Di Marcantonio MC, et al TGF‐beta 1 modulation of IGF‐I signaling pathway in rat thyroid epithelial cells. Exp Cell Res. 2003; 287: 411–23. [DOI] [PubMed] [Google Scholar]
- 24. Hayashi N, Kato H, Kiyosawa T, et al [The change in immunohistochemical localization of basic fibroblast growth factor (b‐FGF) around the lens capsule after extracapsular extraction]. Nihon Ganka Gakkai Zasshi. 1991; 95: 621–4. [PubMed] [Google Scholar]
- 25. Lovicu FJ, McAvoy JW. Localization of acidic fibroblast growth factor, basic fibroblast growth factor, and heparan sulphate proteoglycan in rat lens: implications for lens polarity and growth patterns. Invest Ophthalmol Vis Sci. 1993; 34: 3355–65. [PubMed] [Google Scholar]
- 26. McAvoy JW, Chamberlain CG. Fibroblast growth factor (FGF) induces different responses in lens epithelial cells depending on its concentration. Development. 1989; 107: 221–8. [DOI] [PubMed] [Google Scholar]
- 27. Chamberlain CG, McAvoy JW. Induction of lens fibre differentiation by acidic and basic fibroblast growth factor (FGF). Growth Factors. 1989; 1: 125–34. [DOI] [PubMed] [Google Scholar]
- 28. Nishi O, Nishi K, Fujiwara T, et al Effects of the cytokines on the proliferation of and collagen synthesis by human cataract lens epithelial cells. Br J Ophthalmol. 1996; 80: 63–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kurosaka D, Kato K, Nagamoto T, et al Growth factors influence contractility and alpha‐smooth muscle actin expression in bovine lens epithelial cells. Invest Ophthalmol Vis Sci. 1995; 36: 1701–8. [PubMed] [Google Scholar]
- 30. Nishi O, Hara T, Hara T, et al Refilling the lens with a inflatable endocapsular balloon: surgical procedure in animal eyes. Graefes Arch Clin Exp Ophthalmol. 1992; 230: 47–55. [DOI] [PubMed] [Google Scholar]
- 31. McAvoy JW, Chamberlain CG, de Iongh RU, et al Peter Bishop Lecture: growth factors in lens development and cataract: key roles for fibroblast growth factor and TGF‐beta. Clin Experiment Ophthalmol. 2000; 28: 133–9. [DOI] [PubMed] [Google Scholar]
- 32. Pagot V, Gazagne C, Galiana A, et al [Extracapsular cataract extraction and implantation in the capsular sac during vitrectomy in diabetics]. J Fr Ophtalmol. 1991; 14: 523–8. [PubMed] [Google Scholar]
- 33. Mamuya FA, Wang Y, Roop VH, et al The roles of alphaV integrins in lens EMT and posterior capsular opacification. J Cell Mol Med. 2014; 18: 656–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Serini G, Bochaton‐Piallat ML, Ropraz P, et al The fibronectin domain ED‐A is crucial for myofibroblastic phenotype induction by transforming growth factor‐beta1. J Cell Biol. 1998; 142: 873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meacock WR, Spalton DJ, Stanford MR. Role of cytokines in the pathogenesis of posterior capsule opacification. Br J Ophthalmol. 2000; 84: 332–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhargavan B, Chhunchha B, Fatma N, et al Epigenetic repression of LEDGF during UVB exposure by recruitment of SUV39H1 and HDAC1 to the Sp1‐responsive elements within LEDGF promoter CpG island. Epigenetics. 2013; 8: 268–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF‐beta‐mediated fibrogenesis. Free Radic Biol Med. 2010; 48: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fatma N, Kubo E, Sen M, et al Peroxiredoxin 6 delivery attenuates TNF‐alpha‐and glutamate‐induced retinal ganglion cell death by limiting ROS levels and maintaining Ca2 + homeostasis. Brain Res. 2008; 1233: 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fatma N, Kubo E, Sharma P, et al Impaired homeostasis and phenotypic abnormalities in Prdx6‐/‐mice lens epithelial cells by reactive oxygen species: increased expression and activation of TGFbeta. Cell Death Differ. 2005; 12: 734–50. [DOI] [PubMed] [Google Scholar]
- 40. Tripathi BJ, Tripathi RC, Livingston AM, et al The role of growth factors in the embryogenesis and differentiation of the eye. Am J Anat. 1991; 192: 442–71. [DOI] [PubMed] [Google Scholar]
- 41. Kubo E, Fatma N, Akagi Y, et al TAT‐mediated PRDX6 protein transduction protects against eye lens epithelial cell death and delays lens opacity. Am J Physiol Cell Physiol. 2008; 294: C842–55. [DOI] [PubMed] [Google Scholar]
- 42. Manevich Y, Fisher AB. Peroxiredoxin 6, a 1‐Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic Biol Med. 2005; 38: 1422–32. [DOI] [PubMed] [Google Scholar]
- 43. Wood ZA, Schroder E Robin Harris J, et al Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003; 28: 32–40. [DOI] [PubMed] [Google Scholar]
- 44. Kubo E, Hasanova N, Tanaka Y, et al Protein Expression Profiling of Lens Epithelial Cells from Prdx6‐depleted Mice and Their Vulnerability to UV Radiation Exposure. Am J Physiol Cell Physiol. 2010; 298: 342–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gunning P, O'Neill G, Hardeman E. Tropomyosin‐based regulation of the actin cytoskeleton in time and space. Physiol Rev. 2008; 88: 1–35. [DOI] [PubMed] [Google Scholar]
- 46. Gunning PW, Schevzov G, Kee AJ, et al Tropomyosin isoforms: divining rods for actin cytoskeleton function. Trends Cell Biol. 2005; 15: 333–41. [DOI] [PubMed] [Google Scholar]
- 47. Schevzov G, Whittaker SP, Fath T, et al Tropomyosin isoforms and reagents. Bioarchitecture. 2011; 1: 135–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Geeves MA, Hitchcock‐DeGregori SE, Gunning PW. A systematic nomenclature for mammalian tropomyosin isoforms. J Muscle Res Cell Motil. 2015; 36: 147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wawro B, Greenfield NJ, Wear MA, et al Tropomyosin regulates elongation by formin at the fast‐growing end of the actin filament. Biochemistry. 2007; 46: 8146–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Varga AE, Stourman NV, Zheng Q, et al Silencing of the Tropomyosin‐1 gene by DNA methylation alters tumor suppressor function of TGF‐beta. Oncogene. 2005; 24: 5043–52. [DOI] [PubMed] [Google Scholar]
- 51. Bakin AV, Safina A, Rinehart C, et al A critical role of tropomyosins in TGF‐beta regulation of the actin cytoskeleton and cell motility in epithelial cells. Mol Biol Cell. 2004; 15: 4682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee A, Fischer RS, Fowler VM. Stabilization and remodeling of the membrane skeleton during lens fiber cell differentiation and maturation. Dev Dyn. 2000; 217: 257–70. [DOI] [PubMed] [Google Scholar]
- 53. Zheng Q, Safina A, Bakin AV. Role of high‐molecular weight tropomyosins in TGF‐beta‐mediated control of cell motility. Int J Cancer. 2008; 122: 78–90. [DOI] [PubMed] [Google Scholar]
- 54. Pawlak G, Helfman DM. Cytoskeletal changes in cell transformation and tumorigenesis. Curr Opin Genet Dev. 2001; 11: 41–7. [DOI] [PubMed] [Google Scholar]
- 55. Kubo E, Hasanova N, Fatma N, et al Elevated tropomyosin expression is associated with epithelial‐mesenchymal transition of lens epithelial cells. J Cell Mol Med. 2013; 17: 212–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Singh DP, Ohguro N, Chylack LT Jr, et al Lens epithelium‐derived growth factor: increased resistance to thermal and oxidative stresses. Invest Ophthalmol Vis Sci. 1999; 40: 1444–51. [PubMed] [Google Scholar]
- 57. Kubo E, Miyazawa T, Fatma N, et al Development‐ and age‐associated expression pattern of peroxiredoxin 6, and its regulation in murine ocular lens. Mech Ageing Dev. 2006; 127: 249–56. [DOI] [PubMed] [Google Scholar]
- 58. Kubo E, Singh DP, Fatma N, et al Cellular distribution of lens epithelium‐derived growth factor (LEDGF) in the rat eye: loss of LEDGF from nuclei of differentiating cells. Histochem Cell Biol. 2003; 119: 289–99. [DOI] [PubMed] [Google Scholar]
- 59. Wormstone IM, Tamiya S, Anderson I, et al TGF‐beta2‐induced matrix modification and cell transdifferentiation in the human lens capsular bag. Invest Ophthalmol Vis Sci. 2002; 43: 2301–8. [PubMed] [Google Scholar]
- 60. Jaffe AB, Hall A. Rho GTPases in transformation and metastasis. Adv Cancer Res. 2002; 84: 57–80. [DOI] [PubMed] [Google Scholar]
- 61. Bakin AV, Rinehart C, Tomlinson AK, et al p38 mitogen‐activated protein kinase is required for TGFbeta‐mediated fibroblastic transdifferentiation and cell migration. J Cell Sci. 2002; 115: 3193–206. [DOI] [PubMed] [Google Scholar]
- 62. Edlund S, Landstrom M, Heldin CH, et al Transforming growth factor‐beta‐induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell. 2002; 13: 902–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tojkander S, Gateva G, Lappalainen P. Actin stress fibers–assembly, dynamics and biological roles. J Cell Sci. 2012; 125: 1855–64. [DOI] [PubMed] [Google Scholar]
- 64. Shields JM, Mehta H, Pruitt K, et al Opposing roles of the extracellular signal‐regulated kinase and p38 mitogen‐activated protein kinase cascades in Ras‐mediated downregulation of tropomyosin. Mol Cell Biol. 2002; 22: 2304–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kalluri R, Neilson EG. Epithelial‐mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003; 112: 1776–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thiery JP. Epithelial‐mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003; 15: 740–6. [DOI] [PubMed] [Google Scholar]
- 67. Lee JM, Dedhar S, Kalluri R, et al The epithelial‐mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006; 172: 973–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gervasi M, Bianchi‐Smiraglia A, Cummings M, et al JunB contributes to Id2 repression and the epithelial‐mesenchymal transition in response to transforming growth factor‐beta. J Cell Biol. 2012; 196: 589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Davidson MG, Wormstone M, Morgan D, et al Ex vivo canine lens capsular sac explants. Graefes Arch Clin Exp Ophthalmol. 2000; 238: 708–14. [DOI] [PubMed] [Google Scholar]
- 70. Mansfield KJ, Cerra A, Chamberlain CG. FGF‐2 counteracts loss of TGFbeta affected cells from rat lens explants: implications for PCO (after cataract). Mol Vis. 2004; 10: 521–32. [PubMed] [Google Scholar]
- 71. Symonds JG, Lovicu FJ, Chamberlain CG. Posterior capsule opacification‐like changes in rat lens explants cultured with TGFbeta and FGF: effects of cell coverage and regional differences. Exp Eye Res. 2006; 82: 693–9. [DOI] [PubMed] [Google Scholar]
- 72. Wormstone IM, Del Rio‐Tsonis K, McMahon G, et al FGF: an autocrine regulator of human lens cell growth independent of added stimuli. Invest Ophthalmol Vis Sci. 2001; 42: 1305–11. [PubMed] [Google Scholar]
- 73. Wallentin N, Wickstrom K, Lundberg C. Effect of cataract surgery on aqueous TGF‐beta and lens epithelial cell proliferation. Invest Ophthalmol Vis Sci. 1998; 39: 1410–8. [PubMed] [Google Scholar]
- 74. Govindarajan V, Overbeek PA. Secreted FGFR3, but not FGFR1, inhibits lens fiber differentiation. Development. 2001; 128: 1617–27. [DOI] [PubMed] [Google Scholar]
- 75. Zhao H, Yang T, Madakashira BP, et al Fibroblast growth factor receptor signaling is essential for lens fiber cell differentiation. Dev Biol. 2008; 318: 276–88. [DOI] [PMC free article] [PubMed] [Google Scholar]